
NMR Meets Tau: Insights into Its Function and Pathology

 , , , ,
, , , , 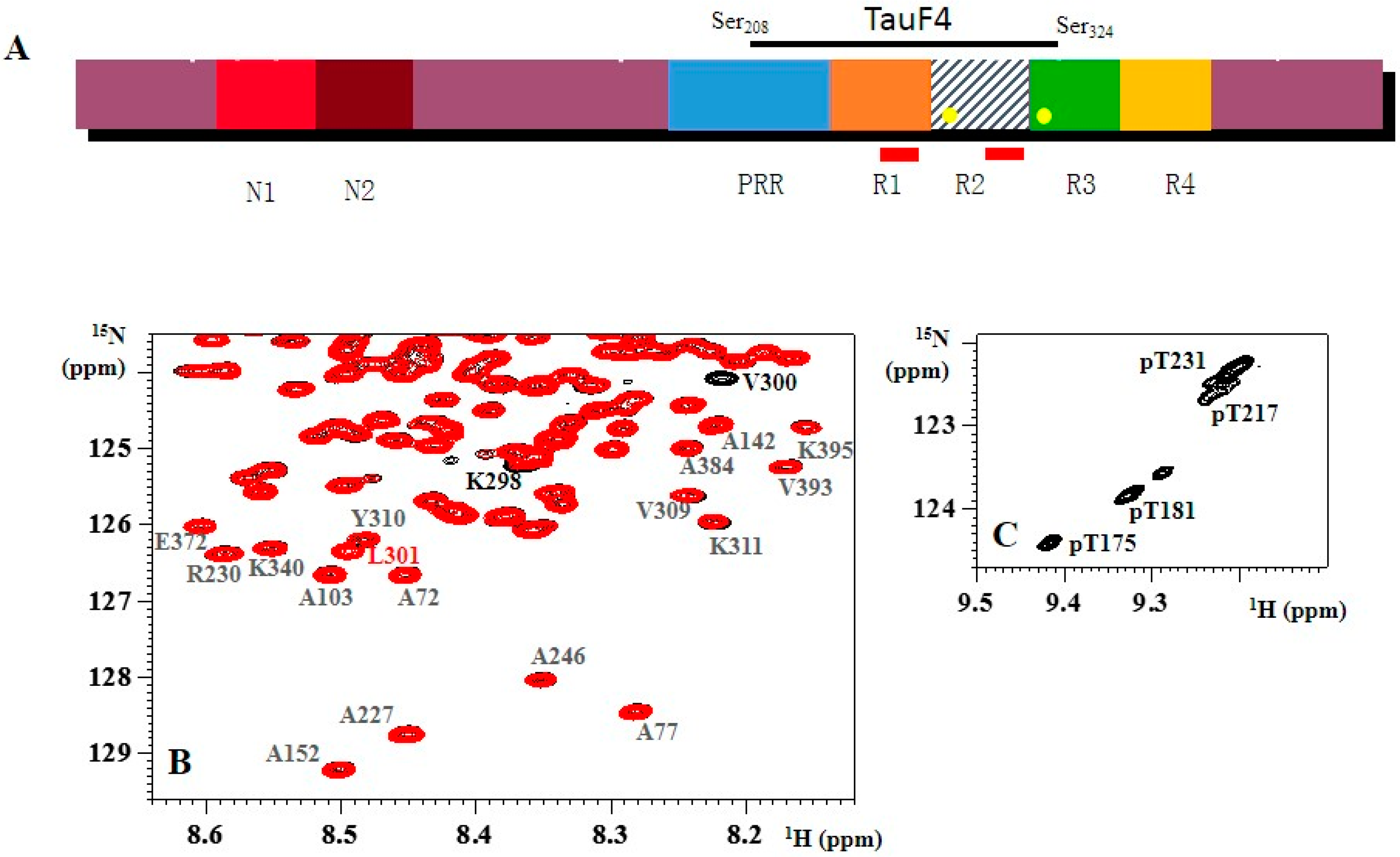{kind=link}
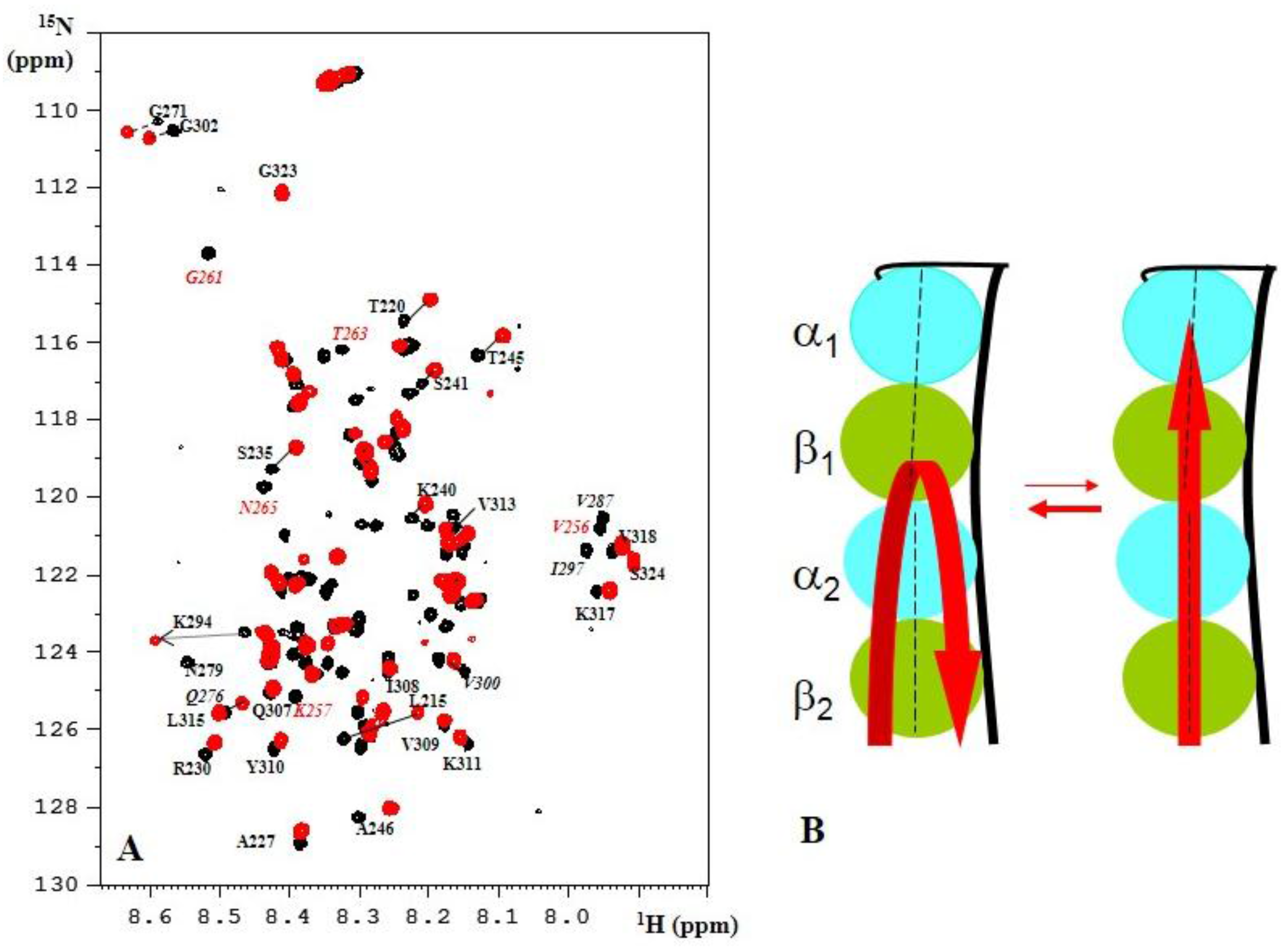{kind=link}
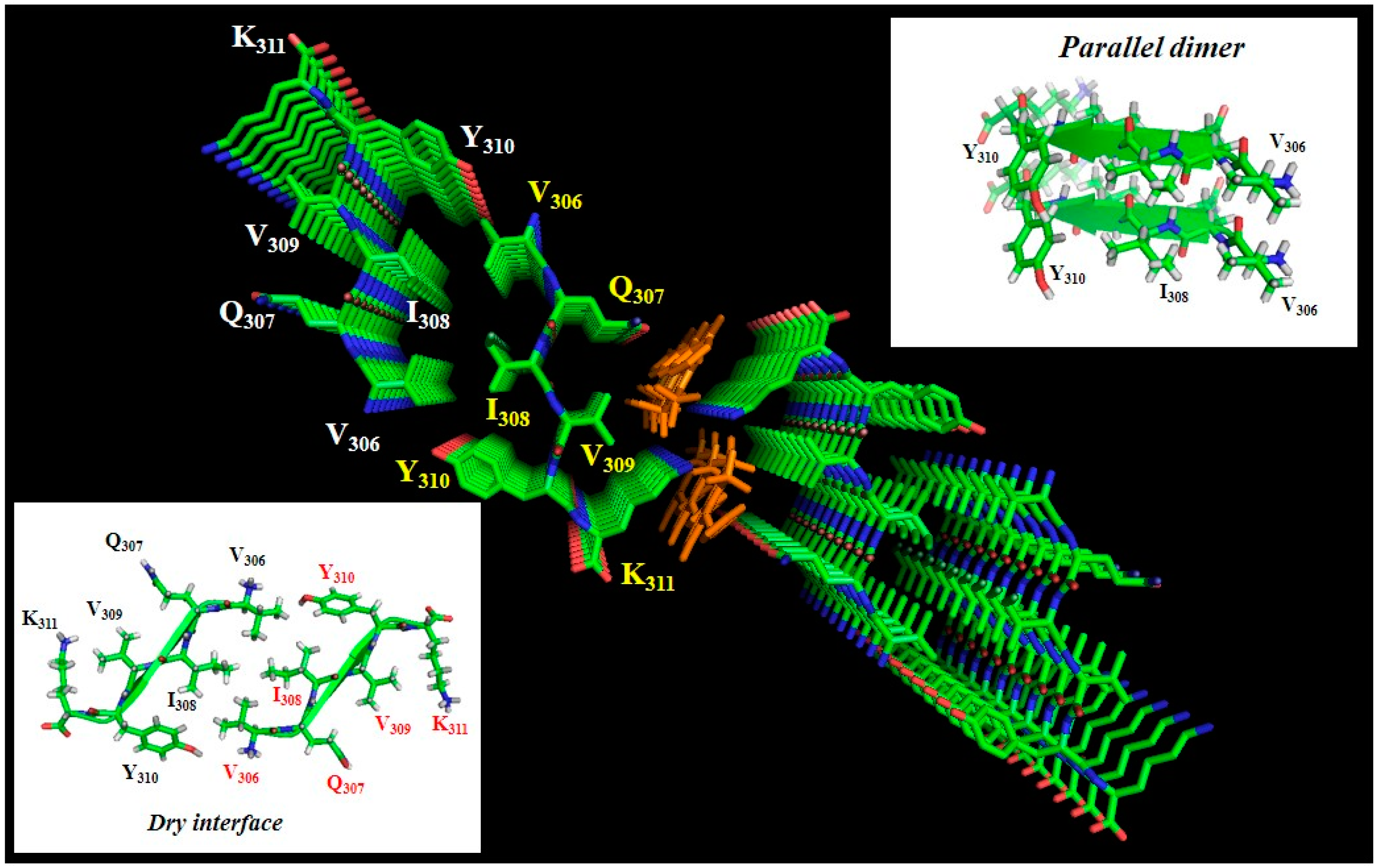{kind=link}
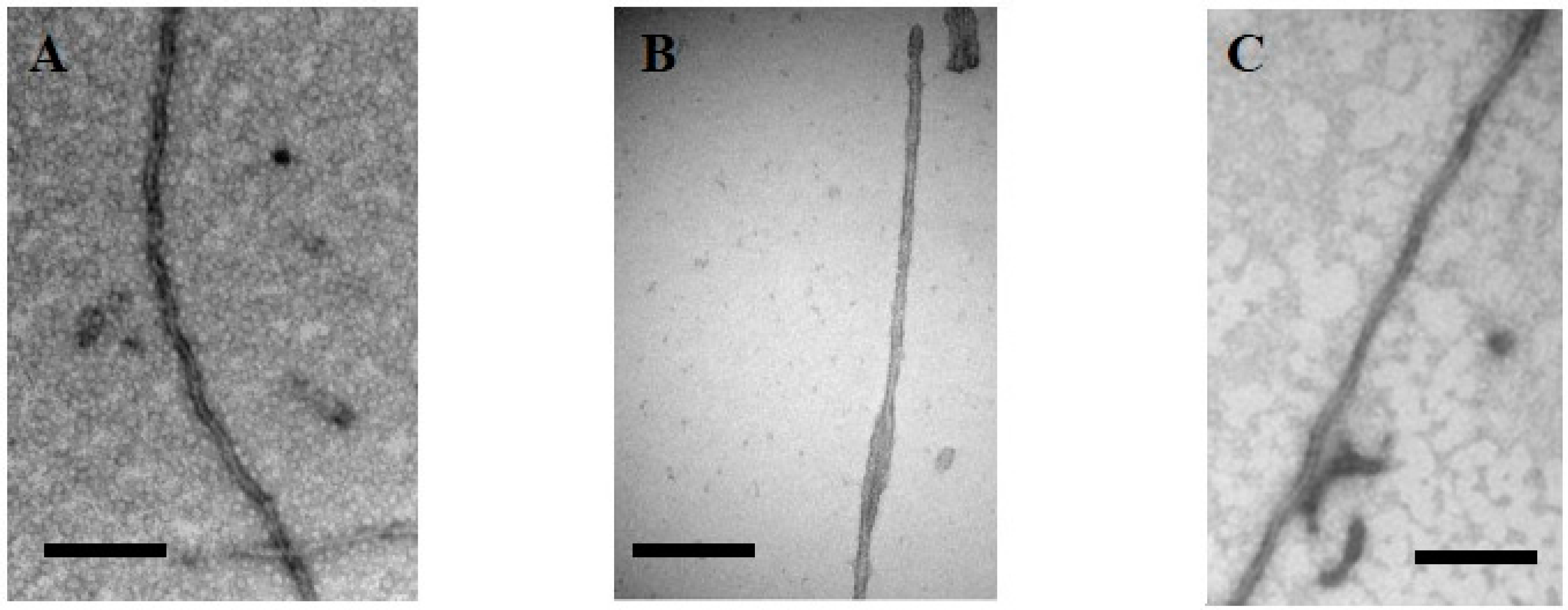{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. NMR Spectroscopy of Isolated Tau
1.2. NMR Spectroscopy of Tau-Tubulin Complexes
1.3. Aggregation of Tau
2. Phosphorylation and Tau
3. Other Protein Interactions and PTMs of Tau
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Brion, J.P.; Flament-Durand, J.; Dustin, P. Alzheimer’s disease and tau proteins. Lancet 1986, 2. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Quinlan, M.; Tung, Y.C.; Zaidi, M.S.; Wisniewski, H.M. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 1986, 261, 6084–6089. [Google Scholar] [PubMed]
- Wood, J.G.; Mirra, S.S.; Pollock, N.J.; Binder, L.I. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau). Proc. Natl. Acad. Sci. USA 1986, 83, 4040–4043. [Google Scholar] [CrossRef] [PubMed]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, D.W.; Hwo, S.Y.; Kirschner, M.W. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Biol. 1977, 116, 227–247. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E.; et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front Cell Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Bukar Maina, M.; Al-Hilaly, Y.K.; Serpell, L.C. Nuclear Tau and Its Potential Role in Alzheimer’s Disease. Biomolecules 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Clavaguera, F.; Tolnay, M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 2010, 33, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.H.; Kumar, S.; Pinotsi, D.; Tunnacliffe, A.; St George-Hyslop, P.; Mandelkow, E.; Mandelkow, E.-M.; Kaminski, C.F.; Kaminski Schierle, G.S. Extracellular monomeric tau protein is sufficient to initiate the spread of tau protein pathology. J. Biol. Chem. 2014, 289, 956–967. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, S.; Lécolle, K.; Caillierez, R.; Bégard, S.; Zommer, N.; Lachaud, C.; Carrier, S.; Dufour, N.; Aurégan, G.; Winderickx, J.; et al. Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: Relevance to sporadic tauopathies. Acta Neuropathol. Commun. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lim, S.; Haque, M.M.; Ryoo, N.; Hong, H.S.; Rhim, H.; Lee, D.-E.; Chang, Y.-T.; Lee, J.-S.; Cheong, E.; et al. Identification of disulfide cross-linked tau dimer responsible for tau propagation. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Carranza, D.L.; Gerson, J.E.; Sengupta, U.; Guerrero-Muñoz, M.J.; Lasagna-Reeves, C.A.; Kayed, R. Specific targeting of tau oligomers in Htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J. Alzheimers Dis. 2014, 40, S97–S111. [Google Scholar] [PubMed]
- Tai, H.-C.; Wang, B.Y.; Serrano-Pozo, A.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. Frequent and symmetric deposition of misfolded tau oligomers within presynaptic and postsynaptic terminals in Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Usenovic, M.; Niroomand, S.; Drolet, R.E.; Yao, L.; Gaspar, R.C.; Hatcher, N.G.; Schachter, J.; Renger, J.J.; Parmentier-Batteur, S. Internalized Tau Oligomers Cause Neurodegeneration by Inducing Accumulation of Pathogenic Tau in Human Neurons Derived from Induced Pluripotent Stem Cells. J. Neurosci. 2015, 35, 14234–14250. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Wegmann, S.; Cho, H.; DeVos, S.L.; Commins, C.; Roe, A.D.; Nicholls, S.B.; Carlson, G.A.; Pitstick, R.; Nobuhara, C.K.; et al. Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Min, S.-W.; Cho, S.-H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M.Y. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Greenwood, A.; Binder, L.; Bigio, E.H.; Denial, S.; Nicholson, L.; Zhou, X.Z.; Lu, K.P. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer’s disease. Cell 2012, 149, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Smet, C.; Leroy, A.; Sillen, A.; Wieruszeski, J.-M.; Landrieu, I.; Lippens, G. Accepting its random coil nature allows a partial NMR assignment of the neuronal Tau protein. Chembiochem 2004, 5, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Lippens, G.; Wieruszeski, J.-M.; Leroy, A.; Smet, C.; Sillen, A.; Buée, L.; Landrieu, I. Proline-directed random-coil chemical shift values as a tool for the NMR assignment of the tau phosphorylation sites. Chembiochem 2004, 5, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Verdegem, D.; Dijkstra, K.; Hanoulle, X.; Lippens, G. Graphical interpretation of Boolean operators for protein NMR assignments. J. Biomol. NMR 2008, 42, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Sibille, N.; Hanoulle, X.; Bonachera, F.; Verdegem, D.; Landrieu, I.; Wieruszeski, J.-M.; Lippens, G. Selective backbone labelling of ILV methyl labelled proteins. J. Biomol. NMR 2009, 43, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.; Ahuja, P.; Gerard, M.; Wieruszeski, J.-M.; Lippens, G. A new strategy for sequential assignment of intrinsically unstructured proteins based on 15N single isotope labelling. J. Magn. Reson. 2013, 236, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; Jeganathan, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009, 7, e34. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, R.L.; Dürr, U.H.N.; Bibow, S.; Biernat, J.; Mandelkow, E.; Zweckstetter, M. Automatic assignment of the intrinsically disordered protein Tau with 441-residues. J. Am. Chem. Soc. 2010, 132, 11906–11907. [Google Scholar] [CrossRef] [PubMed]
