
Organ–Organ Crosstalk and Alcoholic Liver Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
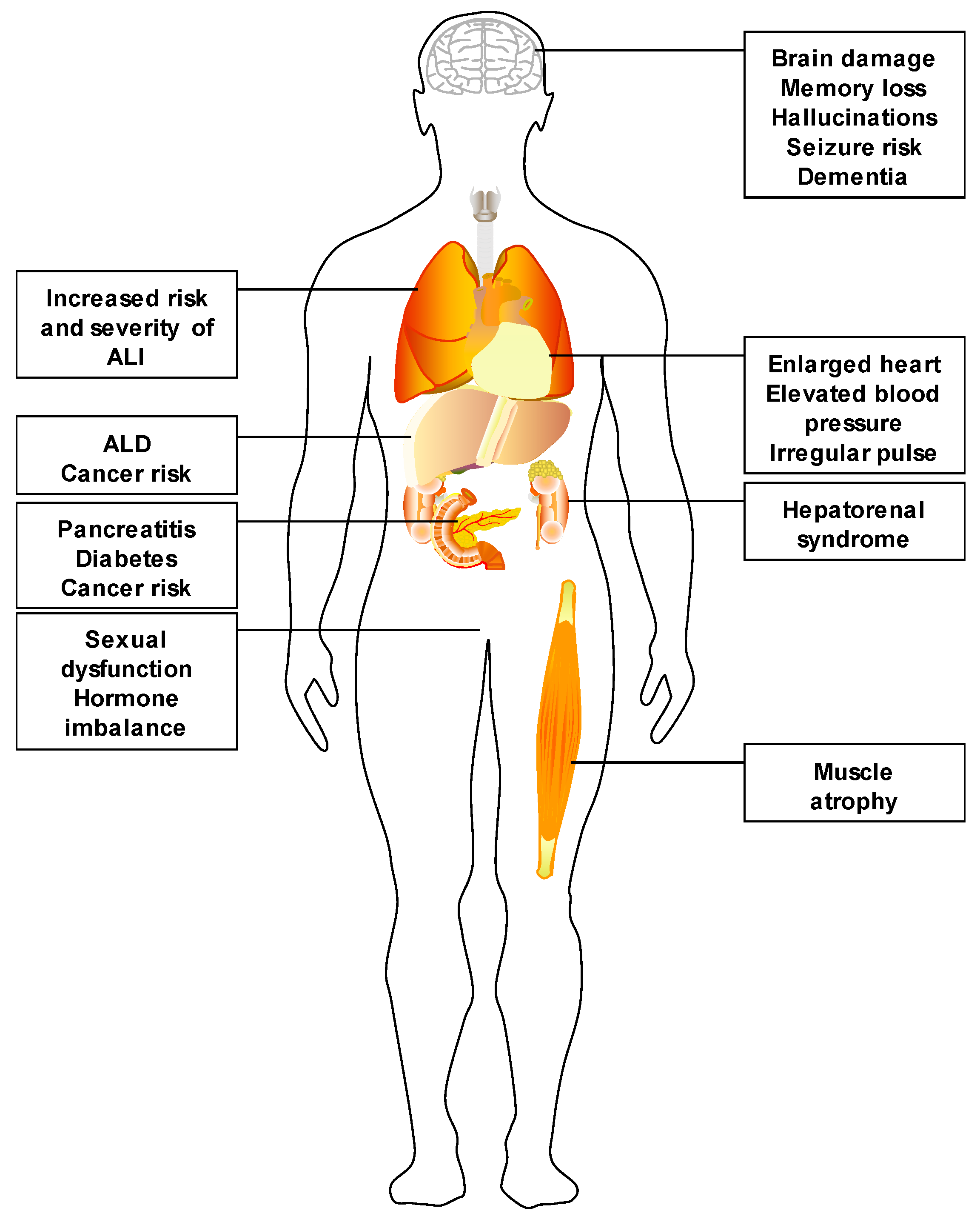
:1. Alcohol Use and Its Impact
2. Alcoholic Liver Disease
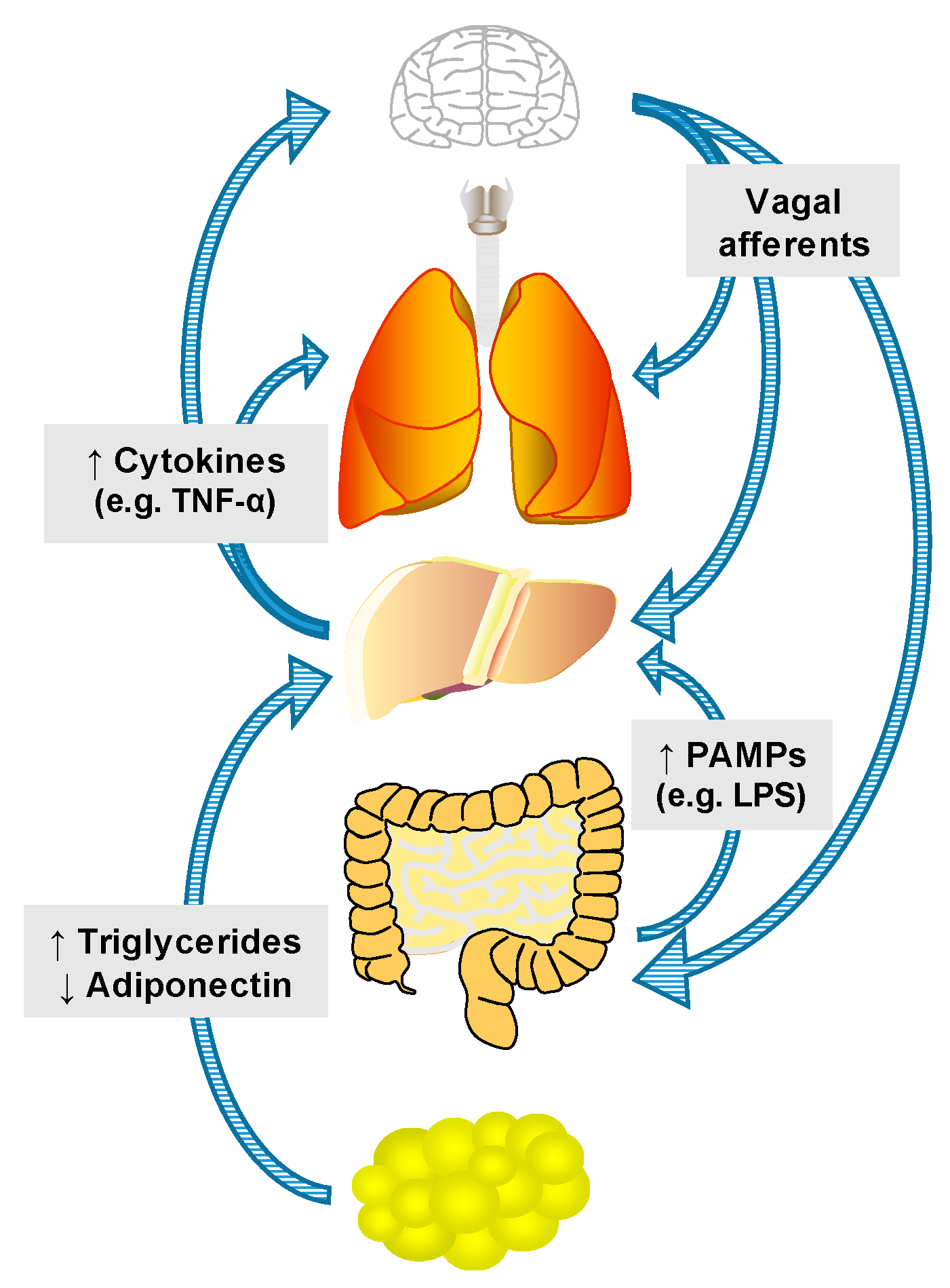
3. Mechanisms of Inter-Organ Communication
3.1. Nutrients, Hormones and Hepatokines
3.2. Innervation
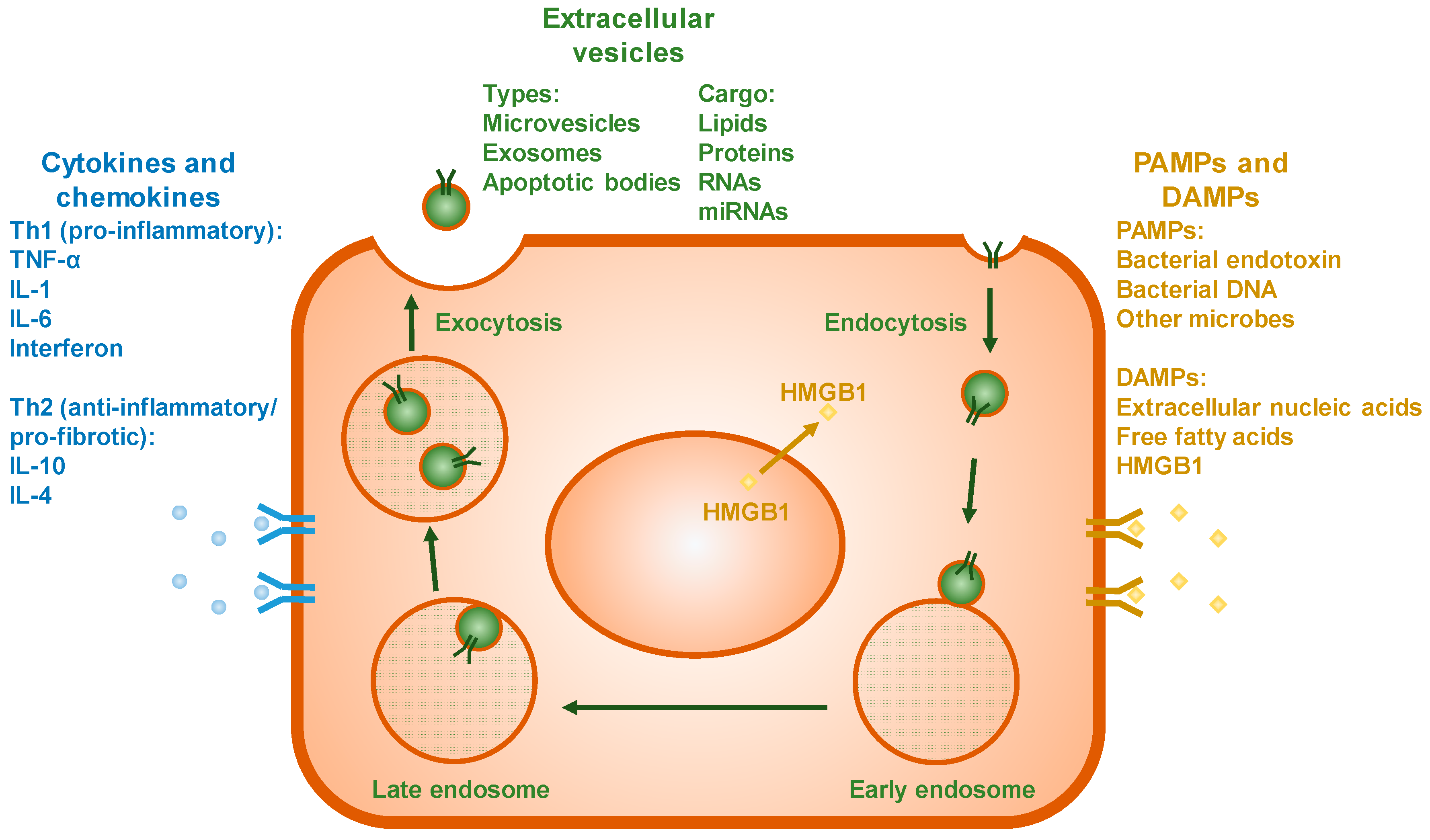
3.3. Extracellular Vesicles
3.4. Cytokines and Chemokines
3.5. PAMPs and DAMPs
4. Known Organ–Organ Interactions in Alcoholic Liver Disease
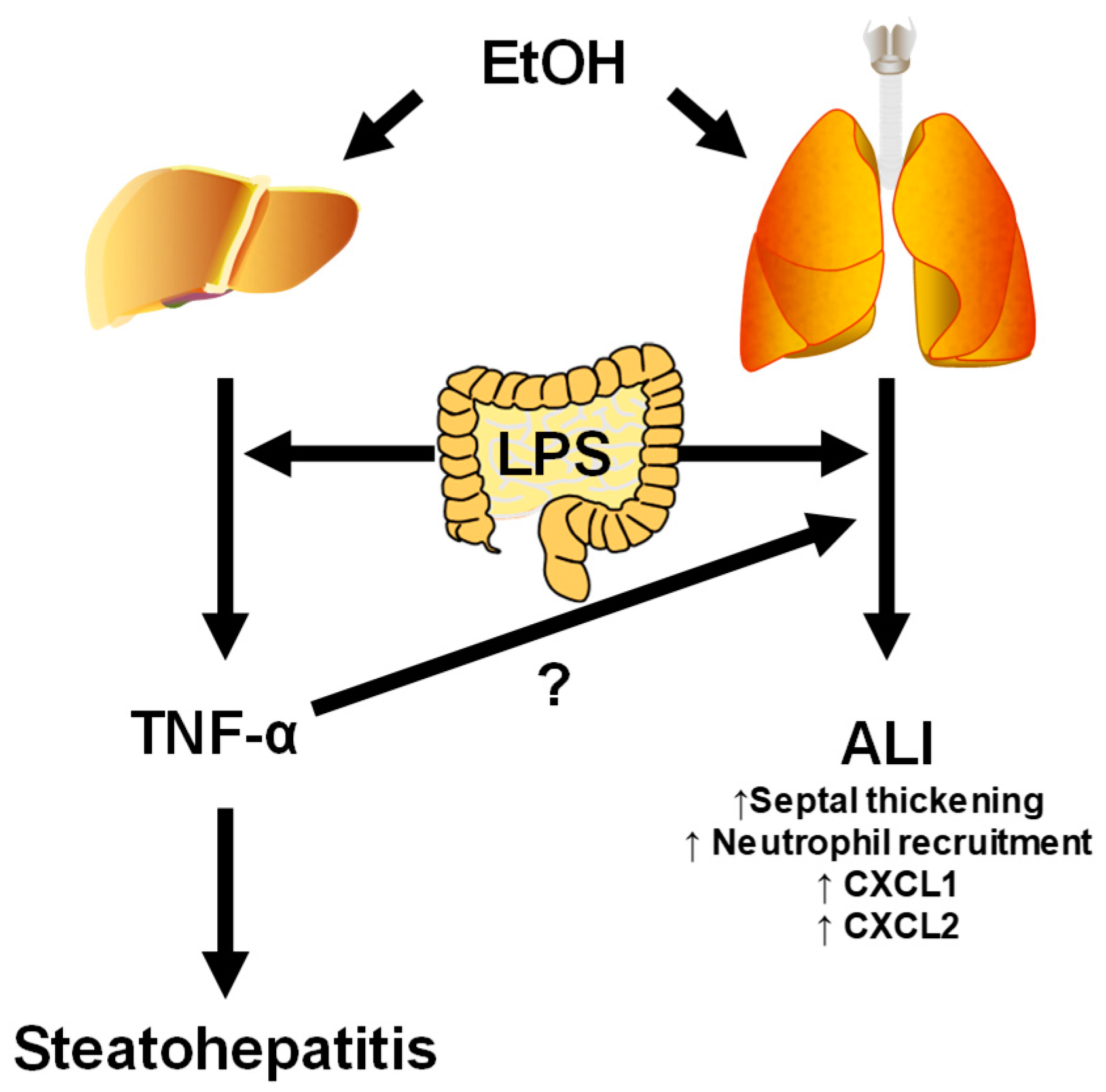
4.1. Gut–Liver Interactions
4.2. Liver–Adipose Interactions
4.3. Liver–Brain Interactions
4.4. Liver–Lung Interactions
5. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Substance Abuse and Mental Health Services Administration. 2015 National Survey on Drug Use and Health; Substance Abuse and Mental Health Services Administration: Rockville, MD, USA, 2015.
- World Health Organization. Global Status Report on Alcohol and Health; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Beier, J.I.; Arteel, G.E.; McClain, C.J. Advances in Alcoholic Liver Disease. Curr. Gastroenterol. Rep. 2011, 13, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Guidot, D.M.; Roman, J. Chronic Ethanol Ingestion Increases Susceptibility to Acute Lung Injury: Role of Oxidative Stress and Tissue Remodeling. Chest 2002, 122, 309S–314S. [Google Scholar] [CrossRef] [PubMed]
- Adachi, J.; Asano, M.; Ueno, Y.; Niemela, O.; Ohlendieck, K.; Peters, T.J.; Preedy, V.R. Alcoholic Muscle Disease and Biomembrane Perturbations (Review). J. Nutr. Biochem. 2003, 14, 616–625. [Google Scholar] [CrossRef]
- Crews, F.T.; Nixon, K. Mechanisms of Neurodegeneration and Regeneration in Alcoholism. Alcohol Alcohol. 2009, 44, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Pandol, S.J.; Raraty, M. Pathobiology of Alcoholic Pancreatitis. Pancreatology 2007, 7, 105–114. [Google Scholar] [CrossRef] [PubMed]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Alcohol Consumption and Ethyl Carbamate. IARC Monogr. Eval. Carcinog. Risks Hum. 2010, 96, 3–1383. [Google Scholar]
- U.S. Burden of Disease Collaborators. The State of U.S. Health, 1990–2010: Burden of Diseases, Injuries, and Risk Factors. JAMA 2013, 310, 591–608. [Google Scholar]
- Levitt, M.D.; Levitt, D.G.; Furne, J.; DeMaster, E.G. Can the Liver Account for First-Pass Metabolism of Ethanol in the Rat? Am. J. Physiol. 1994, 267, G452–G457. [Google Scholar] [PubMed]
- Lelbach, W.K. Liver Damage in Chronic Alcoholism: Results of a Clinical, Clinical-Chemical and Bioptic-Histological Study in 526 Alcoholic Patients During a Low Calorie Diet in an Open Drinking Sanatorium. Acta Hepatosplenol. 1966, 13, 321–349. [Google Scholar] [PubMed]
- Mann, R.E.; Smart, R.G.; Govoni, R. The Epidemiology of Alcoholic Liver Disease. Alcohol Res. Health 2003, 27, 209–219. [Google Scholar] [PubMed]
- Day, C.P. Who Gets Alcoholic Liver Disease: Nature or Nurture? J. R. Coll. Physicians Lond. 2000, 34, 557–562. [Google Scholar] [PubMed]
- Diehl, A.M. Liver Disease in Alcohol Abusers: Clinical Perspective. Alcohol 2002, 27, 7–11. [Google Scholar] [CrossRef]
- Powell, W.J., Jr.; Klatskin, G. Duration of Survival in Patients with Laennec’s Cirrhosis. Influence of Alcohol Withdrawal, and Possible Effects of Recent Changes in General Management of the Disease. Am. J. Med. 1968, 44, 406–420. [Google Scholar] [CrossRef]
- La Vecchia, C.; Negri, E.; D’Avanzo, B.; Boyle, P.; Franceschi, S. Medical History and Primary Liver Cancer. Cancer Res. 1990, 50, 6274–6277. [Google Scholar] [PubMed]
