
Role of Transcription Factors in Steatohepatitis and Hypertension after Ethanol: The Epicenter of Metabolism



{kind=link}
Abstract
:1. Introduction
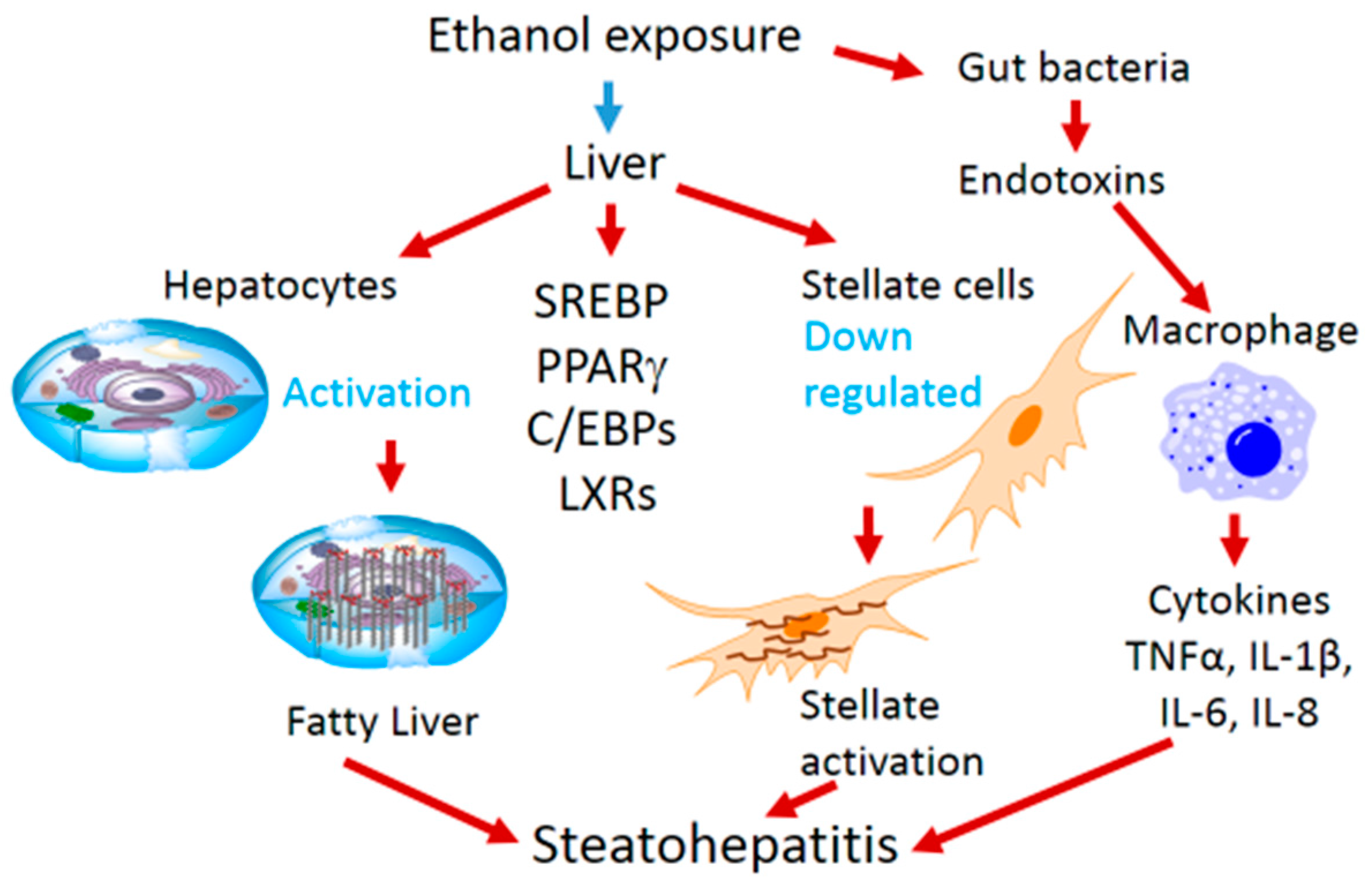
2. Ethanol-Mediated Hepatosteatosis, Steatohepatitis and Role of Transcription Factors
3. Ethanol Mediated Effects on Transcription Factors
4. Ethanol-Mediated Oxidative Stress and Cellular Protection
Chronic Alcohol Intake and Hypertension
5. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McGovern, P.E. Ancient Wine: The Search for Origins of Viniculture; Princeton University Press: Princeton, NJ, USA, 2003; pp. 314–315. [Google Scholar]
- Dietler, M. Alcohol: Anthropological/archaeological perspectives. Annu. Rev. Anthropol. 2006, 35, 229–249. [Google Scholar] [CrossRef]
- Krenz, M.; Korthuis, R.J. Moderate ethanol ingestion and cardiovascular protection: From epidemiologic associations to cellular mechanisms. J. Mol. Cell. Cardiol. 2012, 52, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Miyamae, M.; Kaneda, K.; Domae, N.; Figueredo, V.M. Cardioprotection by regular ethanol consumption: Potential mechanisms and clinical application. Curr. Drug Abuse Rev. 2010, 3, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Hanson, D.J. Preventing Alcohol Abuse: Alcohol, Culture and Control; Greenwood Publishing Group: Westport, CT, USA, 1995. [Google Scholar]
- Husain, K.; Mejia, J.; Lalla, J.; Kazim, S. Dose response of alcohol-induced changes in BP, nitric oxide and antioxidants in rat plasma. Pharmacol. Res. 2005, 51, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Li, T.K.; Hewitt, B.G.; Grant, B.F. Alcohol use disorders and mood disorders: A national institute on alcohol abuse and alcoholism perspective. Biol. Psychiatry 2004, 56, 718–720. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, J.M.; Foege, W.H. Actual causes of death in the United States. JAMA 1993, 270, 2207–2212. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Hepatic and other medical disorders of alcoholism: From pathogenesis to treatment. J. Stud. Alcohol. 1998, 59, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Husain, K.; Ansari, R.A.; Ferder, L. Alcohol-induced hypertension: Mechanism and prevention. World J. Cardiol. 2014, 6, 245–252. [Google Scholar] [PubMed]
- Zhou, Z.; Wang, L.; Song, Z.; Lambert, J.C.; McClain, C.J.; Kang, Y.J. A critical involvement of oxidative stress in acute alcohol-induced hepatic TNF-alpha production. Am. J. Pathol. 2003, 163, 1137–1146. [Google Scholar] [CrossRef]
- Lieber, C.S.; Savolainen, M. Ethanol and lipids. Alcohol. Clin. Exp. Res. 1984, 8, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, M.P.; Jimenez-Lopez, J.M.; Segovia, J.L.; Marco, C. Comparative study of the effects of short- and long-term ethanol treatment and alcohol withdrawal on phospholipid biosynthesis in rat hepatocytes. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2002, 131, 491–497. [Google Scholar] [CrossRef]
- Carrasco, M.P.; Marco, C.; Segovia, J.L. Chronic ingestion of ethanol stimulates lipogenic response in rat hepatocytes. Life Sci. 2001, 68, 1295–1304. [Google Scholar] [CrossRef]
- Shimano, H. SREBPs: Physiology and pathophysiology of the SREBP family. FEBS J. 2009, 276, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Eberle, D.; Hegarty, B.; Bossard, P.; Ferre, P.; Foufelle, F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Shimano, H. Sterol regulatory element-binding proteins (SREBPs): Transcriptional regulators of lipid synthetic genes. Prog. Lipid Res. 2001, 40, 439–452. [Google Scholar] [CrossRef]