- Harbison, N.W.; Bhattacharya, S.; Eliezer, D. Assigning backbone NMR resonances for full length tau isoforms: Efficient compromise between manual assignments and reduced dimensionality. PLoS ONE 2012, 7, e34679. [Google Scholar] [CrossRef] [PubMed]
- Smet-Nocca, C.; Launay, H.; Wieruszeski, J.-M.; Lippens, G.; Landrieu, I. Unraveling a phosphorylation event in a folded protein by NMR spectroscopy: Phosphorylation of the Pin1 WW domain by PKA. J. Biomol. NMR 2013, 55, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Amniai, L.; Barbier, P.; Sillen, A.; Wieruszeski, J.-M.; Peyrot, V.; Lippens, G.; Landrieu, I. Alzheimer disease specific phosphoepitopes of Tau interfere with assembly of tubulin but not binding to microtubules. FASEB J. 2009, 23, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Leroy, A.; Landrieu, I.; Huvent, I.; Legrand, D.; Codeville, B.; Wieruszeski, J.-M.; Lippens, G. Spectroscopic studies of GSK3{beta} phosphorylation of the neuronal tau protein and its interaction with the N-terminal domain of apolipoprotein E. J. Biol. Chem. 2010, 285, 33435–33444. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Prabakaran, S.; Cantrelle, F.C.-X.; Chambraud, B.E.; Gunawardena, J.; Lippens, G.; Landrieu, I. Characterization of Neuronal Tau Protein as a Target of Extracellular-signal-regulated Kinase. J. Biol. Chem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lippens, G.; Sillen, A.; Smet, C.; Wieruszeski, J.-M.; Leroy, A.; Buée, L.; Landrieu, I. Studying the natively unfolded neuronal Tau protein by solution NMR spectroscopy. Protein Pept. Lett. 2006, 13, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Becker, S.; Zweckstetter, M. A six-dimensional alpha proton detection-based APSY experiment for backbone assignment of intrinsically disordered proteins. J. Biomol. NMR 2014, 60, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Von Bergen, M.; Friedhoff, P.; Biernat, J.; Heberle, J.; Mandelkow, E.M.; Mandelkow, E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc. Natl. Acad. Sci. USA 2000, 97, 5129–5134. [Google Scholar] [CrossRef] [PubMed]
- Von Bergen, M.; Barghorn, S.; Li, L.; Marx, A.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta-structure. J. Biol. Chem. 2001, 276, 48165–48174. [Google Scholar] [PubMed]
- Mukrasch, M.D.; Biernat, J.; von Bergen, M.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Sites of tau important for aggregation populate {beta}-structure and bind to microtubules and polyanions. J. Biol. Chem. 2005, 280, 24978–24986. [Google Scholar] [CrossRef] [PubMed]
- Eliezer, D.; Barré, P.; Kobaslija, M.; Chan, D.; Li, X.; Heend, L. Residual structure in the repeat domain of tau: Echoes of microtubule binding and paired helical filament formation. Biochemistry 2005, 44, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.M.; Wong, C.T.; Clarke, J. Coupled folding and binding of the disordered protein PUMA does not require particular residual structure. J. Am. Chem. Soc. 2014, 136, 5197–5200. [Google Scholar] [CrossRef] [PubMed]
- Sogawa, K.; Okuda, R.; In, Y.; Ishida, T.; Taniguchi, T.; Minoura, K.; Tomoo, K. C–H … π interplay between Ile308 and Tyr310 residues in the third repeat of microtubule binding domain is indispensable for self-assembly of three- and four-repeat tau. J. Biochem. 2012, 152, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Sogawa, K.; Minoura, K.; In, Y.; Ishida, T.; Taniguchi, T.; Tomoo, K. CH-π interaction in VQIVYK sequence elucidated by NMR spectroscopy is essential for PHF formation of tau. Biopolymers 2014, 102, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Rosnoblet, C.; Fritzinger, B.; Legrand, D.; Launay, H.; Wieruszeski, J.-M.; Lippens, G.; Hanoulle, X. Hepatitis C virus NS5B and host cyclophilin A share a common binding site on NS5A. J. Biol. Chem. 2012, 287, 44249–44260. [Google Scholar] [CrossRef] [PubMed]
- Bibow, S.; Ozenne, V.; Biernat, J.; Blackledge, M.; Mandelkow, E.; Zweckstetter, M. Structural impact of proline-directed pseudophosphorylation at AT8, AT100, and PHF1 epitopes on 441-residue tau. J. Am. Chem. Soc. 2011, 133, 15842–15845. [Google Scholar] [CrossRef] [PubMed]
- Jeganathan, S.; von Bergen, M.; Brutlach, H.; Steinhoff, H.-J.; Mandelkow, E. Global hairpin folding of tau in solution. Biochemistry 2006, 45, 2283–2293. [Google Scholar] [CrossRef] [PubMed]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.N.; Poorkaj, P.; Wszolek, Z.; Geschwind, D.H.; Nasreddine, Z.S.; Miller, B.; Li, D.; Payami, H.; Awert, F.; Markopoulou, K.; et al. Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc. Natl. Acad. Sci. USA 1998, 95, 13103–13107. [Google Scholar] [CrossRef] [PubMed]
