Molecular Docking of Isolated Alkaloids for Possible α-Glucosidase Inhibition

 ,
,  ,
, 
 ,
,  ,
, 
Abstract
:1. Introduction
2. Methodology
2.1. Target Sequence Retrieval
2.2. Template Selection and Alignment
2.3. Homology Modeling
2.4. Validation of the Modeled Structure
2.5. Active Site Prediction
2.6. Alkaloids Selection
2.7. Preparation of Ligand for Docking Analysis
2.8. Preparation of Protein and Molecular Docking
3. Results
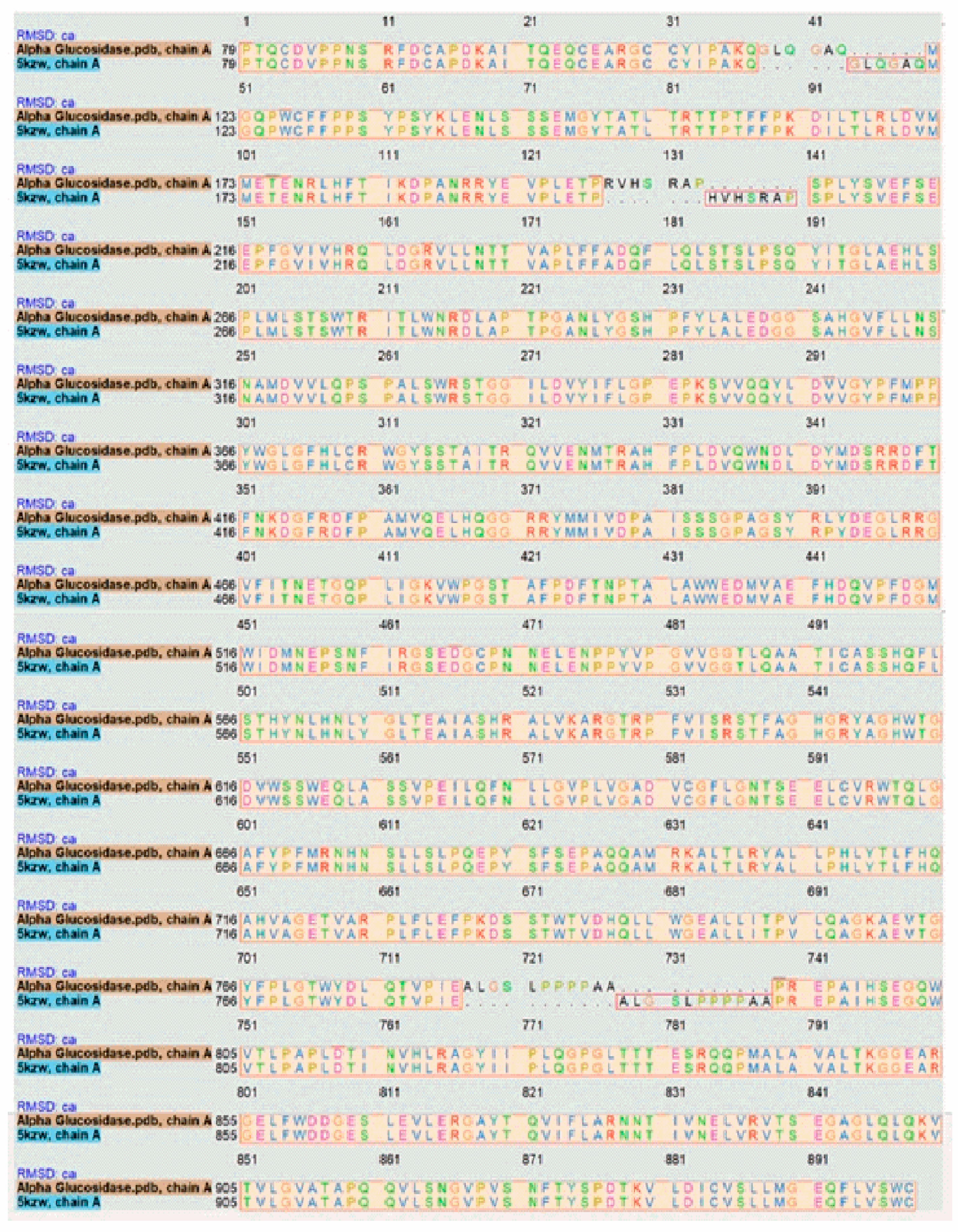
