Rodent Models of Alcoholic Liver Disease: Role of Binge Ethanol Administration


Abstract
:1. Introduction
2. Binge Alcohol Drinking Pattern and Blood Alcohol Concentration
2.1. Binge Drinking in Humans: Definition and Statistics
2.2. Binge Alcohol Administration: Effects on Blood Alcohol Levels in Rodents
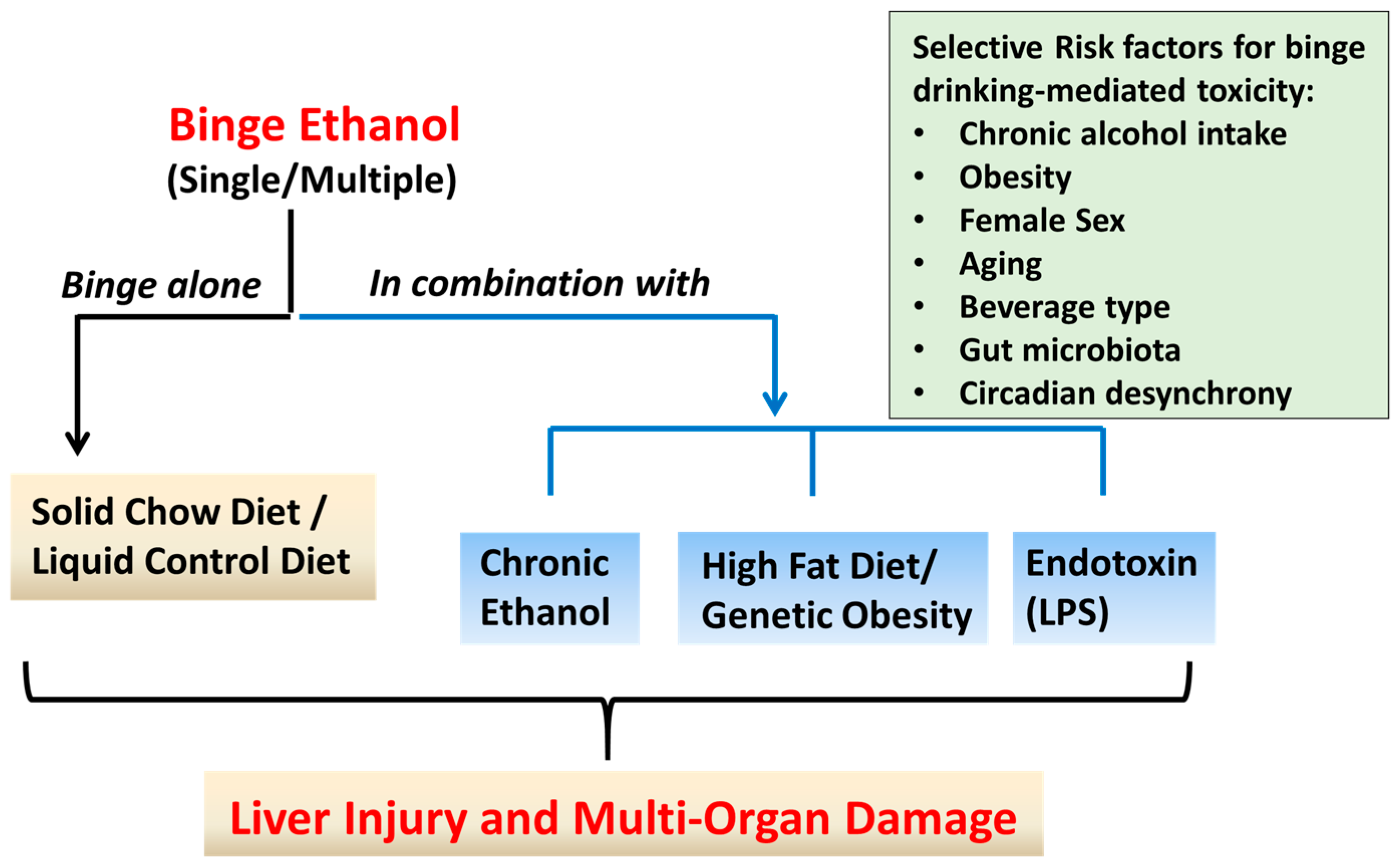
3. Rodents Models of ALD Utilizing Binge Ethanol Administration: Liver Injury Outcomes and Mechanistic Insights
3.1. Single or Multiple Ethanol Binge Administration to Rodents Maintained on Standard Chow Diet
3.2. Rodents Models of Chronic Ethanol Feeding Combined with Binge EtOH Administration
3.2.1. Short-Term Chronic Ethanol Feeding Combined with a Single EtOH Binge (Chronic-Plus-Binge NIAAA Model)
3.2.2. Long-Term Chronic Ethanol Feeding Combined with Single or Multiple EtOH Binges
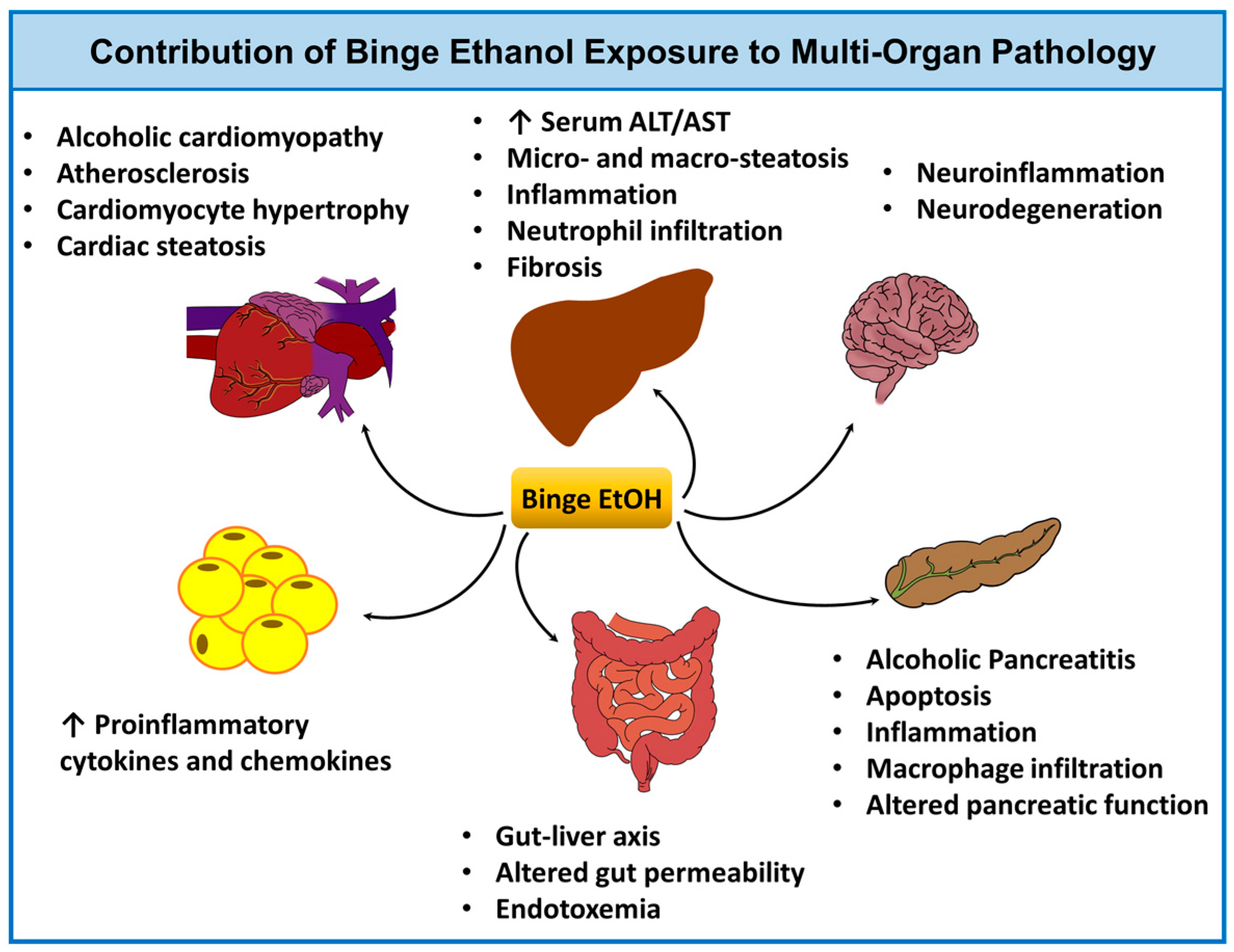
3.2.3. Application of Ethanol Binge Models to Study Multi-Organ Pathology and the Effects of Aging, Sex, and Circadian Rhythm
3.2.4. Chronic Intragastric Ethanol Feeding Combined with Multiple Ethanol Binges (Tsukamoto–French Hybrid Model)
3.3. Rodent Models of High Fat Diet-Induced Liver Injury Combined with Binge Ethanol Administration
3.4. Rodent Model of Combined Binge Ethanol and Lipopolysaccharide Administration
3.5. Technical Considerations
4. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Funding
References
- Becker, U.; Deis, A.; Sorensen, T.I.; Gronbaek, M.; Borch-Johnsen, K.; Muller, C.F.; Schnohr, P.; Jensen, G. Prediction of risk of liver disease by alcohol intake, sex, and age: A prospective population study. Hepatology 1996, 23, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; Amann, M.; Anderson, H.R.; Andrews, K.G.; Aryee, M.; et al. A comparative risk assessment of burden of disease and injury 504 attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A 505 systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef]
- World Heath Organization (WHO). Harmful Use of Alcohol; WHO: Geneva, Switzerland, 2009. [Google Scholar]
- Grant, B.F.; Goldstein, R.B.; Saha, T.D.; Chou, S.P.; Jung, J.; Zhang, H.; Pickering, R.P.; Ruan, W.J.; Smith, S.M.; Huang, B.; et al. Epidemiology of DSM-5 alcohol use disorder: Results from the national epidemiologic survey on alcohol and related conditions III. JAMA Psychiatry 2015, 72, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Lucey, M.R.; Mathurin, P.; Morgan, T.R. Alcoholic hepatitis. N. Engl. J. Med. 2009, 360, 2758–2769. [Google Scholar] [CrossRef] [PubMed]
- Anand, B.S. Cirrhosis of liver. West. J. Med. 1999, 171, 110–115. [Google Scholar] [PubMed]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, R.S.; Dasarathy, S.; McCullough, A.J. Alcoholic liver disease. Hepatology 2010, 51, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Daly, A.K.; Day, C.P. Genetics of alcoholic and nonalcoholic fatty liver disease. Semin. Liver Dis. 2011, 31, 128–146. [Google Scholar] [CrossRef] [PubMed]
- Li, T.K. Quantifying the risk for alcohol-use and alcohol-attributable health disorders: Present findings and future research needs. J. Gastroenterol. Hepatol. 2008, 23 (Suppl. S1), S2–S8. [Google Scholar] [CrossRef] [PubMed]