- Golovanov, A.P.; Blankley, R.T.; Avis, J.M.; Bermel, W. Isotopically discriminated NMR spectroscopy: A tool for investigating complex protein interactions in vitro. J. Am. Chem. Soc. 2007, 129, 6528–6535. [Google Scholar] [CrossRef] [PubMed]
- Bienkiewicz, E.A.; Lumb, K.J. Random-coil chemical shifts of phosphorylated amino acids. J. Biomol. NMR 1999, 15, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Landrieu, I.; Lacosse, L.; Leroy, A.; Wieruszeski, J.-M.; Trivelli, X.; Sillen, A.; Sibille, N.; Schwalbe, H.; Saxena, K.; Langer, T.; et al. NMR analysis of a Tau phosphorylation pattern. J. Am. Chem. Soc. 2006, 128, 3575–3583. [Google Scholar] [CrossRef] [PubMed]
- Peterson, D.W.; Ando, D.M.; Taketa, D.A.; Zhou, H.; Dahlquist, F.W.; Lew, J. No difference in kinetics of tau or histone phosphorylation by CDK5/p25 versus CDK5/p35 in vitro. Proc. Natl. Acad. Sci. USA 2010, 107, 2884–2889. [Google Scholar] [CrossRef] [PubMed]
- Schwalbe, M.; Biernat, J.; Bibow, S.; Ozenne, V.; Jensen, M.R.; Kadavath, H.; Blackledge, M.; Mandelkow, E.; Zweckstetter, M. Phosphorylation of human Tau protein by microtubule affinity-regulating kinase 2. Biochemistry 2013, 52, 9068–9079. [Google Scholar] [CrossRef] [PubMed]
- Sibille, N.; Huvent, I.; Fauquant, C.; Verdegem, D.; Amniai, L.; Leroy, A.; Wieruszeski, J.-M.; Lippens, G.; Landrieu, I. Structural characterization by nuclear magnetic resonance of the impact of phosphorylation in the proline-rich region of the disordered Tau protein. Proteins 2012, 80, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Schwalbe, M.; Kadavath, H.; Biernat, J.; Ozenne, V.; Blackledge, M.; Mandelkow, E.; Zweckstetter, M. Structural Impact of Tau Phosphorylation at Threonine 231. Structure 2015, 23, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
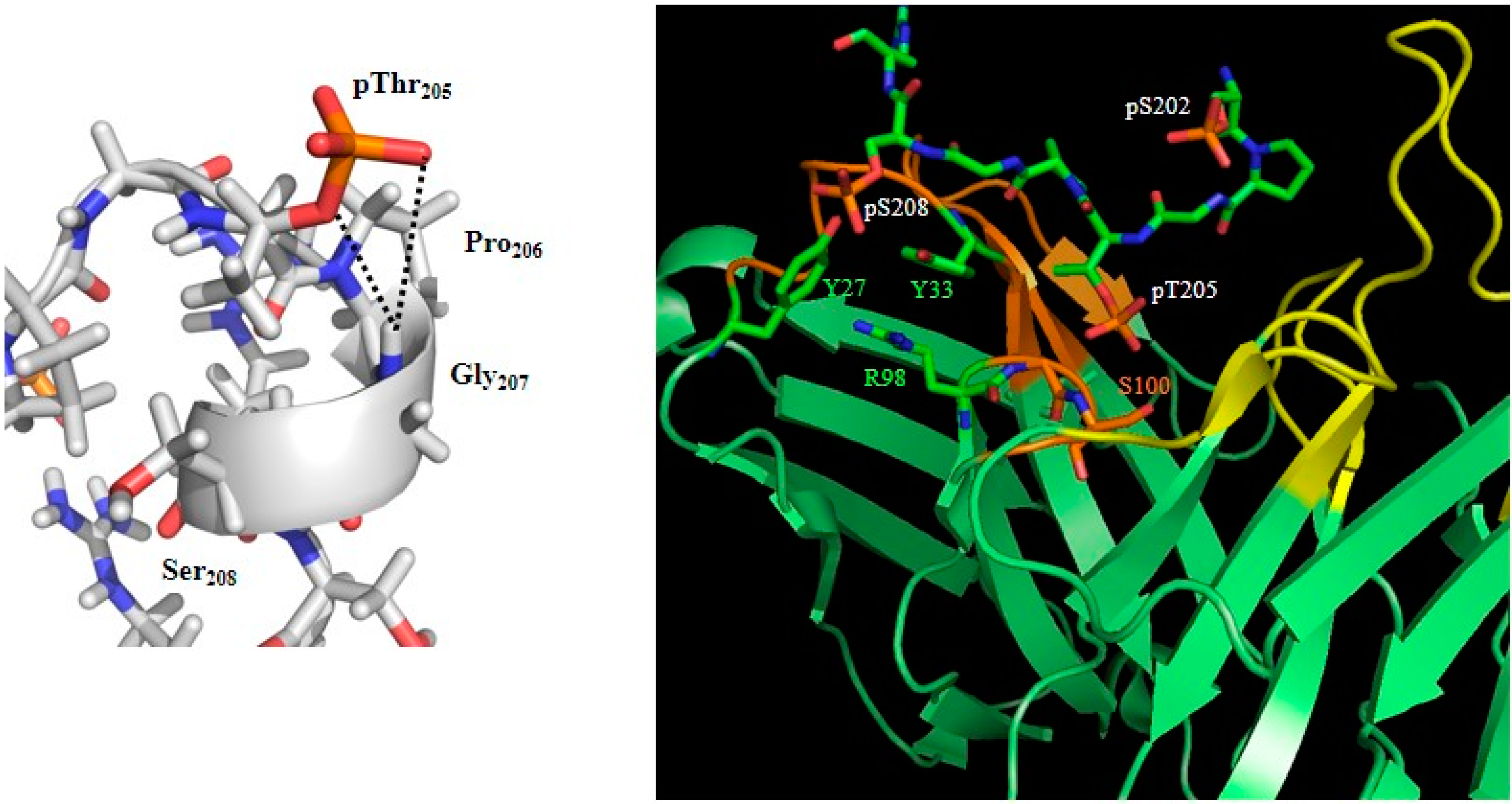
- Gandhi, N.S.; Landrieu, I.; Byrne, C.; Kukic, P.; Amniai, L.; Cantrelle, F.-X.; Wieruszeski, J.-M.; Mancera, R.L.; Jacquot, Y.; Lippens, G. A Phosphorylation-Induced Turn Defines the Alzheimer’s Disease AT8 Antibody Epitope on the Tau Protein. Angew. Chem. Int. Ed. Engl. 2015, 54, 6819–6823. [Google Scholar] [CrossRef] [PubMed]
- Sillen, A.; Barbier, P.; Landrieu, I.; Lefebvre, S.; Wieruszeski, J.-M.; Leroy, A.; Peyrot, V.; Lippens, G. NMR investigation of the interaction between the neuronal protein tau and the microtubules. Biochemistry 2007, 46, 3055–3064. [Google Scholar] [CrossRef] [PubMed]
- Mukrasch, M.D.; von Bergen, M.; Biernat, J.; Fischer, D.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. The “jaws” of the tau-microtubule interaction. J. Biol. Chem. 2007, 282, 12230–12239. [Google Scholar] [CrossRef] [PubMed]
- Butner, K.A.; Kirschner, M.W. Tau protein binds to microtubules through a flexible array of distributed weak sites. J. Cell Biol. 1991, 115, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Panda, D.; Goode, B.L.; Feinstein, S.C.; Wilson, L. Kinetic stabilization of microtubule dynamics at steady state by tau and microtubule-binding domains of tau. Biochemistry 1995, 34, 11117–11127. [Google Scholar] [CrossRef] [PubMed]
- Trinczek, B.; Biernat, J.; Baumann, K.; Mandelkow, E.M.; Mandelkow, E. Domains of tau protein, differential phosphorylation, and dynamic instability of microtubules. Mol. Biol. Cell 1995, 6, 1887–1902. [Google Scholar] [CrossRef] [PubMed]
- Goode, B.L.; Denis, P.E.; Panda, D.; Radeke, M.J.; Miller, H.P.; Wilson, L.; Feinstein, S.C. Functional interactions between the proline-rich and repeat regions of tau enhance microtubule binding and assembly. Mol. Biol. Cell 1997, 8, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Al-Bassam, J.; Ozer, R.S.; Safer, D.; Halpain, S.; Milligan, R.A. MAP2 and tau bind longitudinally along the outer ridges of microtubule protofilaments. J. Cell Biol. 2002, 157, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Santarella, R.A.; Skiniotis, G.; Goldie, K.N.; Tittmann, P.; Gross, H.; Mandelkow, E.-M.; Mandelkow, E.; Hoenger, A. Surface-decoration of microtubules by human tau. J. Mol. Biol. 2004, 339, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Schaap, I.A.T.; Hoffmann, B.; Carrasco, C.; Merkel, R.; Schmidt, C.F. Tau protein binding forms a 1 nm thick layer along protofilaments without affecting the radial elasticity of microtubules. J. Struct. Biol. 2007, 158, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, M.H.; Jalal, A.; Brenner, B.; Mandelkow, E.; Kumar, S.; Scholz, T. Tau protein diffuses along the microtubule lattice. J. Biol. Chem. 2012, 287, 38559–38568. [Google Scholar] [CrossRef] [PubMed]
- Balaram, P.; Bothner-By, A.A.; Dadok, J. Negative nuclear Overhauser effects as probes of macromolecular structure. J. Am. Chem. Soc. 1972, 94, 4015–4017. [Google Scholar] [CrossRef] [PubMed]
- Clore, G.M.; Gronenborn, A.M. Theory and applications of the transferred nuclear overhauser effect to the study of the conformations of small ligands bound to proteins. J. Magn. Reson. (1969) 1982, 48, 402–417. [Google Scholar] [CrossRef]