- Lluis, J.M.; Colell, A.; Garcia-Ruiz, C.; Kaplowitz, N.; Fernandez-Checa, J.C. Acetaldehyde impairs mitochondrial glutathione transport in HepG2 cells through endoplasmic reticulum stress. Gastroenterology 2003, 124, 708–724. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Fischer, M.; Deeg, M.A.; Crabb, D.W. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J. Biol. Chem. 2002, 277, 29342–29347. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Crabb, D.W. Molecular mechanisms of alcoholic fatty liver: Role of sterol regulatory element-binding proteins. Alcohol 2004, 34, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Kaplowitz, N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology 2003, 124, 1488–1499. [Google Scholar] [CrossRef]
- Grygiel-Gorniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014. [Google Scholar] [CrossRef] [PubMed]
- Galli, A.; Pinaire, J.; Fischer, M.; Dorris, R.; Crabb, D.W. The transcriptional and DNA binding activity of peroxisome proliferator-activated receptor alpha is inhibited by ethanol metabolism. A novel mechanism for the development of ethanol-induced fatty liver. J. Biol. Chem. 2001, 276, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; You, M.; Matsumoto, M.; Crabb, D.W. Peroxisome proliferator-activated receptor alpha (PPARalpha) agonist treatment reverses PPARalpha dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice. J. Biol. Chem. 2003, 278, 27997–28004. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Crabb, D.W. Recent advances in alcoholic liver disease II. Minireview: Molecular mechanisms of alcoholic fatty liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1–G6. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Puig, A.J.; Considine, R.V.; Jimenez-Linan, M.; Werman, A.; Pories, W.J.; Caro, J.F.; Flier, J.S. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J. Clin. Investig. 1997, 99, 2416–2422. [Google Scholar] [CrossRef] [PubMed]
- Fajas, L.; Schoonjans, K.; Gelman, L.; Kim, J.B.; Najib, J.; Martin, G.; Fruchart, J.C.; Briggs, M.; Spiegelman, B.M.; Auwerx, J. Regulation of peroxisome proliferator-activated receptor gamma expression by adipocyte differentiation and determination factor 1/sterol regulatory element binding protein 1: Implications for adipocyte differentiation and metabolism. Mol. Cell. Biol. 1999, 19, 5495–5503. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Wright, H.M.; Wright, M.; Spiegelman, B.M. ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proc. Natl. Acad. Sci. USA 1998, 95, 4333–4337. [Google Scholar] [CrossRef] [PubMed]
- Matsusue, K.; Haluzik, M.; Lambert, G.; Yim, S.H.; Gavrilova, O.; Ward, J.M.; Brewer, B., Jr.; Reitman, M.L.; Gonzalez, F.J. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J. Clin. Investig. 2003, 111, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Matsusue, K.; Kashireddy, P.; Cao, W.Q.; Yeldandi, V.; Yeldandi, A.V.; Rao, M.S.; Gonzalez, F.J.; Reddy, J.K. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor gamma1 (PPARgamma1) overexpression. J. Biol. Chem. 2003, 278, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Schadinger, S.E.; Bucher, N.L.; Schreiber, B.M.; Farmer, S.R. PPARgamma2 regulates lipogenesis and lipid accumulation in steatotic hepatocytes. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1195–E1205. [Google Scholar] [CrossRef] [PubMed]
- Gavrilova, O.; Haluzik, M.; Matsusue, K.; Cutson, J.J.; Johnson, L.; Dietz, K.R.; Nicol, C.J.; Vinson, C.; Gonzalez, F.J.; Reitman, M.L. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem. 2003, 278, 34268–34276. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Spiegelman, B.M. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev. 1996, 10, 1096–1107. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Hsu, C.H.; Wang, X.; Sakai, S.; Freeman, M.W.; Gonzalez, F.J.; Spiegelman, B.M. C/EBPalpha induces adipogenesis through PPARgamma: A unified pathway. Genes Dev. 2002, 16, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; She, H.; Hazra, S.; Cheng, J.; Wang, J. Fat paradox of steatohepatitis. J. Gastroenterol. Hepatol. 2008, 23, S104–S107. [Google Scholar] [CrossRef] [PubMed]