3.1. Target-Template Alignment
3.2. Homology Modeling
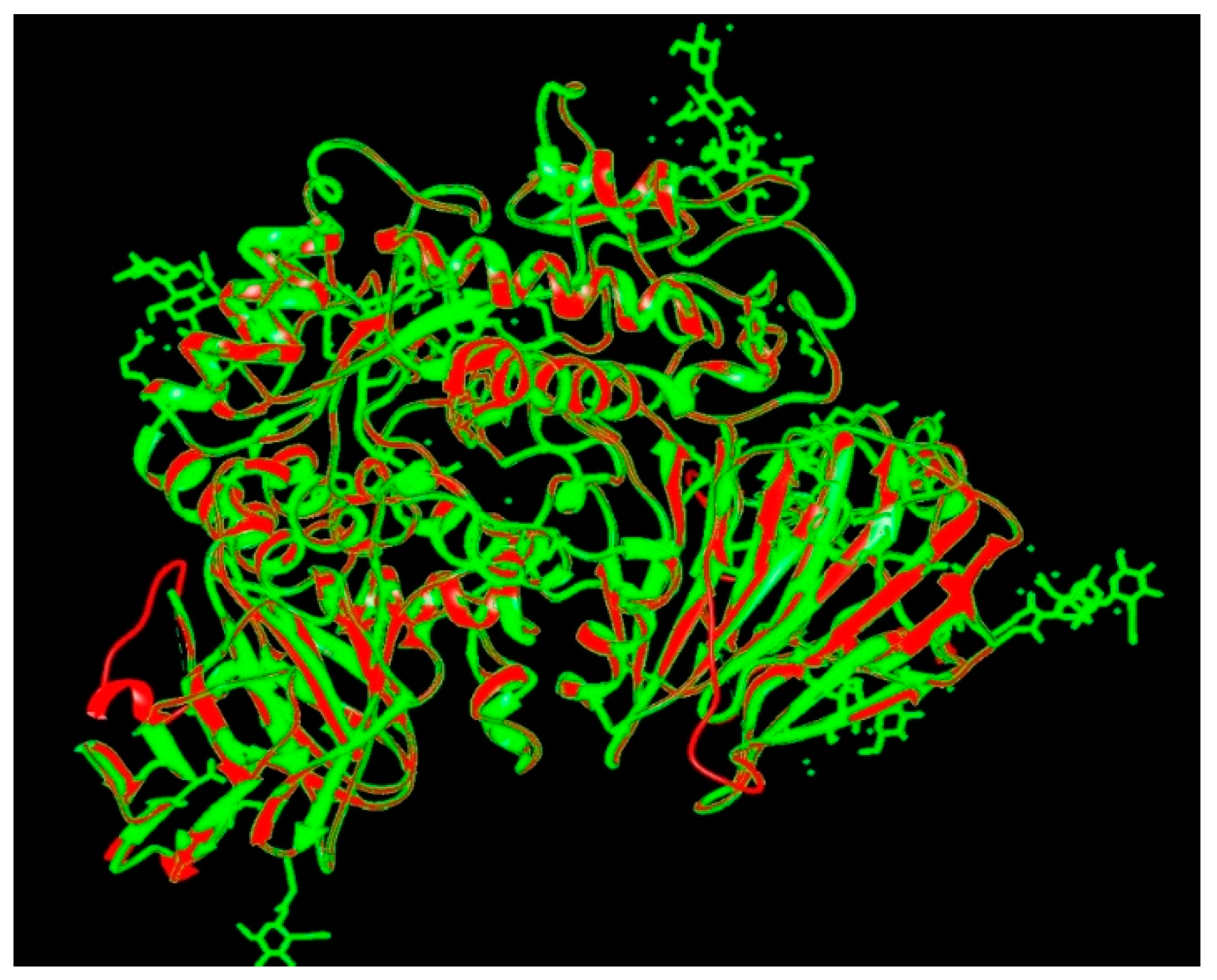
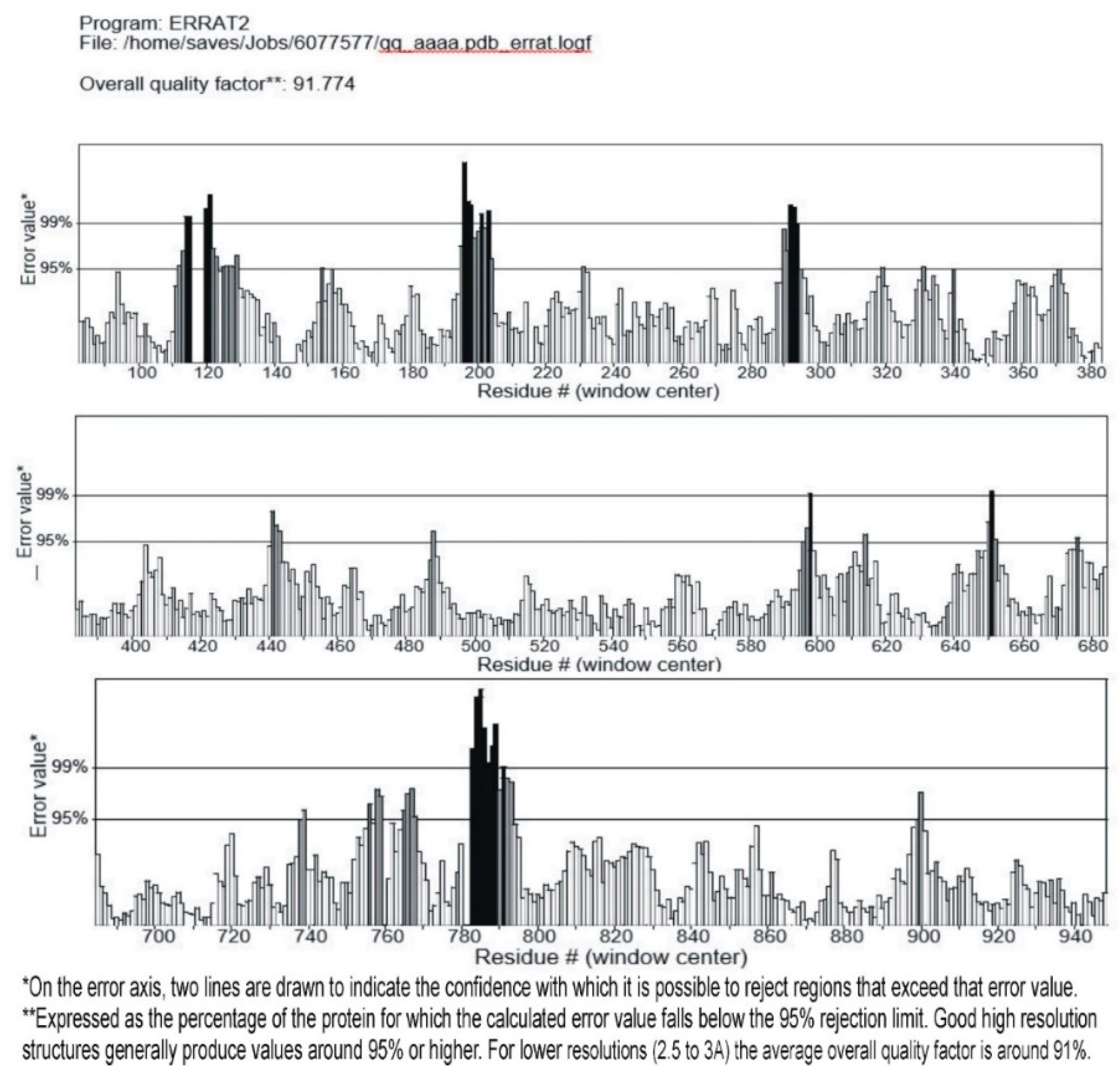
3.3. Validation of the Modeled Structure
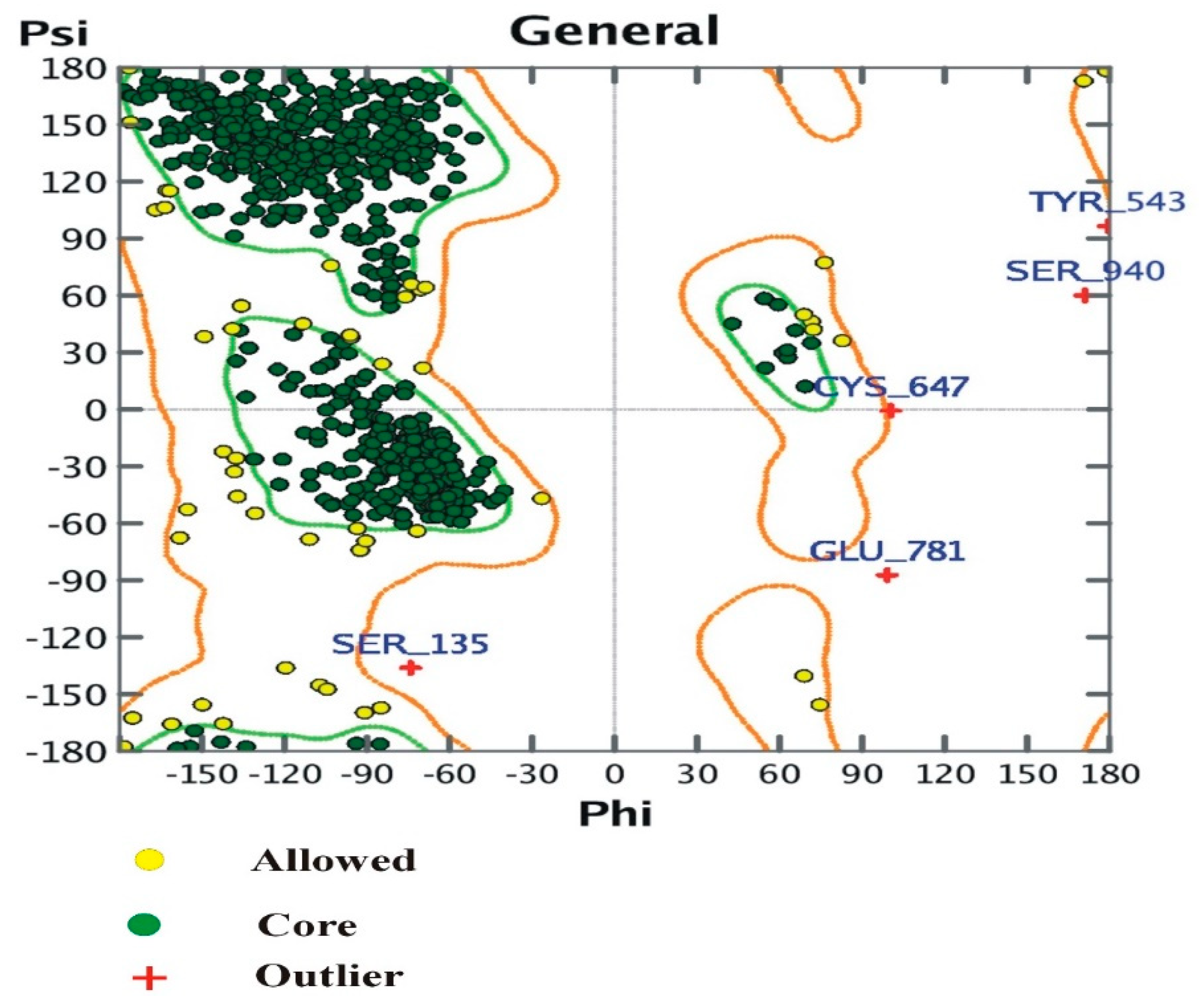
3.4. The Ramachandran Plot
- (i)
- Number of residues in the favored region (~98.0% expected): 898 (94.32%)
- (ii)
- Number of residues in the allowed region (~2.0% expected): 49 (5.1%)
- (iii)
- Number of residues in the outlier region: 5 (0.52%).