- Lippens, R.M.; Cerf, C.; Hallenga, K. Theory and experimental results of transfer-NOE experiments. 1. The influence of the off rate versus cross-relaxation rates. J. Magn. Reson. (1969) 1992, 99, 268–281. [Google Scholar] [CrossRef]
- Kadavath, H.; Jaremko, M.; Jaremko, Ł.; Biernat, J.; Mandelkow, E.; Zweckstetter, M. Folding of the Tau Protein on Microtubules. Angew. Chem. Int. Ed. Engl. 2015, 54, 10347–10351. [Google Scholar] [CrossRef] [PubMed]
- Ackmann, M.; Wiech, H.; Mandelkow, E. Nonsaturable binding indicates clustering of tau on the microtubule surface in a paired helical filament-like conformation. J. Biol. Chem. 2000, 275, 30335–30343. [Google Scholar] [CrossRef] [PubMed]
- Duan, A.R.; Goodson, H.V. Taxol-stabilized microtubules promote the formation of filaments from unmodified full-length Tau in vitro. Mol. Biol. Cell 2012, 23, 4796–4806. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Guo, C.; Hou, G.; Zhang, H.; Lu, X.; Williams, J.C.; Polenova, T. Atomic-resolution structure of the CAP-Gly domain of dynactin on polymeric microtubules determined by magic angle spinning NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, 14611–14616. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Fan, J.; Smith, M.J.; Goedert, M.; Amos, L.A. Repeat motifs of tau bind to the insides of microtubules in the absence of taxol. EMBO J. 2003, 22, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Makrides, V.; Massie, M.R.; Feinstein, S.C.; Lew, J. Evidence for two distinct binding sites for tau on microtubules. Proc. Natl. Acad. Sci. USA 2004, 101, 6746–6751. [Google Scholar] [CrossRef] [PubMed]
- Gigant, B.; Landrieu, I.; Fauquant, C.; Barbier, P.; Huvent, I.; Wieruszeski, J.-M.; Knossow, M.; Lippens, G. Mechanism of Tau-promoted microtubule assembly as probed by NMR spectroscopy. J. Am. Chem. Soc. 2014, 136, 12615–12623. [Google Scholar] [CrossRef] [PubMed]
- Gigant, B.; Curmi, P.A.; Martin-Barbey, C.; Charbaut, E.; Lachkar, S.; Lebeau, L.; Siavoshian, S.; Sobel, A.; Knossow, M. The 4 A X-ray structure of a tubulin: Stathmin-like domain complex. Cell 2000, 102, 809–816. [Google Scholar] [CrossRef]
- Ravelli, R.B.G.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.C.; Raviv, U.; Miller, H.P.; Gaylord, M.R.; Kiris, E.; Ventimiglia, D.; Needleman, D.J.; Kim, M.W.; Wilson, L.; Feinstein, S.C.; et al. Human microtubule-associated-protein tau regulates the number of protofilaments in microtubules: A synchrotron x-ray scattering study. Biophys. J. 2009, 97, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Fauquant, C.; Redeker, V.; Landrieu, I.; Wieruszeski, J.-M.; Verdegem, D.; Laprévote, O.; Lippens, G.; Gigant, B.; Knossow, M. Systematic identification of tubulin-interacting fragments of the microtubule-associated protein Tau leads to a highly efficient promoter of microtubule assembly. J. Biol. Chem. 2011, 286, 33358–33368. [Google Scholar] [CrossRef] [PubMed]
- Mignot, I.; Pecqueur, L.; Dorléans, A.; Karuppasamy, M.; Ravelli, R.B.G.; Dreier, B.; Plückthun, A.; Knossow, M.; Gigant, B. Design and characterization of modular scaffolds for tubulin assembly. J. Biol. Chem. 2012, 287, 31085–31094. [Google Scholar] [CrossRef] [PubMed]
- Drewes, G.; Ebneth, A.; Preuss, U.; Mandelkow, E.M.; Mandelkow, E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell 1997, 89, 297–308. [Google Scholar] [CrossRef]
- Kadavath, H.; Hofele, R.V.; Biernat, J.; Kumar, S.; Tepper, K.; Urlaub, H.; Mandelkow, E.; Zweckstetter, M. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. USA. 2015, 112, 7501–7506. [Google Scholar] [CrossRef] [PubMed]
- Gigant, B.; Wang, C.; Ravelli, R.B.G.; Roussi, F.; Steinmetz, M.O.; Curmi, P.A.; Sobel, A.; Knossow, M. Structural basis for the regulation of tubulin by vinblastine. Nature 2005, 435, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Singer, W.D.; Jordan, M.A.; Wilson, L.; Himes, R.H. Binding of vinblastine to stabilized microtubules. Mol. Pharmacol. 1989, 36, 366–370. [Google Scholar] [PubMed]
- Johnson, V.; Ayaz, P.; Huddleston, P.; Rice, L.M. Design, overexpression, and purification of polymerization-blocked yeast αβ-tubulin mutants. Biochemistry 2011, 50, 8636–8644. [Google Scholar] [CrossRef] [PubMed]
- Minoura, I.; Hachikubo, Y.; Yamakita, Y.; Takazaki, H.; Ayukawa, R.; Uchimura, S.; Muto, E. Overexpression, purification, and functional analysis of recombinant human tubulin dimer. FEBS Lett. 2013, 587, 3450–3455. [Google Scholar] [CrossRef] [PubMed]
- Valenstein, M.L.; Roll-Mecak, A. Graded Control of Microtubule Severing by Tubulin Glutamylation. Cell 2016, 164, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Novak, M.; Edwards, P.C.; Klug, A.; Tichelaar, W.; Crowther, R.A. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1988, 85, 4884–4888. [Google Scholar] [CrossRef] [PubMed]