- Gogel, B.M.; Goldstein, R.M.; Kuhn, J.A.; McCarty, T.M.; Donahoe, A.; Glastad, K. Diagnostic Evaluation of Hepatocellular Carcinoma in a Cirrhotic Liver. Oncology 2000, 14, 15–20. [Google Scholar] [PubMed]
- Moreau, R.; Jalan, R.; Gines, P.; Pavesi, M.; Angeli, P.; Cordoba, J.; Durand, F.; Gustot, T.; Saliba, F.; Domenicali, M.; et al. Acute-on-Chronic Liver Failure Is a Distinct Syndrome That Develops in Patients with Acute Decompensation of Cirrhosis. Gastroenterology 2013, 144, 1426–1437. [Google Scholar] [CrossRef] [PubMed]
- Michelena, J.; Altamirano, J.; Abraldes, J.G.; Affò, S.; Morales-Ibanez, O.; Sancho-Bru, P.; Dominguez, M.; García-Pagán, J.C.; Fernández, J.; Arroyo, V.; et al. Systemic Inflammatory Response and Serum Lipopolysaccharide Levels Predict Multiple Organ Failure and Death in Alcoholic Hepatitis. Hepatology 2015, 62, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Rui, L. Energy Metabolism in the Liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [PubMed]
- Iroz, A.; Couty, J.P.; Postic, C. Hepatokines: Unlocking the Multi-Organ Network in Metabolic Diseases. Diabetologia 2015, 58, 1699–1703. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.J.; Lee, D.S.; Kim, W.K.; Han, B.S.; Lee, S.C.; Bae, K.H. Metabolic Adaptation in Obesity and Type II Diabetes: Myokines, Adipokines and Hepatokines. Int. J. Mol. Sci. 2016, 18, 8. [Google Scholar] [CrossRef] [PubMed]
- Lebensztejn, D.M.; Flisiak-Jackiewicz, M.; Bialokoz-Kalinowska, I.; Bobrus-Chociej, A.; Kowalska, I. Hepatokines and Non-Alcoholic Fatty Liver Disease. Acta Biochim. Pol. 2016, 63, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Oka, Y.; Katagiri, H. Inter-Organ Metabolic Communication Involved in Energy Homeostasis: Potential Therapeutic Targets for Obesity and Metabolic Syndrome. Pharmacol. Ther. 2008, 117, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Ferro, J.M.; Oliveira, S. Neurologic Manifestations of Gastrointestinal and Liver Diseases. Curr. Neurol. Neurosci. Rep. 2014, 14, 487. [Google Scholar] [CrossRef] [PubMed]
- Anthony, D.C.; Couch, Y. The Systemic Response to CNS Injury. Exp. Neurol. 2014, 258, 105–111. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M. Insulin Resistance and Neurodegeneration: Progress towards the Development of New Therapeutics for Alzheimer’s Disease. Drugs 2017, 77, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Kingman, G.I.; Goodall, M. Urinary Epinephrine and Levarterenol Excretion During Acute Sublethal Alcohol Intoxication in Dogs. J. Pharmacol. Exp. Ther. 1957, 121, 313–318. [Google Scholar]
- Bravo, I.R.; Acevedo, C.G.; Gallardo, V. Acute Effects of Ethanol on Liver Blood Circulation in the Anesthetized Dog. Alcohol. Clin. Exp. Res. 1980, 4, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Yuki, T.; Bradford, B.U.; Thurman, R.G. Role of Hormones in the Mechanism of the Swift Increase in Alcohol Metabolism in the Rat. Pharmacol. Biochem. Behav. 1980, 13, 67–71. [Google Scholar] [CrossRef]
- Patterson-Buckendahl, P.; Kubovcakova, L.; Krizanova, O.; Pohorecky, L.A.; Kvetnansky, R. Ethanol Consumption Increases Rat Stress Hormones and Adrenomedullary Gene Expression. Alcohol 2005, 37, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.; Matsuda, A.; Yan, I.K.; Parasramka, M.; Patel, T. Extracellular Vesicles in Liver Diseases. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G194–G200. [Google Scholar] [CrossRef] [PubMed]