- Hatton, J.; Burton, A.; Nash, H.; Munn, E.; Burgoyne, L.; Sheron, N. Drinking patterns, dependency and life-time drinking history in alcohol-related liver disease. Addiction 2009, 104, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Bellentani, S.; Saccoccio, G.; Costa, G.; Tiribelli, C.; Manenti, F.; Sodde, M.; Saveria Croce, L.; Sasso, F.; Pozzato, G.; Cristianini, G.; et al. Drinking habits as cofactors of risk for alcohol induced liver damage. The Dionysos Study Group. Gut 1997, 41, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Stranges, S.; Freudenheim, J.L.; Muti, P.; Farinaro, E.; Russell, M.; Nochajski, T.H.; Trevisan, M. Differential effects of alcohol drinking pattern on liver enzymes in men and women. Alcohol. Clin. Exp. Res. 2004, 28, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Askgaard, G.; Gronbaek, M.; Kjaer, M.S.; Tjonneland, A.; Tolstrup, J.S. Alcohol drinking pattern and risk of alcoholic liver cirrhosis: A prospective cohort study. J. Hepatol. 2015, 62, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Rehm, J.; Roerecke, M. Patterns of drinking and liver cirrhosis—What do we know and where do we go? J. Hepatol. 2015, 62, 1000–1001. [Google Scholar] [CrossRef] [PubMed]
- Mann, R.E.; Smart, R.G.; Govoni, R. The epidemiology of alcoholic liver disease. Alcohol Res. Health 2003, 27, 209–219. [Google Scholar] [PubMed]
- Mathurin, P.; Lucey, M.R. Management of alcoholic hepatitis. J. Hepatol. 2012, 56 (Suppl. S1), S39–S45. [Google Scholar] [CrossRef]
- Choi, G.; Runyon, B.A. Alcoholic hepatitis: A clinician’s guide. Clin. Liver Dis. 2012, 16, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Altamirano, J.; Bataller, R. Alcoholic liver disease: Pathogenesis and new targets for therapy. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Bertola, A.; Park, O.; Gao, B. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: A critical role for E-selectin. Hepatology 2013, 58, 1814–1823. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H. Neutrophil-mediated tissue injury in alcoholic hepatitis. Alcohol 2002, 27, 23–27. [Google Scholar] [CrossRef]
- Bautista, A.P. Neutrophilic infiltration in alcoholic hepatitis. Alcohol 2002, 27, 17–21. [Google Scholar] [CrossRef]
- Dominguez, M.; Miquel, R.; Colmenero, J.; Moreno, M.; Garcia-Pagan, J.C.; Bosch, J.; Arroyo, V.; Gines, P.; Caballeria, J.; Bataller, R. Hepatic expression of CXC chemokines predicts portal hypertension and survival in patients with alcoholic hepatitis. Gastroenterology 2009, 136, 1639–1650. [Google Scholar] [CrossRef] [PubMed]
- Carson, E.J.; Pruett, S.B. Development and characterization of a binge drinking model in mice for evaluation of the immunological effects of ethanol. Alcohol. Clin. Exp. Res. 1996, 20, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Zhou, Z.; Wang, L.; Song, Z.; McClain, C.J.; Kang, Y.J. Prevention of alterations in intestinal permeability is involved in zinc inhibition of acute ethanol-induced liver damage in mice. J. Pharmacol. Exp. Ther. 2003, 305, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Kirpich, I.; Ghare, S.; Zhang, J.; Gobejishvili, L.; Kharebava, G.; Barve, S.J.; Barker, D.; Moghe, A.; McClain, C.J.; Barve, S. Binge alcohol-induced microvesicular liver steatosis and injury are associated with down-regulation of hepatic Hdac 1, 7, 9, 10, 11 and up-regulation of Hdac 3. Alcohol. Clin. Exp. Res. 2012, 36, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Banerjee, A.; Jang, S.; Yoo, S.H.; Yun, J.W.; Gonzalez, F.J.; Keshavarzian, A.; Song, B.J. CYP2E1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis. Free Radic. Biol. Med. 2013, 65, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Ki, S.H.; Park, O.; Zheng, M.; Morales-Ibanez, O.; Kolls, J.K.; Bataller, R.; Gao, B. Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: Role of signal transducer and activator of transcription 3. Hepatology 2010, 52, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, T.; Li, Y.M.; Yin, S.; Xu, M.J.; Feng, D.; Zhou, Z.; Zang, M.; Mukhopadhyay, P.; Varga, Z.V.; Pacher, P.; et al. Aging aggravates alcoholic liver injury and fibrosis in mice by downregulating sirtuin 1 expression. J. Hepatol. 2017, 66, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.D.; Aroor, A.R.; Restrepo, R.; Kharbanda, K.K.; Ibdah, J.A. In vivo acute on chronic ethanol effects in liver: A mouse model exhibiting exacerbated injury, altered metabolic and epigenetic responses. Biomolecules 2015, 5, 3280–3294. [Google Scholar] [CrossRef] [PubMed]
- Aroor, A.R.; Restrepo, R.J.; Kharbanda, K.K.; Shukla, S.D. Epigenetic histone modifications in a clinically relevant rat model of chronic ethanol-binge-mediated liver injury. Hepatol. Int. 2014, 8 (Suppl. S2), 421–430. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.J.; Cai, Y.; Wang, H.; Altamirano, J.; Chang, B.; Bertola, A.; Odena, G.; Lu, J.; Tanaka, N.; Matsusue, K.; et al. Fat-Specific protein 27/CIDEC promotes development of alcoholic steatohepatitis in mice and humans. Gastroenterology 2015, 149, 1030–1041.e6. [Google Scholar] [CrossRef] [PubMed]
- Lazaro, R.; Wu, R.; Lee, S.; Zhu, N.L.; Chen, C.L.; French, S.W.; Xu, J.; Machida, K.; Tsukamoto, H. Osteopontin deficiency does not prevent but promotes alcoholic neutrophilic hepatitis in mice. Hepatology 2015, 61, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Carmiel-Haggai, M.; Cederbaum, A.I.; Nieto, N. Binge ethanol exposure increases liver injury in obese rats. Gastroenterology 2003, 125, 1818–1833. [Google Scholar] [CrossRef] [PubMed]
- Nieto, N.; Rojkind, M. Repeated whiskey binges promote liver injury in rats fed a choline-deficient diet. J. Hepatol. 2007, 46, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Xu, M.J.; Zhou, Z.; Cai, Y.; Li, M.; Wang, W.; Feng, D.; Bertola, A.; Wang, H.; Kunos, G.; et al. Short- or long-term high-fat diet feeding plus acute ethanol binge synergistically induce acute liver injury in mice: An important role for CXCL1. Hepatology 2015, 62, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Beier, J.I.; Luyendyk, J.P.; Guo, L.; von Montfort, C.; Staunton, D.E.; Arteel, G.E. Fibrin accumulation plays a critical role in the sensitization to lipopolysaccharide-induced liver injury caused by ethanol in mice. Hepatology 2009, 49, 1545–1553. [Google Scholar] [CrossRef] [PubMed]
- National Institute on Alcohol Abuse and Alcoholism (NIAAA). NIAAA Drinking Levels Defined. Available online: https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/moderate-binge-drinking (accessed on 1 December 2017).