- Wille, H.; Drewes, G.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein tau in vitro. J. Cell Biol. 1992, 118, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Schweers, O.; Mandelkow, E.M.; Biernat, J.; Mandelkow, E. Oxidation of cysteine-322 in the repeat domain of microtubule-associated protein tau controls the in vitro assembly of paired helical filaments. Proc. Natl. Acad. Sci. USA 1995, 92, 8463–8467. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Edwards, P.C.; Lai, R.Y.; Roth, M.; Harrington, C.R. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc. Natl. Acad. Sci. USA 1996, 93, 11213–11218. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M.; Binder, L.I. Free fatty acids stimulate the polymerization of tau and amyloid beta peptides. In vitro evidence for a common effector of pathogenesis in Alzheimer’s disease. Am. J. Pathol. 1997, 150, 2181–2195. [Google Scholar] [PubMed]
- Gamblin, T.C.; King, M.E.; Kuret, J.; Berry, R.W.; Binder, L.I. Oxidative regulation of fatty acid-induced tau polymerization. Biochemistry 2000, 39, 14203–14210. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R.; Spillantini, M.G.; Hasegawa, M.; Smith, M.J.; Crowther, R.A. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 1996, 383, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.; Valpuesta, J.M.; Medina, M.; Montejo de Garcini, E.; Avila, J. Polymerization of tau into filaments in the presence of heparin: The minimal sequence required for tau-tau interaction. J. Neurochem. 1996, 67, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Kampers, T.; Friedhoff, P.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996, 399, 344–349. [Google Scholar] [CrossRef]
- Friedhoff, P.; Schneider, A.; Mandelkow, E.M.; Mandelkow, E. Rapid assembly of Alzheimer-like paired helical filaments from microtubule-associated protein tau monitored by fluorescence in solution. Biochemistry 1998, 37, 10223–10230. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, S.; Suzuki, N.; Masuda, M.; Hisanaga, S.; Iwatsubo, T.; Goedert, M.; Hasegawa, M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem. 2005, 280, 7614–7623. [Google Scholar] [CrossRef] [PubMed]
- Bulic, B.; Pickhardt, M.; Schmidt, B.; Mandelkow, E.-M.; Waldmann, H.; Mandelkow, E. Development of tau aggregation inhibitors for Alzheimer’s disease. Angew. Chem. Int. Ed. Engl. 2009, 48, 1740–1752. [Google Scholar] [CrossRef] [PubMed]
- Brunden, K.R.; Ballatore, C.; Crowe, A.; Smith, A.B.; Lee, V.M.-Y.; Trojanowski, J.Q. Tau-directed drug discovery for Alzheimer’s disease and related tauopathies: A focus on tau assembly inhibitors. Exp. Neurol. 2010, 223, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Staff, R.T.; Wischik, D.J.; Bentham, P.; Murray, A.D.; Storey, J.M.D.; Kook, K.A.; Harrington, C.R. Tau aggregation inhibitor therapy: An exploratory phase 2 study in mild or moderate Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 705–720. [Google Scholar] [PubMed]
- Akoury, E.; Pickhardt, M.; Gajda, M.; Biernat, J.; Mandelkow, E.; Zweckstetter, M. Mechanistic basis of phenothiazine-driven inhibition of Tau aggregation. Angew. Chem. Int. Ed. Engl. 2013, 52, 3511–3515. [Google Scholar] [CrossRef] [PubMed]
- Crowe, A.; James, M.J.; Lee, V.M.-Y.; Smith, A.B.; Trojanowski, J.Q.; Ballatore, C.; Brunden, K.R. Aminothienopyridazines and methylene blue affect Tau fibrillization via cysteine oxidation. J. Biol. Chem. 2013, 288, 11024–11037. [Google Scholar] [CrossRef] [PubMed]
- Baddeley, T.C.; McCaffrey, J.; Storey, J.M.D.; Cheung, J.K.S.; Melis, V.; Horsley, D.; Harrington, C.R.; Wischik, C.M. Complex disposition of methylthioninium redox forms determines efficacy in tau aggregation inhibitor therapy for Alzheimer’s disease. J. Pharmacol. Exp. Ther. 2015, 352, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Sillen, A.; Leroy, A.; Wieruszeski, J.-M.; Loyens, A.; Beauvillain, J.-C.; Buée, L.; Landrieu, I.; Lippens, G. Regions of tau implicated in the paired helical fragment core as defined by NMR. Chembiochem 2005, 6, 1849–1856. [Google Scholar] [CrossRef] [PubMed]
- Sillen, A.; Wieruszeski, J.-M.; Leroy, A.; Younes, A.B.; Landrieu, I.; Lippens, G. High-resolution magic angle spinning NMR of the neuronal tau protein integrated in Alzheimer’s-like paired helical fragments. J. Am. Chem. Soc. 2005, 127, 10138–10139. [Google Scholar] [CrossRef] [PubMed]
- Bibow, S.; Mukrasch, M.D.; Chinnathambi, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. The dynamic structure of filamentous tau. Angew. Chem. Int. Ed. Engl. 2011, 50, 11520–11524. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, H.; Hasegawa, M.; Tamaoka, A. Fibrillogenic nuclei composed of P301L mutant tau induce elongation of P301L tau but not wild-type tau. J. Biol. Chem. 2007, 282, 20309–20318. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.; Udgaonkar, J.B. Difference in fibril core stability between two tau four-repeat domain proteins: A hydrogen-deuterium exchange coupled to mass spectrometry study. Biochemistry 2013, 52, 8787–8789. [Google Scholar] [CrossRef] [PubMed]
- Tycko, R. Solid-state NMR studies of amyloid fibril structure. Annu. Rev. Phys. Chem. 2011, 62, 279–299. [Google Scholar] [CrossRef] [PubMed]