- Buzas, E.I.; Gyorgy, B.; Nagy, G.; Falus, A.; Gay, S. Emerging Role of Extracellular Vesicles in Inflammatory Diseases. Nat. Rev. Rheumatol. 2014, 10, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Fusegawa, H.; Shiraishi, K.; Ogasawara, F.; Shimizu, M.; Haruki, Y.; Miyachi, H.; Matsuzaki, S.; Ando, Y. Platelet Activation in Patients with Chronic Hepatitis C. Tokai J. Exp. Clin. Med. 2002, 27, 101–106. [Google Scholar] [PubMed]
- Hirsova, P.; Ibrahim, S.H.; Verma, V.K.; Morton, L.A.; Shah, V.H.; LaRusso, N.F.; Gores, G.J.; Malhi, H. Extracellular Vesicles in Liver Pathobiology: Small Particles with Big Impact. Hepatology 2016, 64, 2219–2233. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, F.F. Platelet Activation in Patients with Alcoholic Liver Disease. Tokai J. Exp. Clin. Med. 2005, 30, 41–48. [Google Scholar] [PubMed]
- Eguchi, A.; Lazaro, R.G.; Wang, J.; Kim, J.; Povero, D.; Willliams, B.; Ho, S.B.; Stärkel, P.; Schnabl, B.; Ohno-Machado, L.; et al. Extracellular Vesicles Released by Hepatocytes from Gastric Infusion Model of Alcoholic Liver Disease Contain a MicroRNA Barcode That Can Be Detected in Blood. Hepatology 2017, 65, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.K.; Li, H.; Wang, R.; Hirsova, P.; Mushref, M.; Liu, Y.; Cao, S.; Contreras, P.C.; Malhi, H.; Kamath, P.S.; et al. Alcohol Stimulates Macrophage Activation through Caspase Dependent Hepatocyte Derived Release of CD40L Containing Extracellular Vesicles. J. Hepatol. 2016, 64, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, Y.; Katsuda, T.; Ochiya, T. Circulating MicroRNAs as Hormones: Intercellular and Inter-Organ Conveyors of Epigenetic Information? EXS. 2015, 106, 255–267. [Google Scholar] [PubMed]
- Deng, Z.B.; Poliakov, A.; Hardy, R.W.; Clements, R.; Liu, C.; Liu, Y.; Wang, J.; Xiang, X.; Zhang, S.; Zhuang, X.; et al. Adipose Tissue Exosome-Like Vesicles Mediate Activation of Macrophage-Induced Insulin Resistance. Diabetes 2009, 58, 2498–2505. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Liu, H.; Shen, Y.; Zhang, Y.; Pan, S. Platelet-Derived Microvesicles Are Involved in Cardio-Protective Effects of Remote Preconditioning. Int. J. Clin. Exp. Pathol. 2015, 8, 10832–10839. [Google Scholar] [PubMed]
- Giricz, Z.; Varga, Z.V.; Baranyai, T.; Sipos, P.; Paloczi, K.; Kittel, A.; Buzas, E.I.; Ferdinandy, P. Cardioprotection by Remote Ischemic Preconditioning of the Rat Heart Is Mediated by Extracellular Vesicles. J. Mol. Cell. Cardiol. 2014, 68, 75–78. [Google Scholar] [CrossRef] [PubMed]
- McClain, C.J.; Barve, S.; Deaciuc, I.; Kugelmas, M.; Hill, D. Cytokines in Alcoholic Liver Disease. Semin. Liver Dis. 1999, 19, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Bode, C.H.; Kugler, V.; Bode, J.C.H. Endotoxemia in Patients with Alcoholic and Non-Alcoholic Cirrhosis and in Subjects with No Evidence of Chronic Liver Disease Following Acute Alcohol Excess. J. Hepatol. 1987, 4, 8–14. [Google Scholar] [CrossRef]
- Nolan, J.P. The Role of Endotoxin in Liver Injury. Gastroenterology 1975, 69, 1346–1356. [Google Scholar] [PubMed]
- Khoruts, A.; Stahnke, L.; McClain, C.J.; Logan, G.; Allen, J.I. Circulating Tumor Necrosis Factor, Interleukin-1 and Interleukin-6 Concentrations in Chronic Alcoholic Patients. Hepatology 1991, 13, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Iimuro, Y.; Gallucci, R.M.; Luster, M.I.; Kono, H.; Thurman, R.G. Antibodies to Tumor Necrosis Factor-α Attenuate Hepatic Necrosis and Inflammation Due to Chronic Exposure to Ethanol in the Rat. Hepatology 1997, 26, 1530–1537. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Wheeler, M.D.; Kono, H.; Bradford, B.U.; Gallucci, R.M.; Luster, M.I.; Thurman, R.G. Essential Role of Tumor Necrosis Factor α in Alcohol-Induced Liver Injury. Gastroenterology 1999, 117, 942–952. [Google Scholar] [CrossRef]