- National Institute on Alcohol Abuse and Alcoholism (NIAAA). What Is A Standard Drink? Description of Different Alcoholic Beverages and Their Alcohol Content. Available online: https://niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/what-standard-drink (accessed on December 2017).
- Bouchery, E.E.; Harwood, H.J.; Sacks, J.J.; Simon, C.J.; Brewer, R.D. Economic costs of excessive alcohol consumption in the US, 2006. Am. J. Prev. Med. 2011, 41, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Stahre, M.; Roeber, J.; Kanny, D.; Brewer, R.D.; Zhang, X. Contribution of excessive alcohol consumption to deaths and years of potential life lost in the United States. Prev. Chronic Dis. 2014, 11, E109. [Google Scholar] [CrossRef] [PubMed]
- Gronbaek, M.; Jensen, M.K.; Johansen, D.; Sorensen, T.I.; Becker, U. Intake of beer, wine and spirits and risk of heavy drinking and alcoholic cirrhosis. Biol. Res. 2004, 37, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Sacks, J.J.; Gonzales, K.R.; Bouchery, E.E.; Tomedi, L.E.; Brewer, R.D. 2010 National and State Costs of Excessive Alcohol Consumption. Am. J. Prev. Med. 2015, 49, e73–E79. [Google Scholar] [CrossRef] [PubMed]
- Substance Abuse and Mental Health Services Administration (SAMHSA). 2015 National Survey on Drug Use and Health (NSDUH); Table 6.84D—Tobacco Product and Alcohol Use in Past Month among Persons Aged 18 to 22, by College Enrollment Status: Percentages 2014 and 2015; SAMHSA: Rockville, MD, USA, 2015.
- Holmes, R.S.; Duley, J.A.; Algar, E.M.; Mather, P.B.; Rout, U.K. Biochemical and genetic studies on enzymes of alcohol metabolism: The mouse as a model organism for human studies. Alcohol Alcohol. 1986, 21, 41–56. [Google Scholar] [PubMed]
- Roychowdhury, S.; McMullen, M.R.; Pritchard, M.T.; Hise, A.G.; van Rooijen, N.; Medof, M.E.; Stavitsky, A.B.; Nagy, L.E. An early complement-dependent and TLR-4—independent phase in the pathogenesis of ethanol-induced liver injury in mice. Hepatology 2009, 49, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Hritz, I.; Mandrekar, P.; Velayudham, A.; Catalano, D.; Dolganiuc, A.; Kodys, K.; Kurt-Jones, E.; Szabo, G. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology 2008, 48, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Zhong, W.; Liu, J.; Song, Z.; McClain, C.J.; Kang, Y.J.; Zhou, Z. Zinc supplementation reverses alcohol-induced steatosis in mice through reactivating hepatocyte nuclear factor-4α and peroxisome proliferator-activated receptor-α. Hepatology 2009, 50, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Liang, X.; Rogers, C.Q.; Rideout, D.; You, M. Involvement of adiponectin-SIRT1-AMPK signaling in the protective action of rosiglitazone against alcoholic fatty liver in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G364–G374. [Google Scholar] [CrossRef] [PubMed]
- Leo, M.A.; Lieber, C.S. Hepatic fibrosis after long-term administration of ethanol and moderate vitamin A supplementation in the rat. Hepatology 1983, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; DeCarli, L.M. The feeding of alcohol in liquid diets: Two decades of applications and 1982 update. Alcohol. Clin. Exp. Res. 1982, 6, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; DeCarli, L.M. Liquid diet technique of ethanol administration: 1989 update. Alcohol Alcohol. 1989, 24, 197–211. [Google Scholar] [PubMed]
- Tsukamoto, H.; French, S.W.; Reidelberger, R.D.; Largman, C. Cyclical pattern of blood alcohol levels during continuous intragastric ethanol infusion in rats. Alcohol. Clin. Exp. Res. 1985, 9, 31–37. [Google Scholar] [CrossRef] [PubMed]
- French, S.W. Intragastric ethanol infusion model for cellular and molecular studies of alcoholic liver disease. J. Biomed. Sci. 2001, 8, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Mkrtchyan, H.; Dynnyk, A. Intragastric ethanol infusion model in rodents. Methods Mol. Biol. 2008, 447, 33–48. [Google Scholar] [PubMed]
- Ueno, A.; Lazaro, R.; Wang, P.Y.; Higashiyama, R.; Machida, K.; Tsukamoto, H. Mouse intragastric infusion (iG) model. Nat. Protoc. 2012, 7, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.J.; Wolcott, R.M.; Pruett, S.B. Ethanol decreases the number and activity of splenic natural killer cells in a mouse model for binge drinking. J. Pharmacol. Exp. Ther. 1994, 271, 722–729. [Google Scholar] [PubMed]
- Matyas, C.; Varga, Z.V.; Mukhopadhyay, P.; Paloczi, J.; Lajtos, T.; Erdelyi, K.; Nemeth, B.T.; Nan, M.; Hasko, G.; Gao, B.; et al. Chronic plus binge ethanol feeding induces myocardial oxidative stress, mitochondrial and cardiovascular dysfunction, and steatosis. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1658–H1670. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Wang, X.; Xu, M.; Yang, F.; Frank, J.A.; Ke, Z.J.; Luo, J. Binge ethanol exposure causes endoplasmic reticulum stress, oxidative stress and tissue injury in the pancreas. Oncotarget 2016, 7, 54303–54316. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Yang, F.; Wang, X.; Wang, Y.; Xu, M.; Frank, J.A.; Ke, Z.J.; Zhang, Z.; Shi, X.; Luo, J. Chronic plus binge ethanol exposure causes more severe pancreatic injury and inflammation. Toxicol. Appl. Pharmacol. 2016, 308, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Aroor, A.R.; Jackson, D.E.; Shukla, S.D. Elevated activation of ERK1 and ERK2 accompany enhanced liver injury following alcohol binge in chronically ethanol-fed rats. Alcohol. Clin. Exp. Res. 2011, 35, 2128–2138. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Miyamoto, Y.; Mazagova, M.; Lee, K.C.; Eckmann, L.; Schnabl, B. Microbiota protects mice against acute alcohol-induced liver injury. Alcohol. Clin. Exp. Res. 2015, 39, 2313–2323. [Google Scholar] [CrossRef] [PubMed]