- Martin, H. Role of PPAR-gamma in inflammation. Prospects for therapeutic intervention by food components. Mutat. Res. 2010, 690, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Onishi, Y.; Honda, M.; Ogihara, T.; Sakoda, H.; Anai, M.; Fujishiro, M.; Ono, H.; Shojima, N.; Fukushima, Y.; Inukai, K.; et al. Ethanol feeding induces insulin resistance with enhanced PI 3-kinase activation. Biochem. Biophys. Res. Commun. 2003, 303, 788–794. [Google Scholar] [CrossRef]
- Enomoto, N.; Takei, Y.; Hirose, M.; Konno, A.; Shibuya, T.; Matsuyama, S.; Suzuki, S.; Kitamura, K.I.; Sato, N. Prevention of ethanol-induced liver injury in rats by an agonist of peroxisome proliferator-activated receptor-gamma, pioglitazone. J. Pharmacol. Exp. Ther. 2003, 306, 846–854. [Google Scholar] [CrossRef] [PubMed]
- McClain, C.J.; Cohen, D.A. Increased tumor necrosis factor production by monocytes in alcoholic hepatitis. Hepatology 1989, 9, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, P.; Szabo, G. Signalling pathways in alcohol-induced liver inflammation. J. Hepatol. 2009, 50, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Khoruts, A.; Stahnke, L.; McClain, C.J.; Logan, G.; Allen, J.I. Circulating tumor necrosis factor, interleukin-1 and interleukin-6 concentrations in chronic alcoholic patients. Hepatology 1991, 13, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.I.; Park, O.; Gao, B. Abrogation of the antifibrotic effects of natural killer cells/interferon-gamma contributes to alcohol acceleration of liver fibrosis. Gastroenterology 2008, 134, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Wang, Y.; Keshaw, H.; Xu, L.Y.; Lam, K.S.; Cooper, G.J. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J. Clin. Investig. 2003, 112, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colgan, S.M.; Tang, D.; Werstuck, G.H.; Austin, R.C. Endoplasmic reticulum stress causes the activation of sterol regulatory element binding protein-2. Int. J. Biochem. Cell Biol. 2007, 39, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Ji, C. Mechanisms of alcohol-induced endoplasmic reticulum stress and organ injuries. Biochem. Res. Int. 2012. [Google Scholar] [CrossRef] [PubMed]
- Howarth, D.L.; Vacaru, A.M.; Tsedensodnom, O.; Mormone, E.; Nieto, N.; Costantini, L.M.; Snapp, E.L.; Sadler, K.C. Alcohol disrupts endoplasmic reticulum function and protein secretion in hepatocytes. Alcohol. Clin. Exp. Res. 2012, 36, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.E. Molecular aspects of alcohol metabolism: Transcription factors involved in early ethanol-induced liver injury. Annu. Rev. Nutr. 2004, 24, 55–78. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Wang, F.; Li, X.; Rogers, C.Q.; Liang, X.; Finck, B.N.; You, M. Regulation of hepatic lipin-1 by ethanol: Role of AMP-activated protein kinase/sterol regulatory element-binding protein 1 signaling in mice. Hepatology 2012, 55, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Lawler, J.F., Jr.; Yin, M.; Diehl, A.M.; Roberts, E.; Chatterjee, S. Tumor necrosis factor-alpha stimulates the maturation of sterol regulatory element binding protein-1 in human hepatocytes through the action of neutral sphingomyelinase. J. Biol. Chem. 1998, 273, 5053–5059. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Takase, I.; Hakucho, A.; Okamura, N.; Fujimiya, T. Carvedilol attenuates the progression of alcohol fatty liver disease in rats. Alcohol. Clin. Exp. Res. 2012, 36, 1587–1599. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Dudenhausen, E.E.; Pan, Y.X.; Zhong, C.; Kilberg, M.S. Human CCAAT/enhancer-binding protein beta gene expression is activated by endoplasmic reticulum stress through an unfolded protein response element downstream of the protein coding sequence. J. Biol. Chem. 2004, 279, 27948–27956. [Google Scholar] [CrossRef] [PubMed]