- Massey, V.L.; Poole, L.G.; Siow, D.L.; Torres, E.; Warner, N.L.; Schmidt, R.H.; Ritzenthaler, J.D.; Roman, J.; Arteel, G.E. Chronic Alcohol Exposure Enhances Lipopolysaccharide-Induced Lung Injury in Mice: Potential Role of Systemic Tumor Necrosis Factor-α. Alcohol. Clin. Exp. Res. 2015, 39, 1978–1988. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.S.; Knapp, D.J.; Crews, F.T. Systemic LPS Causes Chronic Neuroinflammation and Progressive Neurodegeneration. GLIA 2007, 55, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R.; Szabo, G. Interleukin-1 and Inflammasomes in Alcoholic Liver Disease/Acute Alcoholic Hepatitis and Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. Hepatology 2016, 64, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Hirsiger, S.; Simmen, H.P.; Werner, C.M.; Wanner, G.A.; Rittirsch, D. Danger Signals Activating the Immune Response After Trauma. Mediat. Inflamm. 2012, 2012, 315941. [Google Scholar] [CrossRef] [PubMed]
- Gustot, T.; Lemmers, A.; Moreno, C.; Nagy, N.; Quertinmont, E.; Nicaise, C.; Franchimont, D.; Louis, H.; Deviere, J.; Le, M.O. Differential Liver Sensitization to Toll-Like Receptor Pathways in Mice with Alcoholic Fatty Liver. Hepatology 2006, 43, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, M. The Anti-Toxic Function of the Liver. Lancet 1893, 2, 1092. [Google Scholar]
- Rutenburg, A.M.; Sonnenblick, E.; Koven, I.; Aprahamian, H.A.; Reiner, L.; Fine, J. The Role of Intestinal Bacteria in the Development of Dietary Cirrhosis in Rats. J. Exp. Med. 1957, 106, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nolan, J.P.; Ali, M.V. Endotoxin and the Liver. II Effect of Tolerance on Carbon Tetrachloride Induced Injury. J. Med. 1973, 4, 28–38. [Google Scholar] [PubMed]
- Nolan, J.P.; Leibowitz, A.I. Endotoxin and the Liver. III. Modification of Acute Carbon Tetrachloride Injury by Polymyxin B—An Antiendotoxin. Gastroenterology 1978, 75, 445–449. [Google Scholar] [PubMed]
- Wilkinson, S.P.; Arroyo, V.; Gazzard, B.G.; Moodie, H.; Williams, R. Relation of Renal Impairment and Haemorrhagic Diathesis to Endotoxaemia in Fulminant Hepatic Failure. Lancet 1974, 1, 521–524. [Google Scholar] [CrossRef]
- Tarao, K.; Moroi, T.; Nagakura, Y.; Ikeuchi, T.; Suyama, T.; Endo, O.; Fukushima, K. Relationship Between Endotoxaemia and Protein Concentration of Ascites in Cirrhotic Patients. Gut 1979, 20, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Kirpich, I.A.; Solovieva, N.V.; Leikhter, S.N.; Shidakova, N.A.; Lebedeva, O.V.; Sidorov, P.I.; Bazhukova, T.A.; Soloviev, A.G.; Barve, S.S.; McClain, C.J.; et al. Probiotics Restore Bowel Flora and Improve Liver Enzymes in Human Alcohol-Induced Liver Injury: A Pilot Study. Alcohol 2008, 42, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Zhou, Z.; Wang, L.; Song, Z.; McClain, C.J.; Kang, Y.J. Prevention of Alterations in Intestinal Permeability Is Involved in Zinc Inhibition of Acute Ethanol-Induced Liver Damage in Mice. J. Pharmacol. Exp. Ther. 2003, 305, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Segawa, S.; Wakita, Y.; Hirata, H.; Watari, J. Oral Administration of Heat-Killed Lactobacillus Brevis SBC8803 Ameliorates Alcoholic Liver Disease in Ethanol-Containing Diet-Fed C57BL/6N Mice. Int. J. Food Microbiol. 2008, 128, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Antoine, D.J.; Lu, Y.; Arriazu, E.; Leung, T.M.; Klepper, A.L.; Branch, A.D.; Fiel, M.I.; Nieto, N. High Mobility Group Box-1 (HMGB1) Participates in the Pathogenesis of Alcoholic Liver Disease (ALD). J. Biol. Chem. 2014, 289, 22672–22691. [Google Scholar] [CrossRef] [PubMed]