- Bala, S.; Marcos, M.; Gattu, A.; Catalano, D.; Szabo, G. Acute binge drinking increases serum endotoxin and bacterial DNA levels in healthy individuals. PLoS ONE 2014, 9, e96864. [Google Scholar] [CrossRef] [PubMed]
- Kirpich, I.A.; McClain, C.J.; Vatsalya, V.; Schwandt, M.; Phillips, M.; Falkner, K.C.; Zhang, L.; Harwell, C.; George, D.T.; Umhau, J.C. Liver injury and endotoxemia in male and female alcohol-dependent individuals admitted to an alcohol treatment program. Alcohol. Clin. Exp. Res. 2017, 41, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G. Gut-liver axis in alcoholic liver disease. Gastroenterology 2015, 148, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Bala, S.; Petrasek, J.; Gattu, A. Gut-liver axis and sensing microbes. Dig. Dis. 2010, 28, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Goslawski, M.; Piano, M.R.; Bian, J.T.; Church, E.C.; Szczurek, M.; Phillips, S.A. Binge drinking impairs vascular function in young adults. J. Am. Coll. Cardiol. 2013, 62, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Redmond, E.M.; Morrow, D.; Cullen, J.P. Differential effects of daily-moderate versus weekend-binge alcohol consumption on atherosclerotic plaque development in mice. Atherosclerosis 2011, 219, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Pletcher, M.J.; Varosy, P.; Kiefe, C.I.; Lewis, C.E.; Sidney, S.; Hulley, S.B. Alcohol consumption, binge drinking, and early coronary calcification: Findings from the coronary artery risk development in young adults (cardia) study. Am. J. Epidemiol. 2005, 161, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Waszkiewicz, N.; Szulc, A.; Zwierz, K. Binge drinking-induced subtle myocardial injury. Alcohol. Clin. Exp. Res. 2013, 37, 1261–1263. [Google Scholar] [CrossRef] [PubMed]
- Zagrosek, A.; Messroghli, D.; Schulz, O.; Dietz, R.; Schulz-Menger, J. Effect of binge drinking on the heart as assessed by cardiac magnetic resonance imaging. JAMA 2010, 304, 1328–1330. [Google Scholar] [CrossRef] [PubMed]
- Fulham, M.A.; Mandrekar, P. Sexual dimorphism in alcohol induced adipose inflammation relates to liver injury. PLoS ONE 2016, 11, e0164225. [Google Scholar] [CrossRef] [PubMed]
- Bank, S.; Indaram, A. Causes of acute and recurrent pancreatitis. Clinical considerations and clues to diagnosis. Gastroenterol. Clin. N. Am. 1999, 28, 571–589. [Google Scholar] [CrossRef]
- Deng, L.; Xue, P.; Huang, L.; Yang, X.; Wan, M.; Xia, Q. Binge drinking aggravates the outcomes of first-attack severe acute pancreatitis. Pancreas 2010, 39, 149–152. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Kril, J.J. Human alcohol-related neuropathology. Acta Neuropathol. 2014, 127, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Coehlo, M.; McGregor, H.A.; Waltermire, R.S.; Szumlinski, K.K. Binge alcohol drinking elicits persistent negative affect in mice. Behav. Brain Res. 2015, 291, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Nixon, K.; Crews, F.T. Binge ethanol exposure decreases neurogenesis in adult rat hippocampus. J. Neurochem. 2002, 83, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Shukla, S.D. Acute in vivo effect of ethanol (binge drinking) on histone H3 modifications in rat tissues. Alcohol Alcohol. 2006, 41, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Bardag-Gorce, F.; Oliva, J.; Wong, W.; Fong, S.; Li, J.; French, B.A.; French, S.W. S-Adenosylmethionine decreases the peak blood alcohol levels 3 h after an acute bolus of ethanol by inducing alcohol metabolizing enzymes in the liver. Exp. Mol. Pathol. 2010, 89, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Landmann, M.; Wagnerberger, S.; Kanuri, G.; Ziegenhardt, D.; Bergheim, I. Beer is less harmful for the liver than plain ethanol: Studies in male mice using a binge-drinking model. Alcohol Alcohol. 2015, 50, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Landmann, M.; Sellmann, C.; Engstler, A.J.; Ziegenhardt, D.; Jung, F.; Brombach, C.; Bergheim, I. Hops (humulus lupulus) content in beer modulates effects of beer on the liver after acute ingestion in female mice. Alcohol Alcohol. 2017, 52, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Cui, K.; Yan, G.; Xu, C.; Chen, Y.; Wang, J.; Zhou, R.; Bai, L.; Lian, Z.; Wei, H.; Sun, R.; et al. Invariant NKT cells promote alcohol-induced steatohepatitis through interleukin-1β in mice. J. Hepatol. 