- Poli, V. The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J. Biol. Chem. 1998, 273, 29279–29282. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Ronis, M.J.; Badger, T.M. Ethanol induction of class I alcohol dehydrogenase expression in the rat occurs through alterations in CCAAT/enhancer binding proteins beta and gamma. J. Biol. Chem. 2002, 277, 43572–43577. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N.; Dominitz, J.A.; Weiss, N.S.; Heagerty, P.J.; Kowdley, K.V. The effect of alcohol consumption on the prevalence of iron overload, iron deficiency, and iron deficiency anemia. Gastroenterology 2004, 126, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Lin, M.; Ohata, M.; Giulivi, C.; French, S.W.; Brittenham, G. Iron primes hepatic macrophages for NF-kappaB activation in alcoholic liver injury. Am. J. Physiol. 1999, 277, G1240–G1250. [Google Scholar] [PubMed]
- Xiong, S.; She, H.; Sung, C.K.; Tsukamoto, H. Iron-dependent activation of NF-kappaB in Kupffer cells: A priming mechanism for alcoholic liver disease. Alcohol 2003, 30, 107–113. [Google Scholar] [CrossRef]
- She, H.; Xiong, S.; Lin, M.; Zandi, E.; Giulivi, C.; Tsukamoto, H. Iron activates NF-kappaB in Kupffer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G719–G726. [Google Scholar] [CrossRef] [PubMed]
- Feierman, D.E.; Winston, G.W.; Cederbaum, A.I. Ethanol oxidation by hydroxyl radicals: Role of iron chelates, superoxide, and hydrogen peroxide. Alcohol. Clin. Exp. Res. 1985, 9, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Heritage, M.L.; Murphy, T.L.; Bridle, K.R.; Anderson, G.J.; Crawford, D.H.; Fletcher, L.M. Hepcidin regulation in wild-type and Hfe knockout mice in response to alcohol consumption: Evidence for an alcohol-induced hypoxic response. Alcohol. Clin. Exp. Res. 2009, 33, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol Res. Health 2006, 29, 245–254. [Google Scholar] [PubMed]
- Aaltonen, T.; Abazov, V.M.; Abbott, B.; Acharya, B.S.; Adams, M.; Adams, T.; Agnew, J.P.; Alexeev, G.D.; Alkhazov, G.; Alton, A.; et al. Observation of s-channel production of single top quarks at the tevatron. Phys. Rev. Lett. 2014. [Google Scholar] [CrossRef] [PubMed]
- Arteel, G.E.; Iimuro, Y.; Yin, M.; Raleigh, J.A.; Thurman, R.G. Chronic enteral ethanol treatment causes hypoxia in rat liver tissue in vivo. Hepatology 1997, 25, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Zelickson, B.R.; Benavides, G.A.; Johnson, M.S.; Chacko, B.K.; Venkatraman, A.; Landar, A.; Betancourt, A.M.; Bailey, S.M.; Darley-Usmar, V.M. Nitric oxide and hypoxia exacerbate alcohol-induced mitochondrial dysfunction in hepatocytes. Biochim. Biophys. Acta 2011, 1807, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Aaltonen, T.; Amerio, S.; Amidei, D.; Anastassov, A.; Annovi, A.; Antos, J.; Apollinari, G.; Appel, J.A.; Arisawa, T.; Artikov, A.; et al. Search for s-channel single-top-quark production in events with missing energy plus jets in pp collisions at sqrt[s]=1.96 TeV. Phys. Rev. Lett. 2014. [Google Scholar] [CrossRef]
- Arteel, G.E.; Raleigh, J.A.; Bradford, B.U.; Thurman, R.G. Acute alcohol produces hypoxia directly in rat liver tissue in vivo: Role of Kupffer cells. Am. J. Physiol. 1996, 271, G494–G500. [Google Scholar] [PubMed]