- Coleman, L.G.; Zou, J.; Crews, F.T. Microglial-Derived miRNA let-7 and HMGB1 Contribute to Ethanol-Induced Neurotoxicity via TLR7. J. Neuroinflamm. 2017, 14, 22. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.Y.; Crews, F.T. Release of Neuronal HMGB1 by Ethanol through Decreased HDAC Activity Activates Brain Neuroimmune Signaling. PLoS ONE 2014, 9, e87915. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Wang, X.; Xu, M.; Yang, F.; Frank, J.A.; Ke, Z.; Luo, J. Binge Ethanol Exposure Causes Endoplasmic Reticulum Stress, Oxidative Stress and Tissue Injury in the Pancreas. Oncotarget 2016, 7, 54303–54316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating Mitochondrial DAMPs Cause Inflammatory Responses to Injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhao, C.; Dong, Y.; Zhang, M.; Wang, Y.; Li, F.; Li, X.; McClain, C.; Yang, S.; Feng, W. Inhibition of miR122a by Lactobacillus Rhamnosus GG Culture Supernatant Increases Intestinal Occludin Expression and Protects Mice from Alcoholic Liver Disease. Toxicol. Lett. 2015, 234, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhang, L.; Forsyth, C.B.; Shaikh, M.; Song, S.; Keshavarzian, A. The Role of miR-212 and iNOS in Alcohol-Induced Intestinal Barrier Dysfunction and Steatohepatitis. Alcohol. Clin. Exp. Res. 2015, 39, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Pettinelli, P.; Videla, L.A. Up-Regulation of PPAR-γ mRNA Expression in the Liver of Obese Patients: An Additional Reinforcing Lipogenic Mechanism to SREBP-1c Induction. J. Clin. Endocrinol. Metab. 2011, 96, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Dasarathy, S.; Brown, J.M. Alcoholic Liver Disease on the Rise: Interorgan Cross Talk Driving Liver Injury. Alcohol. Clin. Exp. Res. 2017, 41, 880–882. [Google Scholar] [CrossRef] [PubMed]
- Stärkel, P.; Schnabl, B. Bidirectional Communication between Liver and Gut during Alcoholic Liver Disease. Semin. Liver Dis. 2016, 36, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Bull-Otterson, L.; Feng, W.; Kirpich, I.; Wang, Y.; Qin, X.; Liu, Y.; Gobejishvili, L.; Joshi-Barve, S.; Ayvaz, T.; Petrosino, J.; et al. Metagenomic Analyses of Alcohol Induced Pathogenic Alterations in the Intestinal Microbiome and the Effect of Lactobacillus Rhamnosus GG Treatment. PLoS ONE 2013, 8, e53028. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, E.A.; Gillevet, P.M.; Rangwala, H.; Sikaroodi, M.; Naqvi, A.; Engen, P.A.; Kwasny, M.; Lau, C.K.; Keshavarzian, A. Colonic Microbiome Is Altered in Alcoholism. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G966–G978. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, S.; Matamoros, S.; Cani, P.D.; Neyrinck, A.M.; Jamar, F.; Stärkel, P.; Windey, K.; Tremaroli, V.; Bäckhed, F.; Verbeke, K.; et al. Intestinal Permeability, Gut-Bacterial Dysbiosis, and Behavioral Markers of Alcohol-Dependence Severity. Proc. Natl. Acad. Sci. USA 2014, 111, E4485–E4493. [Google Scholar] [CrossRef] [PubMed]