2015, 62, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Mathews, S.; Feng, D.; Maricic, I.; Ju, C.; Kumar, V.; Gao, B. Invariant natural killer T cells contribute to chronic-plus-binge ethanol-mediated liver injury by promoting hepatic neutrophil infiltration. Cell. Mol. Immunol. 2016, 13, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Maricic, I.; Sheng, H.; Marrero, I.; Seki, E.; Kisseleva, T.; Chaturvedi, S.; Molle, N.; Mathews, S.A.; Gao, B.; Kumar, V. Inhibition of type I natural killer T cells by retinoids or following sulfatide-mediated activation of type II natural killer T cells attenuates alcoholic liver disease in mice. Hepatology 2015, 61, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Xu, M.J.; Koritzinsky, E.H.; Zhou, Z.; Wang, W.; Cao, H.; Yuen, P.S.; Ross, R.A.; Star, R.A.; Liangpunsakul, S.; et al. Mitochondrial DNA-enriched microparticles promote acute-on-chronic alcoholic neutrophilia and hepatotoxicity. JCI Insight 2017, 20, 2. [Google Scholar] [CrossRef] [PubMed]
- Aroor, A.R.; Roy, L.J.; Restrepo, R.J.; Mooney, B.P.; Shukla, S.D. A proteomic analysis of liver after ethanol binge in chronically ethanol treated rats. Proteome Sci. 2012, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Crews, F.T. NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J. Neuroinflamm. 2012, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Kane, C.J.; Phelan, K.D.; Douglas, J.C.; Wagoner, G.; Johnson, J.W.; Xu, J.; Phelan, P.S.; Drew, P.D. Effects of ethanol on immune response in the brain: Region-specific changes in adolescent versus adult mice. Alcohol. Clin. Exp. Res. 2014, 38, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Stickel, F. Alcoholic liver disease in the elderly. Clin. Geriatr. Med. 2007, 23, 905–921. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Xu, Y.; Simanowski, U.A.; Osswald, B. Effect of age and gender on in vivo ethanol elimination, hepatic alcohol dehydrogenase activity, and NAD+ availability in F344 rats. Res. Exp. Med. 1992, 192, 205–212. [Google Scholar] [CrossRef]
- Seitz, H.K.; Meydani, M.; Ferschke, I.; Simanowski, U.A.; Boesche, J.; Bogusz, M.; Hoepker, W.W.; Blumberg, J.B.; Russell, R.M. Effect of aging on in vivo and in vitro ethanol metabolism and its toxicity in F344 rats. Gastroenterology 1989, 97, 446–456. [Google Scholar] [CrossRef]
- Choudhury, M.; Jonscher, K.R.; Friedman, J.E. Reduced mitochondrial function in obesity-associated fatty liver: SIRT3 takes on the fat. Aging 2011, 3, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Meier, P.; Seitz, H.K. Age, alcohol metabolism and liver disease. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Lindros, K.O.; Baraona, E.; Ikejima, K.; Mezey, E.; Jarvelainen, H.A.; Ramchandani, V.A. Sex difference in alcohol-related organ injury. Alcohol. Clin. Exp. Res. 2001, 25, 40s–45s. [Google Scholar] [CrossRef] [PubMed]
- Frezza, M.; di Padova, C.; Pozzato, G.; Terpin, M.; Baraona, E.; Lieber, C.S. High blood alcohol levels in women. The role of decreased gastric alcohol dehydrogenase activity and first-pass metabolism. N. Engl. J. Med. 1990, 322, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Lelbach, W.K. Epidemiology of alcoholic liver disease. Prog. Liver Dis. 1976, 5, 494–515. [Google Scholar] [PubMed]
- Iimuro, Y.; Frankenberg, M.V.; Arteel, G.E.; Bradford, B.U.; Wall, C.A.; Thurman, R.G. Female rats exhibit greater susceptibility to early alcohol-induced liver injury than males. Am. J. Physiol. 1997, 272, G1186–G1194. [Google Scholar] [CrossRef] [PubMed]
- Kono, H.; Wheeler, M.D.; Rusyn, I.; Lin, M.; Seabra, V.; Rivera, C.A.; Bradford, B.U.; Forman, D.T.; Thurman, R.G. Gender differences in early alcohol-induced liver injury: Role of CD14, NF-κB, and TNF-α. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G652–G661. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Ikejima, K.; Wheeler, M.D.; Bradford, B.U.; Seabra, V.; Forman, D.T.; Sato, N.; Thurman, R.G. Estrogen is involved in early alcohol-induced liver injury in a rat enteral feeding model. Hepatology 2000, 31, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Inamine, T.; Yang, A.M.; Wang, L.; Lee, K.C.; Llorente, C.; Schnabl, B. Genetic loss of immunoglobulin a does not influence development of alcoholic steatohepatitis in mice. Alcohol. Clin. Exp. Res. 2016, 40, 2604–2613. [Google Scholar] [CrossRef] [PubMed]
- Filiano, A.N.; Millender-Swain, T.; Johnson, R., Jr.; Young, M.E.; Gamble, K.L.; Bailey, S.M. Chronic ethanol consumption disrupts the core molecular clock and diurnal rhythms of metabolic genes in the liver without affecting the suprachiasmatic nucleus. PLoS ONE 2013, 8, e71684. [Google Scholar] [CrossRef] [PubMed]