- Meier, B.H.; Böckmann, A. The structure of fibrils from “misfolded” proteins. Curr. Opin. Struct. Biol. 2015, 30, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Eichner, T.; Radford, S.E. A diversity of assembly mechanisms of a generic amyloid fold. Mol. Cell 2011, 43, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Tycko, R.; Wickner, R.B. Molecular structures of amyloid and prion fibrils: Consensus versus controversy. Acc. Chem. Res. 2013, 46, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.-X.; Qiang, W.; Yau, W.-M.; Schwieters, C.D.; Meredith, S.C.; Tycko, R. Molecular structure of β-amyloid fibrils in Alzheimer’s disease brain tissue. Cell 2013, 154, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Morozova, O.A.; March, Z.M.; Robinson, A.S.; Colby, D.W. Conformational features of tau fibrils from Alzheimer’s disease brain are faithfully propagated by unmodified recombinant protein. Biochemistry 2013, 52, 6960–6967. [Google Scholar] [CrossRef] [PubMed]
- Meyer, V.; Dinkel, P.D.; Rickman Hager, E.; Margittai, M. Amplification of Tau fibrils from minute quantities of seeds. Biochemistry 2014, 53, 5804–5809. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.W.; McFarlane, H.T.; et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Sievers, S.A.; Karanicolas, J.; Chang, H.W.; Zhao, A.; Jiang, L.; Zirafi, O.; Stevens, J.T.; Münch, J.; Baker, D.; Eisenberg, D. Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature 2011, 475, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Landau, M.; Sawaya, M.R.; Faull, K.F.; Laganowsky, A.; Jiang, L.; Sievers, S.A.; Liu, J.; Barrio, J.R.; Eisenberg, D. Towards a pharmacophore for amyloid. PLoS Biol. 2011, 9, e1001080. [Google Scholar] [CrossRef] [PubMed]
- Andronesi, O.C.; von Bergen, M.; Biernat, J.; Seidel, K.; Griesinger, C.; Mandelkow, E.; Baldus, M. Characterization of Alzheimer’s-like paired helical filaments from the core domain of tau protein using solid-state NMR spectroscopy. J. Am. Chem. Soc. 2008, 130, 5922–5928. [Google Scholar] [CrossRef] [PubMed]
- Daebel, V.; Chinnathambi, S.; Biernat, J.; Schwalbe, M.; Habenstein, B.; Loquet, A.; Akoury, E.; Tepper, K.; Müller, H.; Baldus, M.; et al. β-Sheet core of tau paired helical filaments revealed by solid-state NMR. J. Am. Chem. Soc. 2012, 134, 13982–13989. [Google Scholar] [CrossRef] [PubMed]
- Meyer, V.; Margittai, M. Spin Labeling and Characterization of Tau Fibrils Using Electron Paramagnetic Resonance (EPR). Methods Mol. Biol. 2016, 1345, 185–199. [Google Scholar] [PubMed]
- Margittai, M.; Langen, R. Template-assisted filament growth by parallel stacking of tau. Proc. Natl. Acad. Sci. USA 2004, 101, 10278–10283. [Google Scholar] [CrossRef] [PubMed]
- Abraha, A.; Ghoshal, N.; Gamblin, T.C.; Cryns, V.; Berry, R.W.; Kuret, J.; Binder, L.I. C-terminal inhibition of tau assembly in vitro and in Alzheimer’s disease. J. Cell. Sci. 2000, 113, 3737–3745. [Google Scholar] [PubMed]
- Huvent, I.; Kamah, A.; Cantrelle, F.-X.; Barois, N.; Slomianny, C.; Smet-Nocca, C.; Landrieu, I.; Lippens, G. A functional fragment of Tau forms fibers without the need for an intermolecular cysteine bridge. Biochem. Biophys. Res. Commun. 2014, 445, 299–303. [Google Scholar] [CrossRef] [PubMed]
- King, M.E.; Ghoshal, N.; Wall, J.S.; Binder, L.I.; Ksiezak-Reding, H. Structural analysis of Pick’s disease-derived and in vitro-assembled tau filaments. Am. J. Pathol. 2001, 158, 1481–1490. [Google Scholar] [CrossRef]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda-Diaz, J.E.; Alavi Naini, S.M.; Huynh, M.B.; Ouidja, M.O.; Yanicostas, C.; Chantepie, S.; Villares, J.; Lamari, F.; Jospin, E.; van Kuppevelt, T.H.; et al. HS3ST2 expression is critical for the abnormal phosphorylation of tau in Alzheimer’s disease-related tau pathology. Brain 2015, 138, 1339–1354. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Crowther, R.A.; Jakes, R.; Goedert, M. Alzheimer-like changes in microtubule-associated protein Tau induced by sulfated glycosaminoglycans. Inhibition of microtubule binding, stimulation of phosphorylation, and filament assembly depend on the degree of sulfation. J. Biol. Chem. 1997, 272, 33118–33124. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.-L.; Fernández, C.; Fan, J.-B.; Shewmaker, F.; Chen, J.; Minton, A.P.; Liang, Y. Quantitative characterization of heparin binding to Tau protein: Implication for inducer-mediated Tau filament formation. J. Biol. Chem. 2010, 285, 3592–3599. [Google Scholar] [CrossRef] [PubMed]
- Sibille, N.; Sillen, A.; Leroy, A.; Wieruszeski, J.-M.; Mulloy, B.; Landrieu, I.; Lippens, G. Structural impact of heparin binding to full-length Tau as studied by NMR spectroscopy. Biochemistry 2006, 45, 12560–12572. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Morishima-Kawashima, M.; Takio, K.; Suzuki, M.; Titani, K.; Ihara, Y. Protein sequence and mass spectrometric analyses of tau in the Alzheimer’s disease brain. J. Biol. Chem. 1992, 267, 17047–17054. [Google Scholar] [PubMed]