- Temko, J.E.; Bouhlal, S.; Farokhnia, M.; Lee, M.R.; Cryan, J.F.; Leggio, L. The Microbiota, the Gut and the Brain in Eating and Alcohol Use Disorders: A ‘Menage a Trois’? Alcohol Alcohol. 2017, 52, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Kim, S.W.; Hong, M.; Suk, K.T. Microbiota-Based Treatments in Alcoholic Liver Disease. World J. Gastroenterol. 2016, 22, 6673–6682. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Chi, F.; Wang, G.; Liu, X.; Zhang, Q.; Chen, Y.; Zhang, H.; Chen, W. Lactobacillus Rhamnosus CCFM1107 Treatment Ameliorates Alcohol-Induced Liver Injury in a Mouse Model of Chronic Alcohol Feeding. J. Microbiol. 2015, 53, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Sidhu, A.; Ma, Z.; McClain, C.; Feng, W. Lactobacillus Rhamnosus GG Culture Supernatant Ameliorates Acute Alcohol-Induced Intestinal Permeability and Liver Injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G32–G41. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Kirpich, I.; Ma, Z.; Wang, C.; Zhang, M.; Suttles, J.; McClain, C.; Feng, W. Lactobacillus Rhamnosus GG Reduces Hepatic TNFα Production and Inflammation in Chronic Alcohol-Induced Liver Injury. J. Nutr. Biochem. 2013, 24, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.C.; Xu, L.M.; Du, S.J.; Huang, S.S.; Wu, H.; Dong, J.J.; Huang, J.R.; Wang, X.D.; Feng, W.K.; Chen, Y.P. Lactobacillus Rhamnosus GG Supernatant Promotes Intestinal Barrier Function, Balances Treg and TH17 Cells and Ameliorates Hepatic Injury in a Mouse Model of Chronic-Binge Alcohol Feeding. Toxicol. Lett. 2016, 241, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Forsyth, C.B.; Banan, A.; Fields, J.Z.; Keshavarzian, A. Oats Supplementation Prevents Alcohol-Induced Gut Leakiness in Rats by Preventing Alcohol-Induced Oxidative Tissue Damage. J. Pharmacol. Exp. Ther. 2009, 329, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, H.; Yin, P.; Fan, H.; Sun, L.; Liu, Y. Flaxseed Oil Ameliorates Alcoholic Liver Disease via Anti-Inflammation and Modulating Gut Microbiota in Mice. Lipids Health Dis. 2017, 16, 44. [Google Scholar] [CrossRef] [PubMed]
- Kalambokis, G.N.; Mouzaki, A.; Rodi, M.; Tsianos, E.V. Rifaximin Improves Thrombocytopenia in Patients with Alcoholic Cirrhosis in Association with Reduction of Endotoxaemia. Liver Int. 2012, 32, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Miyamoto, Y.; Mazagova, M.; Lee, K.C.; Eckmann, L.; Schnabl, B. Microbiota Protects Mice Against Acute Alcohol-Induced Liver Injury. Alcohol. Clin. Exp. Res. 2015, 39, 2313–2323. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Zhong, W.; Li, H. Alteration of Bile Acid Metabolism in the Rat Induced by Chronic Ethanol Consumption. FASEB J. 2013, 27, 3583–3593. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Moschetta, A.; Lee, Y. Regulation of Antibacterial Defense in the Small Intestine by the Nuclear Bile Acid Receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [PubMed]
- Moro-Sibilot, L.; Blanc, P.; Taillardet, M. Mouse and Human Liver Contain Immunoglobulin a-Secreting Cells Originating from Peyer’s Patches and Directed Against Intestinal Antigens. Gastroenterology 2016, 151, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.; Zeng, Z.; O’Neil, M.; Zhao, J.; Whitener, M.; Yu-Jui, W.Y.; Mitchell, E.K.; Handler, M.; Weinman, S.A. The Interaction of Rs738409, Obesity, and Alcohol: A Population-Based Autopsy Study. Am. J. Gastroenterol. 2012, 107, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Ruhl, C.E.; Everhart, J.E. Joint Effects of Body Weight and Alcohol on Elevated Serum Alanine Aminotransferase in the United States Population. Clin. Gastroenterol. Hepatol. 2005, 3, 1260–1268. [Google Scholar] [CrossRef]