- Sato, N.; Kamada, T.; Kawano, S.; Hayashi, N.; Kishida, Y.; Meren, H.; Yoshihara, H.; Abe, H. Effect of acute and chronic ethanol consumption on hepatic tissue oxygen tension in rats. Pharmacol. Biochem. Behav. 1983, 18, 443–447. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [PubMed]
- Furuta, E.; Pai, S.K.; Zhan, R.; Bandyopadhyay, S.; Watabe, M.; Mo, Y.Y.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 2008, 68, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Rao, S.; Reddy, J.K. Peroxisome proliferator-activated receptors, fatty acid oxidation, steatohepatitis and hepatocarcinogenesis. Curr. Mol. Med. 2003, 3, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Zollner, G.; Trauner, M. Nuclear receptors in liver disease. Hepatology 2011, 53, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhuge, J.; Wang, X.; Bai, J.; Cederbaum, A.I. Cytochrome P450 2E1 contributes to ethanol-induced fatty liver in mice. Hepatology 2008, 47, 1483–1494. [Google Scholar] [CrossRef] [PubMed]
- Lebrun, V.; Molendi-Coste, O.; Lanthier, N.; Sempoux, C.; Cani, P.D.; van Rooijen, N.; Stärkel, P.; Horsmans, Y.; Leclercq, I.A. Impact of PPAR-alpha induction on glucose homoeostasis in alcohol-fed mice. Clin. Sci. 2013, 125, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.Z.; Usher, M.G.; Mortensen, R.M. Peroxisome proliferator-activated receptor-gamma-mediated effects in the vasculature. Circ. Res. 2008, 102, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Boelsterli, U.A.; Bedoucha, M. Toxicological consequences of altered peroxisome proliferator-activated receptor gamma (PPARgamma) expression in the liver: Insights from models of obesity and type 2 diabetes. Biochem. Pharmacol. 2002, 63, 1–10. [Google Scholar] [CrossRef]
- Memon, R.A.; Tecott, L.H.; Nonogaki, K.; Beigneux, A.; Moser, A.H.; Grunfeld, C.; Feingold, K.R. Up-regulation of peroxisome proliferator-activated receptors (PPAR-alpha) and PPAR-gamma messenger ribonucleic acid expression in the liver in murine obesity: Troglitazone induces expression of PPAR-gamma-responsive adipose tissue-specific genes in the liver of obese diabetic mice. Endocrinology 2000, 141, 4021–4031. [Google Scholar] [PubMed]
- Rahimian, R.; Masih-Khan, E.; Lo, M.; van Breemen, C.; McManus, B.M.; Dube, G.P. Hepatic over-expression of peroxisome proliferator activated receptor gamma2 in the ob/ob mouse model of non-insulin dependent diabetes mellitus. Mol. Cell. Biochem. 2001, 224, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.J.; Morimoto, M.; Thurman, R.G.; Bojes, H.K.; French, S.W. Expression of the peroxisome proliferator-activated receptor gene is decreased in experimental alcoholic liver disease. Life Sci. 1995, 56, 307–317. [Google Scholar] [CrossRef]
- Liangpunsakul, S.; Ross, R.A.; Crabb, D.W. Activation of carbohydrate response element-binding protein by ethanol. J. Investig. Med. 2013, 61, 270–277. [Google Scholar] [CrossRef]
- Liangpunsakul, S.; Sozio, M.S.; Shin, E.; Zhao, Z.; Xu, Y.; Ross, R.A.; Zeng, Y.; Crabb, D.W. Inhibitory effect of ethanol on AMPK phosphorylation is mediated in part through elevated ceramide levels. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G1004–G1012. [Google Scholar] [CrossRef] [PubMed]
- Liangpunsakul, S.; Wou, S.E.; Zeng, Y.; Ross, R.A.; Jayaram, H.N.; Crabb, D.W. Effect of ethanol on hydrogen peroxide-induced AMPK phosphorylation. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G1173–G1181. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Iizuka, K.; Miller, B.C.; Uyeda, K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 15597–15602. [Google Scholar] [CrossRef] [PubMed]