- Summa, K.C.; Voigt, R.M.; Forsyth, C.B.; Shaikh, M.; Cavanaugh, K.; Tang, Y.; Vitaterna, M.H.; Song, S.; Turek, F.W.; Keshavarzian, A. Disruption of the circadian clock in mice increases intestinal permeability and promotes alcohol-induced hepatic pathology and inflammation. PLoS ONE 2013, 8, e67102. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Ross, R.A.; Pywell, C.M.; Liangpunsakul, S.; Duffield, G.E. Disturbances in the murine hepatic circadian clock in alcohol-induced hepatic steatosis. Sci. Rep. 2014, 4, 3725. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.B.; Voigt, R.M.; Burgess, H.J.; Swanson, G.R.; Keshavarzian, A. Circadian rhythms, alcohol and gut interactions. Alcohol 2015, 49, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Udoh, U.S.; Valcin, J.A.; Gamble, K.L.; Bailey, S.M. The molecular circadian clock and alcohol-induced liver injury. Biomolecules 2015, 5, 2504–2537. [Google Scholar] [CrossRef] [PubMed]
- Damaggio, A.S.; Gorman, M.R. The circadian timing system in ethanol consumption and dependence. Behav. Neurosci. 2014, 128, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Hasler, B.P.; Clark, D.B. Circadian misalignment, reward-related brain function, and adolescent alcohol involvement. Alcohol. Clin. Exp. Res. 2013, 37, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Rosenwasser, A.M. Circadian clock genes: Non-circadian roles in sleep, addiction, and psychiatric disorders? Neurosci. Biobehav. Rev. 2010, 34, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Zhang, D.; Arthurs, B.; Li, P.; Durudogan, L.; Gupta, N.; Yin, L. Palmitate inhibits SIRT1-dependent BMAL1/CLOCK interaction and disrupts circadian gene oscillations in hepatocytes. PLoS ONE 2015, 10, e0130047. [Google Scholar] [CrossRef] [PubMed]
- Kudo, T.; Tamagawa, T.; Shibata, S. Effect of chronic ethanol exposure on the liver of Clock-mutant mice. J. Circadian Rhythm. 2009, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.; Yang, Z.; Liangpunsakul, S.; Wang, L. Metabolomics analysis revealed distinct cyclic changes of metabolites altered by chronic ethanol-plus-binge and Shp deficiency. Alcohol. Clin. Exp. Res. 2016, 40, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Tsuchiya, H.; Zhang, Y.; Lee, S.; Liu, C.; Huang, Y.; Vargas, G.M.; Wang, L. REV-ERBα activates C/EBP homologous protein to control small heterodimer partner-mediated oscillation of alcoholic fatty liver. Am. J. Pathol. 2016, 186, 2909–2920. [Google Scholar] [CrossRef] [PubMed]
- Naveau, S.; Giraud, V.; Borotto, E.; Aubert, A.; Capron, F.; Chaput, J.C. Excess weight risk factor for alcoholic liver disease. Hepatology 1997, 25, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Iturriaga, H.; Bunout, D.; Hirsch, S.; Ugarte, G. Overweight as a risk factor or a predictive sign of histological liver damage in alcoholics. Am. J. Clin. Nutr. 1988, 47, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Naveau, S.; Dobrin, A.S.; Balian, A.; Njike-Nakseu, M.; Nohra, P.; Asnacios, A.; Prevot, S.; Perlemuter, G. Body fat distribution and risk factors for fibrosis in patients with alcoholic liver disease. Alcohol. Clin. Exp. Res. 2013, 37, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Ruhl, C.E.; Everhart, J.E. Joint effects of body weight and alcohol on elevated serum alanine aminotransferase in the United States population. Clin. Gastroenterol. Hepatol. 2005, 3, 1260–1268. [Google Scholar] [CrossRef]
- Alatalo, P.I.; Koivisto, H.M.; Hietala, J.P.; Puukka, K.S.; Bloigu, R.; Niemela, O.J. Effect of moderate alcohol consumption on liver enzymes increases with increasing body mass index. Am. J. Clin. Nutr. 2008, 88, 1097–1103. [Google Scholar] [PubMed]
- Shen, Z.; Li, Y.; Yu, C.; Shen, Y.; Xu, L.; Xu, C.; Xu, G. A cohort study of the effect of alcohol consumption and obesity on serum liver enzyme levels. Eur. J. Gastroenterol. Hepatol. 2010, 22, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Bettencourt, R.; Barrett-Connor, E. Synergistic association between alcohol intake and body mass index with serum alanine and aspartate aminotransferase levels in older adults: The Rancho Bernardo Study. Aliment. Pharmacol. Ther. 2009, 30, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Hart, C.L.; Morrison, D.S.; Batty, G.D.; Mitchell, R.J.; Davey Smith, G. Effect of body mass index and alcohol consumption on liver disease: Analysis of data from two prospective cohort studies. BMJ 2010, 340, c1240. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Brauner, B.; Bode, J.C.; Bode, C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: Reevaluation with an improved chromogenic assay. J. Hepatol. 1991, 12, 162–169. [Google Scholar] [CrossRef]
- Soares, J.B.; Pimentel-Nunes, P.; Roncon-Albuquerque, R.; Leite-Moreira, A. The role of lipopolysaccharide/toll-like receptor 4 signaling in chronic liver diseases. Hepatol. Int. 2010, 4, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Soto, M.; Chaumontet, C.; Even, P.C.; Azzout-Marniche, D.; Tome, D.; Fromentin, G. Metabolic effects of intermittent access to caloric or non-caloric sweetened solutions in mice fed a high-caloric diet. Physiol. Behav. 2017, 175, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, M.; Ji, C.; Kosyk, O.; Shymonyak, S.; Melnyk, S.; Kono, H.; Tryndyak, V.; Muskhelishvili, L.; Pogribny, I.P.; Kaplowitz, N.; et al. Interstrain differences in liver injury and one-carbon metabolism in alcohol-fed mice. Hepatology 2012, 56, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Wei, V.L.; Singh, S.M. Genetically determined response of hepatic aldehyde dehydrogenase activity to ethanol exposures may be associated with alcohol sensitivity in mouse genotypes. Alcohol. Clin. Exp. Res. 1988, 12, 39–45. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Strain | Age | Sex | Binge Model | Liver Injury Outcomes and Mechanistic Insights | Comments | Ref. |