- Hart, C.L.; Morrison, D.S.; Batty, G.D.; Mitchell, R.J.; Davey, S.G. Effect of Body Mass Index and Alcohol Consumption on Liver Disease: Analysis of Data from Two Prospective Cohort Studies. BMJ 2010, 340, c1240. [Google Scholar] [CrossRef] [PubMed]
- Alatalo, P.I.; Koivisto, H.M.; Hietala, J.P.; Puukka, K.S.; Bloigu, R.; Niemela, O.J. Effect of Moderate Alcohol Consumption on Liver Enzymes Increases with Increasing Body Mass Index. Am. J. Clin. Nutr. 2008, 88, 1097–1103. [Google Scholar] [PubMed]
- Steiner, J.L.; Lang, C.H. Alcohol, Adipose Tissue, and Lipid Dysregulation. Biomolecules 2017, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Addolorato, G.; Capristo, E.; Greco, A.V.; Stefanini, G.F.; Gasbarrini, G. Energy Expenditure, Substrate Oxidation, and Body Composition in Subjects with Chronic Alcoholism: New Findings from Metabolic Assessment. Alcohol. Clin. Exp. Res. 1997, 21, 962–967. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Zhong, W.; Liu, J.; Song, Z.; McClain, C.J.; Kang, Y.J.; Zhou, Z. Zinc Supplementation Reverses Alcohol-Induced Steatosis in Mice Through Reactivating Hepatocyte Nuclear Factor-4α and Peroxisome Proliferators Activated Receptor-α. Hepatology 2009, 50, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Zhao, Y.; Tang, Y.; Wei, X.; Shi, X.; Sun, W.; Sun, X.; Yin, X.; Sun, X.; Kim, S.; et al. Chronic Alcohol Exposure Stimulates Adipose Tissue Lipolysis in Mice: Role of Reverse Triglyceride Transport in the Pathogenesis of Alcoholic Steatosis. Am. J. Pathol. 2012, 180, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Shi, X.; Zhong, W.; Zhao, Y.; Tang, Y.; Sun, W.; Yin, X.; Bogdanov, B.; Kim, S.; McClain, C.; et al. Chronic Alcohol Exposure Disturbs Lipid Homeostasis at the Adipose Tissue-Liver Axis in Mice: Analysis of Triacylglycerols Using High-Resolution Mass Spectrometry in Combination with in Vivo Metabolite Deuterium Labeling. PLoS ONE 2013, 8, e55382. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sebastian, B.M.; Tang, H.; McMullen, M.M.; Axhemi, A.; Jacobsen, D.W.; Nagy, L.E. Taurine Supplementation Prevents Ethanol-Induced Decrease in Serum Adiponectin and Reduces Hepatic Steatosis in Rats. Hepatology 2009, 49, 1554–1562. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Li, M.; Zheng, D.; Chen, Q.; Liu, W.; Feng, L. Adipose Tissue Hypoxia and Low-Grade Inflammation: A Possible Mechanism for Ethanol-Related Glucose Intolerance? Br. J. Nutr. 2015, 113, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Zappalà, G.; Rechler, M.M. IGFBP-3, Hypoxia and TNF-α Inhibit Adiponectin Transcription. Biochem. Biophys. Res. Commun. 2009, 382, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Wang, Y.; Keshaw, H.; Xu, L.Y.; Lam, K.S.; Cooper, G.J. The Fat-Derived Hormone Adiponectin Alleviates Alcoholic and Nonalcoholic Fatty Liver Diseases in Mice. J. Clin. Investig. 2003, 112, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; He, J.; Hanes, R.N.; Pluzarev, O.; Hong, J.S.; Crews, F.T. Increased Systemic and Brain Cytokine Production and Neuroinflammation by Endotoxin Following Ethanol Treatment. J. Neuroinflamm. 2008, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Mayfield, J.; Ferguson, L.; Harris, R.A. Neuroimmune Signaling: A Key Component of Alcohol Abuse. Curr. Opin. Neurobiol. 2013, 23, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Zakhari, S.; Jung, M.K. Alcohol, Inflammation, and Gut–Liver–Brain Interactions in Tissue Damage and Disease Development. World J. Gastroenterol. 