- Benhamed, F.; Denechaud, P.D.; Lemoine, M.; Robichon, C.; Moldes, M.; Bertrand-Michel, J.; Ratziu, V.; Serfaty, L.; Housset, C.; Capeau, J.; et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Investig. 2012, 122, 2176–2194. [Google Scholar] [CrossRef] [PubMed]
- Bettermann, K.; Hohensee, T.; Haybaeck, J. Steatosis and steatohepatitis: Complex disorders. Int. J. Mol. Sci. 2014, 15, 9924–9944. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Horikawa, Y. ChREBP: A glucose-activated transcription factor involved in the development of metabolic syndrome. Endocr J. 2008, 55, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Anandatheerthavarada, H.K.; Shankar, S.K.; Bhamre, S.; Boyd, M.R.; Song, B.J.; Ravindranath, V. Induction of brain cytochrome P-450IIE1 by chronic ethanol treatment. Brain Res. 1993, 601, 279–285. [Google Scholar] [CrossRef]
- Terpe, K. Overview of tag protein fusions: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2003, 60, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.M.; Pietsch, E.C.; Cunningham, C.C. Ethanol stimulates the production of reactive oxygen species at mitochondrial complexes I and III. Free Radic. Biol. Med. 1999, 27, 891–900. [Google Scholar] [CrossRef]
- Fernandez-Checa, J.C.; Kaplowitz, N.; Garcia-Ruiz, C.; Colell, A.; Miranda, M.; Mari, M.; Ardite, E.; Morales, A. GSH transport in mitochondria: Defense against TNF-induced oxidative stress and alcohol-induced defect. Am. J. Physiol. 1997, 273, G7–G17. [Google Scholar] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.K.; Kaspar, J.W.; Shen, J.; Jaiswal, A.K. NRF2 signaling and cell survival. Toxicol. Appl. Pharmacol. 2010, 244, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.R.; Johnson, J.A. The NRF2-ARE cytoprotective pathway in astrocytes. Expert Rev. Mol. Med. 2009. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of NRF2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.M.; Yang, J.H.; Ki, S.H. Role of the NRF2-ARE pathway in liver diseases. Oxid. Med. Cell. Longev. 2013. [Google Scholar] [CrossRef] [PubMed]
- Lamle, J.; Marhenke, S.; Borlak, J.; von Wasielewski, R.; Eriksson, C.J.; Geffers, R.; Manns, M.P.; Yamamoto, M.; Vogel, A. Nuclear factor-eythroid 2-related factor 2 prevents alcohol-induced fulminant liver injury. Gastroenterology 2008, 134, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Bhakta, A.; Wang, S.; Li, Z.; Manley, S.; Huang, H.; Copple, B.; Ding, W.X. Role of hypoxia inducing factor-1beta in alcohol-induced autophagy, steatosis and liver injury in mice. PLoS ONE 2014, 9, e115849. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, D.; Yang, L.; Gan, L.; Cederbaum, A.I. Cytochrome P450 2E1 potentiates ethanol induction of hypoxia and HIF-1alpha in vivo. Free Radic. Biol. Med. 2013, 63, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Kietzmann, T.; Gorlach, A. Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Semin. Cell Dev. Biol. 2005, 16, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Mole, D.R.; Blancher, C.; Copley, R.R.; Pollard, P.J.; Gleadle, J.M.; Ragoussis, J.; Ratcliffe, P.J. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J. Biol. Chem. 2009, 284, 16767–16775. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, Y.; Goda, N.; Kanai, M.; Niwa, D.; Osanai, K.; Yamamoto, Y.; Senoo-Matsuda, N.; Johnson, R.S.; Miura, S.; Kabe, Y.; et al. HIF-1alpha induction suppresses excessive lipid accumulation in alcoholic fatty liver in mice. J. Hepatol. 2012, 56, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Mehal, W.Z. HIF-1alpha is a major and complex player in alcohol induced liver diseases. J. Hepatol. 2012, 56, 311–312. [Google Scholar] [CrossRef] [PubMed]