- Morishima-Kawashima, M.; Hasegawa, M.; Takio, K.; Suzuki, M.; Yoshida, H.; Titani, K.; Ihara, Y. Proline-directed and non-proline-directed phosphorylation of PHF-tau. J. Biol. Chem. 1995, 270, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Knudsen, G.M.; Maeda, S.; Trinidad, J.C.; Ioanoviciu, A.; Burlingame, A.L.; Mucke, L. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 2015, 18, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Biernat, J.; von Bergen, M.; Mandelkow, E.; Mandelkow, E.M. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry 1999, 38, 3549–3558. [Google Scholar] [CrossRef] [PubMed]
- Serber, Z.; Ferrell, J.E. Tuning bulk electrostatics to regulate protein function. Cell 2007, 128, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Biernat, J.; Mandelkow, E.M.; Schröter, C.; Lichtenberg-Kraag, B.; Steiner, B.; Berling, B.; Meyer, H.; Mercken, M.; Vandermeeren, A.; Goedert, M. The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. EMBO J. 1992, 11, 1593–1597. [Google Scholar] [PubMed]
- Goedert, M.; Jakes, R.; Vanmechelen, E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci. Lett. 1995, 189, 167–169. [Google Scholar] [CrossRef]
- Porzig, R.; Singer, D.; Hoffmann, R. Epitope mapping of mAbs AT8 and Tau5 directed against hyperphosphorylated regions of the human tau protein. Biochem. Biophys. Res. Commun. 2007, 358, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Malia, T.J.; Teplyakov, A.; Ernst, R.; Wu, S.-J.; Lacy, E.R.; Liu, X.; Vandermeeren, M.; Mercken, M.; Luo, J.; Sweet, R.W.; et al. Epitope mapping and structural basis for the recognition of phosphorylated tau by the anti-tau antibody AT8. Proteins 2016, 84, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R.; Crowther, R.A.; Six, J.; Lübke, U.; Vandermeeren, M.; Cras, P.; Trojanowski, J.Q.; Lee, V.M. The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proc. Natl. Acad. Sci. USA 1993, 90, 5066–5070. [Google Scholar] [CrossRef] [PubMed]
- Amniai, L.; Lippens, G.; Landrieu, I. Characterization of the AT180 epitope of phosphorylated Tau protein by a combined nuclear magnetic resonance and fluorescence spectroscopy approach. Biochem. Biophys. Res. Commun. 2011, 412, 743–746. [Google Scholar] [CrossRef] [PubMed]
- Rosseels, J.; Van den Brande, J.; Violet, M.; Jacobs, D.; Grognet, P.; Lopez, J.; Huvent, I.; Caldara, M.; Swinnen, E.; Papegaey, A.; et al. Tau monoclonal antibody generation based on humanized yeast models: Impact on Tau oligomerization and diagnostics. J. Biol. Chem. 2015, 290, 4059–4074. [Google Scholar] [CrossRef] [PubMed]
- Bah, A.; Vernon, R.M.; Siddiqui, Z.; Krzeminski, M.; Muhandiram, R.; Zhao, C.; Sonenberg, N.; Kay, L.E.; Forman-Kay, J.D. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature 2015, 519, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.C.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 5562–5566. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.C.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 1996, 2, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar] [CrossRef] [PubMed]
- Tepper, K.; Biernat, J.; Kumar, S.; Wegmann, S.; Timm, T.; Hübschmann, S.; Redecke, L.; Mandelkow, E.-M.; Müller, D.J.; Mandelkow, E. Oligomer formation of tau protein hyperphosphorylated in cells. J. Biol. Chem. 2014, 289, 34389–34407. [Google Scholar] [CrossRef] [PubMed]
- Despres, C.; Lippens, G.; Smet-Nocca, C. Unité de Glycobiologie Structurale et Fontionnelle, University of Lille (UGSF) Lille, France. Unpublished data. 2016. [Google Scholar]
- Chapuis, J.; Hansmannel, F.; Gistelinck, M.; Mounier, A.; Van Cauwenberghe, C.; Kolen, K.V.; Geller, F.; Sottejeau, Y.; Harold, D.; Dourlen, P.; et al. GERAD consortium Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol. Psychiatry 2013, 18, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Zelenika, D.; Hiltunen, M.; Chouraki, V.; Combarros, O.; Bullido, M.J.; Tognoni, G.; Fiévet, N.; Boland, A.; Arosio, B.; et al. Evidence of the association of BIN1 and PICALM with the AD risk in contrasting European populations. Neurobiol. Aging 2011, 32. [Google Scholar] [CrossRef] [PubMed]
- Sottejeau, Y.; Bretteville, A.; Cantrelle, F.-X.; Malmanche, N.; Demiaute, F.; Mendes, T.; Delay, C.; Alves Dos Alves, H.; Flaig, A.; Davies, P.; et al. Tau phosphorylation regulates the interaction between BIN1’s SH3 domain and Tau’s proline-rich domain. Acta Neuropathol. Commun. 2015, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, Y.; Schumacher, B.; Landrieu, I.; Bartel, M.; Smet-Nocca, C.; Jang, A.; Choi, H.S.; Jeon, N.L.; Chang, K.-A.; Kim, H.-S.; et al. Involvement of 14-3-3 in tubulin instability and impaired axon development is mediated by Tau. FASEB J. 2015, 29, 4133–4144. [Google Scholar] [CrossRef] [PubMed]
- Milroy, L.-G.; Bartel, M.; Henen, M.A.; Leysen, S.; Adriaans, J.M.C.; Brunsveld, L.; Landrieu, I.; Ottmann, C. Stabilizer-Guided Inhibition of Protein-Protein Interactions. Angew. Chem. Int. Ed. Engl. 2015, 54, 15720–15724. [Google Scholar] [CrossRef] [PubMed]
- Chambraud, B.; Sardin, E.; Giustiniani, J.; Dounane, O.; Schumacher, M.; Goedert, M.; Baulieu, E.-E. A role for FKBP52 in Tau protein function. Proc. Natl. Acad. Sci. USA 2010, 107, 2658–2663. [Google Scholar] [CrossRef] [PubMed]