2010, 16, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Richardson, H.N.; Lee, S.Y.; O’Dell, L.E.; Koob, G.F.; Rivier, C.L. Alcohol Self-Administration Acutely Stimulates the Hypothalamic-Pituitary-Adrenal Axis, but Alcohol Dependence Leads to a Dampened Neuroendocrine State. Eur. J. Neurosci. 2008, 28, 1641–1653. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, S.; de Timary, P.; Delzenne, N.M.; Starkel, P. The Link Between Inflammation, Bugs, the Intestine and the Brain in Alcohol Dependence. Transl. Psychiatry 2017, 7, e1048. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; Dinan, T.G. Mind-Altering Microorganisms: The Impact of the Gut Microbiota on Brain and Behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Raison, C.L.; Rutherford, R.E.; Woolwine, B.J.; Shuo, C.; Schettler, P.; Drake, D.F.; Haroon, E.; Miller, A.H. A Randomized Controlled Trial of the Tumor Necrosis Factor-α Antagonist Infliximab in Treatment Resistant Depression: Role of Baseline Inflammatory Biomarkers. JAMA Psychiatry 2013, 70, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Afshar, M.; Smith, G.S.; Terrin, M.L.; Barrett, M.; Lissauer, M.E.; Mansoor, S.; Jeudy, J.; Netzer, G. Blood Alcohol Content, Injury Severity and Acute Respiratory Distress Syndrome. J. Trauma Acute Care Surg. 2014, 76, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.; Bucher, B.; Moore, F.A.; Moore, E.E.; Parsons, P.E. The Role of Chronic Alcohol Abuse in the Development of Acute Respiratory Distress Syndrome in Adults. JAMA 1996, 275, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.; Parsons, P.E.; Steinberg, K.P.; Hudson, L.D.; Guidot, D.M.; Burnham, E.L.; Eaton, S.; Cotsonis, G.A. Chronic Alcohol Abuse Is Associated with an Increased Incidence of Acute Respiratory Distress Syndrome and Severity of Multiple Organ Dysfunction in Patients with Septic Shock. Crit. Care Med. 2003, 31, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Siore, A.M.; Parker, R.E.; Stecenko, A.A.; Cuppels, C.; McKean, M.; Christman, B.W.; Cruz-Gervis, R.; Brigham, K.L. Endotoxin-Induced Acute Lung Injury Requires Interaction with the Liver. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L769–L776. [Google Scholar] [CrossRef] [PubMed]
- Bautista, A.P.; Skrepnik, N.; Niesman, M.R.; Bagby, G.J. Elimination of Macrophages by Liposome-Encapsulated Dichlorovethylene Diphosphonate Suppresses the Endotoxin-Induced Priming of Kupffer Cells. J. Leukoc. Biol. 1994, 55, 321–327. [Google Scholar] [PubMed]
- Patterson, E.K.; Yao, L.J.; Ramic, N.; Lewis, J.F.; Cepinskas, G.; McCaig, L.; Veldhuizen, R.A.; Yamashita, C.M. Lung-Derived Mediators Induce Cytokine Production in Downstream Organs via an NF-κB-Dependent Mechanism. Mediat. Inflamm. 2013, 2013, 586895. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poole, L.G.; Dolin, C.E.; Arteel, G.E. Organ–Organ Crosstalk and Alcoholic Liver Disease. Biomolecules 2017, 7, 62. https://doi.org/10.3390/biom7030062
Poole LG, Dolin CE, Arteel GE. Organ–Organ Crosstalk and Alcoholic Liver Disease. Biomolecules. 2017; 7(3):62. https://doi.org/10.3390/biom7030062
Chicago/Turabian StylePoole, Lauren G., Christine E. Dolin, and Gavin E. Arteel. 2017. "Organ–Organ Crosstalk and Alcoholic Liver Disease" Biomolecules 7, no. 3: 62. https://doi.org/10.3390/biom7030062
APA StylePoole, L. G., Dolin, C. E., & Arteel, G. E. (2017). Organ–Organ Crosstalk and Alcoholic Liver Disease. Biomolecules, 7(3), 62. https://doi.org/10.3390/biom7030062