- Nath, B.; Levin, I.; Csak, T.; Petrasek, J.; Mueller, C.; Kodys, K.; Szabo, G. Hepatocyte-specific hypoxia-inducible factor-1alpha is a determinant of lipid accumulation and liver injury in alcohol-induced steatosis in mice. Hepatology 2011, 53, 1526–1537. [Google Scholar] [CrossRef] [PubMed]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.O.; Sanchez-Ramos, C.; Prieto-Arroyo, I.; Urbanek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Oskoui, P.R.; Banko, M.R.; Maniar, J.M.; Gygi, M.P.; Gygi, S.P.; Brunet, A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 2007, 282, 30107–30119. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Du, K.; You, M.; Ding, W.X. Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. Am. J. Pathol. 2013, 183, 1815–1825. [Google Scholar] [CrossRef] [PubMed]
- Tikhanovich, I.; Kuravi, S.; Campbell, R.V.; Kharbanda, K.K.; Artigues, A.; Villar, M.T.; Weinman, S.A. Regulation of FOXO3 by phosphorylation and methylation in hepatitis C virus infection and alcohol exposure. Hepatology 2014, 59, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.D.; Liu, C.; Chung, J.; Stickel, F.; Seitz, H.K.; Russell, R.M. Chronic alcohol intake reduces retinoic acid concentration and enhances AP-1 (c-Jun and c-Fos) expression in rat liver. Hepatology 1998, 28, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Yeligar, S.M.; Machida, K.; Tsukamoto, H.; Kalra, V.K. Ethanol augments RANTES/CCL5 expression in rat liver sinusoidal endothelial cells and human endothelial cells via activation of NF-kappa B, HIF-1 alpha, and AP-1. J. Immunol. 2009, 183, 5964–5976. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, N.M. Alcohol and hypertension. Lancet 1995, 345, 1588–1589. [Google Scholar] [CrossRef]
- Beilin, L.J.; Puddey, I.B. Alcohol and hypertension: An update. Hypertension 2006, 47, 1035–1038. [Google Scholar] [CrossRef] [PubMed]
- Klatsky, A.L. Alcohol-associated hypertension: When one drinks makes a difference. Hypertension 2004, 44, 805–806. [Google Scholar] [CrossRef] [PubMed]
- MacMahon, S. Alcohol consumption and hypertension. Hypertension 1987, 9, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Grogan, J.R.; Kochar, M.S. Alcohol and hypertension. Arch. Fam. Med. 1994, 3, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Randin, D.; Vollenweider, P.; Tappy, L.; Jequier, E.; Nicod, P.; Scherrer, U. Suppression of alcohol-induced hypertension by dexamethasone. N. Engl. J. Med. 1995, 332, 1733–1737. [Google Scholar] [CrossRef] [PubMed]
- Resstel, L.B.; Scopinho, A.A.; Lopes da Silva, A.; Antunes-Rodrigues, J.; Correa, F.M. Increased circulating vasopressin may account for ethanol-induced hypertension in rats. Am. J. Hypertens. 2008, 21, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Soardo, G.; Donnini, D.; Moretti, M.; Milocco, C.; Catena, C.; Sechi, L.A. Effects of antihypertensive drugs on alcohol-induced functional responses of cultured human endothelial cells. Hypertens. Res. 2008, 31, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Gould, A.B.; Green, D. Kinetics of the human renin and human substrate reaction. Cardiovasc. Res. 1971, 5, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Fasola, A.F.; Martz, B.L.; Helmer, O.M. Renin activity during supine exercise in normotensives and hypertensives. J. Appl. Physiol. 1966, 21, 1709–1712. [Google Scholar] [PubMed]
- Watt, G.C.; Harrap, S.B.; Foy, C.J.; Holton, D.W.; Edwards, H.V.; Davidson, H.R.; Fraser, R. Abnormalities of glucocorticoid metabolism and the renin-angiotensin system: A four-corners approach to the identification of genetic determinants of blood pressure. J. Hypertens. 1992, 10, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.J.; Habener, J.F. Angiotensinogen gene is expressed and differentially regulated in multiple tissues of the rat. J. Clin. Investig. 1986, 78, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, K.; Sugiyama, F.; Goto, Y.; Ishida, J.; Takimoto, E.; Yagami, K.; Murakami, K. Angiotensinogen-deficient mice with hypotension. J. Biol. Chem. 1994, 269, 31334–31337. [Google Scholar] [PubMed]