|---|---|---|---|---|---|---|
| EtOH binge alone (single or n; ad libitum standard chow diet) | ||||||
| C57BL/6 x C3HF1 mice | 8 weeks | Females | 1 binge (3, 4, 5, 6, and 7 g/kg) |
| Shows dose- and time-dependent relationship of binge with BAC and ALT | [24] |
| 129/SvPC J mice | 8–10 weeks | Males | 1 binge (6 g/kg) |
| Shows effect of single binge on liver injury, and intestinal permeability | [25] |
| C57BL/6J mice | 8 weeks | Males | 3 binges (4.5 g/kg) at 12 h intervals |
| Shows effect of multiple binges on liver injury, and HDAC as a mechanism | [26] |
| Mice on 129/Svj background | Females | 3 binges (6 g/kg) at 12 h intervals |
| Shows effect of multiple binges on liver injury, and intestinal permeability in female mice | [27] | |
| Chronic (short- or long-term) EtOH diet plus EtOH binge (single or multiple) | ||||||
| C57BL/6N mice | 8–10 weeks | Males | 10 d + 1 B (5 g/kg) |
| Shows effect of NIAAA 10 d + 1 B model on liver injury in male mice | [28] |
| C57BL/6J mice | 8–12 weeks | Females | 10 d + 1 B (5 g/kg) |
| Shows effect of 10 d + 1 B model on liver injury in female mice, and hepatic neutrophils as a mechanism | [20] |
| C57BL/6N mice | Young (y; 8–12 weeks), Middle-age (m; 12–14 months), Old (o; > 16 months) | Females | 10 d + 1 B (5 g/kg) 8 w + nB | 10 d + 1 B: y < m < o mice
8 w + nB: y < m < o mice
| Shows effect of long-term chronic EtOH + multiple binges in mice; Examines effect of aging; Shows that model also recapitulates features of AH and achieves hepatic fibrosis | [29] |
| C57BL/6J mice | 7 weeks | Males | Chronic 4 weeks + 3 binges (3.5 g/kg) at 12 h intervals |
| Shows effect of long-term chronic EtOH + multiple binges in mice; Shows histone modification as a common mechanism similar to single binge alone | [30] |
| Sprague Dawley rats | 7 weeks | Males | Chronic 4 weeks + 3 binges (5 g/kg) at 12 h intervals |
| Shows effect of long-term chronic EtOH + multiple binges in rats; Shows histone modification as a common mechanism | [31] |
| C57BL/6N mice | 8–10 weeks | Males and Females | 1 binge 10 d + 1 B Chronic 4, 8, and 12 weeks + 1 binge (E8 w; E4 w + 1 B; E8 w + 1 B; E12 w + 1 B) Chronic 8 weeks + biweekly binges (5 g/kg; E8 w + nB) |
E8 w + 1 B, E8 w + nB > E8 w
| Shows effect of varying lengths of long-term chronic EtOH + single/multiple binges; Shows model achieves fibrosis and recapitulates advanced ALD | [32] |
| C57BL/6 mice | Males | Hybrid model: ad libitum Western diet (cholesterol and saturated fat; HCFD, 2 weeks) combined with IG EtOH liquid diet (27 g/kg/day, 8 weeks) + weekly EtOH binges (4~5 g/kg) started from second week of IG feeding |
| Shows model recapitulates features of advanced ASH | [33] | |
| High-fat diet plus EtOH binge (single or multiple) | ||||||
| fa/fa obese Zucker rats | 15 weeks | Males | Binges (4 g/kg) every 12 h for 3 days |
| Shows effect of multiple binges in a model of genetic obesity in rats | [34] |
| Lewis rats | 12 weeks | Males | Choline-deficient diet + 3 binges of whiskey (1.5 mL/100 g) per week for 3 months |
| Shows effect of multiple whiskey binges in a model of diet-induced obesity in rats | [35] |
| C57BL/6J mice | 8–12 weeks | Males | HFD (60% kcal fat) for 3 days or 3 months + 1 binge (5 g/kg) [3 d-HFD + 1 B; 3 m-HFD + 1 B] | 3 m-HFD + 1 B > 3 d-HFD + 1 B > 3 d/3 m-HFD alone
| Shows effect of a single binge on short- or long-term HFD-induced obesity | [36] |
| Multiple EtOH binges plus lipopolysaccharide injection | ||||||
| C57BL/6J mice | 8 weeks | Males | 3 binges (6 g/kg) in 3 days + LPS (10 mg/kg i.p.) 24 h post-final binge |
| Shows effect of multiple binges combined with LPS injection | [37] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghosh Dastidar, S.; Warner, J.B.; Warner, D.R.; McClain, C.J.; Kirpich, I.A. Rodent Models of Alcoholic Liver Disease: Role of Binge Ethanol Administration. Biomolecules 2018, 8, 3. https://doi.org/10.3390/biom8010003
Ghosh Dastidar S, Warner JB, Warner DR, McClain CJ, Kirpich IA. Rodent Models of Alcoholic Liver Disease: Role of Binge Ethanol Administration. Biomolecules. 2018; 8(1):3. https://doi.org/10.3390/biom8010003
Chicago/Turabian StyleGhosh Dastidar, Shubha, Jeffrey B. Warner, Dennis R. Warner, Craig J. McClain, and Irina A. Kirpich. 2018. "Rodent Models of Alcoholic Liver Disease: Role of Binge Ethanol Administration" Biomolecules 8, no. 1: 3. https://doi.org/10.3390/biom8010003
APA StyleGhosh Dastidar, S., Warner, J. B., Warner, D. R., McClain, C. J., & Kirpich, I. A. (2018). Rodent Models of Alcoholic Liver Disease: Role of Binge Ethanol Administration. Biomolecules, 8(1), 3. https://doi.org/10.3390/biom8010003