- Giustiniani, J.; Chambraud, B.; Sardin, E.; Dounane, O.; Guillemeau, K.; Nakatani, H.; Paquet, D.; Kamah, A.; Landrieu, I.; Lippens, G.; et al. Immunophilin FKBP52 induces Tau-P301L filamentous assembly in vitro and modulates its activity in a model of tauopathy. Proc. Natl. Acad. Sci. USA 2014, 111, 4584–4589. [Google Scholar] [CrossRef] [PubMed]
- Kamah, A.; Cantrelle, F.X.; Huvent, I.; Giustiniani, J.; Guillemeau, K.; Byrne, C.; Jacquot, Y.; Landrieu, I.; Baulieu, E.E.; Smet, C.; et al. Isomerisation and oligomerization of truncated and mutated tau forms by FKBP52 are independent processes. J. Mol. Biol. 2016, 6, 1080–1090. [Google Scholar] [CrossRef] [PubMed]
- Jinwal, U.K.; Akoury, E.; Abisambra, J.F.; O’Leary, J.C.; Thompson, A.D.; Blair, L.J.; Jin, Y.; Bacon, J.; Nordhues, B.A.; Cockman, M.; et al. Imbalance of Hsp70 family variants fosters tau accumulation. FASEB J. 2013, 27, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, S.N.; Rauch, J.N.; Nordhues, B.A.; Assimon, V.A.; Stothert, A.R.; Jinwal, U.K.; Sabbagh, J.J.; Chang, L.; Stevens, S.M.; Zuiderweg, E.R.P.; et al. Isoform-selective Genetic Inhibition of Constitutive Cytosolic Hsp70 Activity Promotes Client Tau Degradation Using an Altered Co-chaperone Complement. J. Biol. Chem. 2015, 290, 13115–13127. [Google Scholar] [CrossRef] [PubMed]
- Karagöz, G.E.; Duarte, A.M.S.; Akoury, E.; Ippel, H.; Biernat, J.; Morán Luengo, T.; Radli, M.; Didenko, T.; Nordhues, B.A.; Veprintsev, D.B.; et al. Hsp90-Tau complex reveals molecular basis for specificity in chaperone action. Cell 2014, 156, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Smet-Nocca, C.; Broncel, M.; Wieruszeski, J.-M.; Tokarski, C.; Hanoulle, X.; Leroy, A.; Landrieu, I.; Rolando, C.; Lippens, G.; Hackenberger, C.P.R. Identification of O-GlcNAc sites within peptides of the Tau protein and their impact on phosphorylation. Mol. Biosyst. 2011, 7, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Kamah, A.; Huvent, I.; Cantrelle, F.-X.; Qi, H.; Lippens, G.; Landrieu, I.; Smet-Nocca, C. Nuclear magnetic resonance analysis of the acetylation pattern of the neuronal Tau protein. Biochemistry 2014, 53, 3020–3032. [Google Scholar] [CrossRef] [PubMed]
- Kondo, A.; Shahpasand, K.; Mannix, R.; Qiu, J.; Moncaster, J.; Chen, C.-H.; Yao, Y.; Lin, Y.-M.; Driver, J.A.; Sun, Y.; et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 2015, 523, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, P.; Cantrelle, F.-X.; Huvent, I.; Hanoulle, X.; Lopez, J.; Smet, C.; Wieruszeski, J.-M.; Landrieu, I.; Lippens, G. Proline Conformation in a Functional Tau Fragment. J. Mol. Biol. 2016, 428, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.H.; Tu, C.; Cao, W.; Klein, A.; Ramsey, R.; Fennell, B. J.; Lambert, M.; Ní Shúilleabháin, D.; Autin, B.; Kouranova, E.; et al. An ultra-specific avian antibody to phosphorylated tau protein reveals a unique mechanism for phosphoepitope recognition. J. Biol. Chem. 2012, 287, 44425–44434. [Google Scholar] [CrossRef] [PubMed]
- Giustiniani, J.; Guillemeau, K.; Dounane, O.; Sardin, E.; Huvent, I.; Schmitt, A.; Hamdane, M.; Buée, L.; Landrieu, I.; Lippens, G.; et al. The FK506-binding protein FKBP52 in vitro induces aggregation of truncated Tau forms with prion-like behavior. FASEB J. 2015, 29, 3171–3181. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.J.; Wulf, G.; Zhou, X.Z.; Davies, P.; Lu, K.P. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 1999, 399, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Liou, Y.-C.; Sun, A.; Ryo, A.; Zhou, X.Z.; Yu, Z.-X.; Huang, H.-K.; Uchida, T.; Bronson, R.; Bing, G.; Li, X.; et al. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature 2003, 424, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Landrieu, I.; Smet-Nocca, C.; Amniai, L.; Louis, J.V.; Wieruszeski, J.-M.; Goris, J.; Janssens, V.; Lippens, G. Molecular implication of PP2A and Pin1 in the Alzheimer’s disease specific hyperphosphorylation of Tau. PLoS ONE 2011, 6, e21521. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lippens, G.; Landrieu, I.; Smet, C.; Huvent, I.; Gandhi, N.S.; Gigant, B.; Despres, C.; Qi, H.; Lopez, J. NMR Meets Tau: Insights into Its Function and Pathology. Biomolecules 2016, 6, 28. https://doi.org/10.3390/biom6020028
Lippens G, Landrieu I, Smet C, Huvent I, Gandhi NS, Gigant B, Despres C, Qi H, Lopez J. NMR Meets Tau: Insights into Its Function and Pathology. Biomolecules. 2016; 6(2):28. https://doi.org/10.3390/biom6020028
Chicago/Turabian StyleLippens, Guy, Isabelle Landrieu, Caroline Smet, Isabelle Huvent, Neha S. Gandhi, Benoît Gigant, Clément Despres, Haoling Qi, and Juan Lopez. 2016. "NMR Meets Tau: Insights into Its Function and Pathology" Biomolecules 6, no. 2: 28. https://doi.org/10.3390/biom6020028
APA StyleLippens, G., Landrieu, I., Smet, C., Huvent, I., Gandhi, N. S., Gigant, B., Despres, C., Qi, H., & Lopez, J. (2016). NMR Meets Tau: Insights into Its Function and Pathology. Biomolecules, 6(2), 28. https://doi.org/10.3390/biom6020028