- Kim, H.S.; Krege, J.H.; Kluckman, K.D.; Hagaman, J.R.; Hodgin, J.B.; Best, C.F.; Jennette, J.C.; Coffman, T.M.; Maeda, N.; Smithies, O. Genetic control of blood pressure and the angiotensinogen locus. Proc. Natl. Acad. Sci. USA 1995, 92, 2735–2739. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Mohuczy, D.; Zhang, Y.C.; Kimura, B.; Galli, S.M.; Phillips, M.I. Intravenous angiotensinogen antisense in AAV-based vector decreases hypertension. Am. J. Physiol. 1999, 277, H2392–H2399. [Google Scholar] [PubMed]
- Krege, J.H.; Kim, H.S.; Moyer, J.S.; Jennette, J.C.; Peng, L.; Hiller, S.K.; Smithies, O. Angiotensin-converting enzyme gene mutations, blood pressures, and cardiovascular homeostasis. Hypertension 1997, 29, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Aroor, A.R.; Shukla, S.D. Binge ethanol intake in chronically exposed rat liver decreases LDL-receptor and increases angiotensinogen gene expression. World J. Hepatol. 2011, 3, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, N.; Katsurada, A.; Miyata, K.; Satou, R.; Saito, T.; Urushihara, M.; Kobori, H. Activation of reactive oxygen species and the renin-angiotensin system in IgA nephropathy model mice. Clin. Exp. Pharmacol. Physiol. 2009, 36, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Husain, K.; Vazquez, M.; Ansari, R.A.; Malafa, M.P.; Lalla, J. Chronic alcohol-induced oxidative endothelial injury relates to angiotensin II levels in the rat. Mol. Cell. Biochem. 2008, 307, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Wright, J. Fetal alcohol syndrome. Nurs. Times 1986, 82, 34–35. [Google Scholar] [PubMed]
- Okuno, F.; Arai, M.; Ishii, H.; Shigeta, Y.; Ebihara, Y.; Takagi, S.; Tsuchiya, M. Mild but prolonged elevation of serum angiotensin converting enzyme (ACE) activity in alcoholics. Alcohol 1986, 3, 357–359. [Google Scholar] [CrossRef]
- Cheng, C.P.; Cheng, H.J.; Cunningham, C.; Shihabi, Z.K.; Sane, D.C.; Wannenburg, T.; Little, W.C. Angiotensin II type 1 receptor blockade prevents alcoholic cardiomyopathy. Circulation 2006, 114, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Ansari, R.A.; Clark, M.A. Regulation of angiotensinogen gene after ethanol in hepatocytes. In Proceedings of the 50th Anniversary and Annual Meeting, Society of Toxicology, Washington, DC, USA, 6–10 March 2011.
- Ansari, R.A.; Rizvi, S.A.A.; Clark, M.A. Angiotensinogen gene regulation after ethanol exposure in hepatocytes. In Proceedings of the World Congress on Gastroenterology and Urology, Omaha, NE, USA, 12–14 March 2012.

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ansari, R.A.; Husain, K.; Rizvi, S.A.A. Role of Transcription Factors in Steatohepatitis and Hypertension after Ethanol: The Epicenter of Metabolism. Biomolecules 2016, 6, 29. https://doi.org/10.3390/biom6030029
Ansari RA, Husain K, Rizvi SAA. Role of Transcription Factors in Steatohepatitis and Hypertension after Ethanol: The Epicenter of Metabolism. Biomolecules. 2016; 6(3):29. https://doi.org/10.3390/biom6030029
Chicago/Turabian StyleAnsari, Rais A., Kazim Husain, and Syed A. A. Rizvi. 2016. "Role of Transcription Factors in Steatohepatitis and Hypertension after Ethanol: The Epicenter of Metabolism" Biomolecules 6, no. 3: 29. https://doi.org/10.3390/biom6030029
APA StyleAnsari, R. A., Husain, K., & Rizvi, S. A. A. (2016). Role of Transcription Factors in Steatohepatitis and Hypertension after Ethanol: The Epicenter of Metabolism. Biomolecules, 6(3), 29. https://doi.org/10.3390/biom6030029