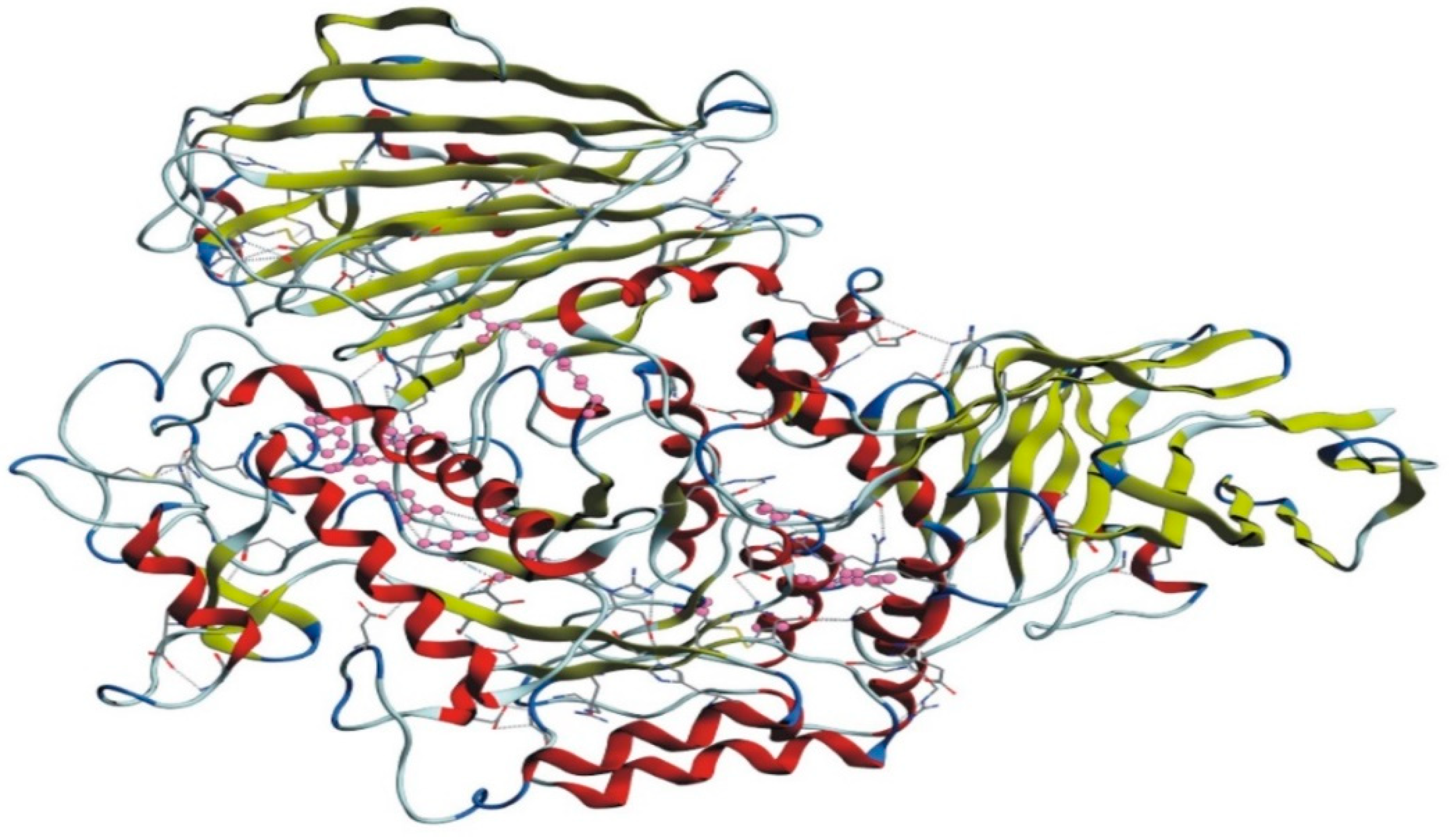
3.5. Active Site Prediction
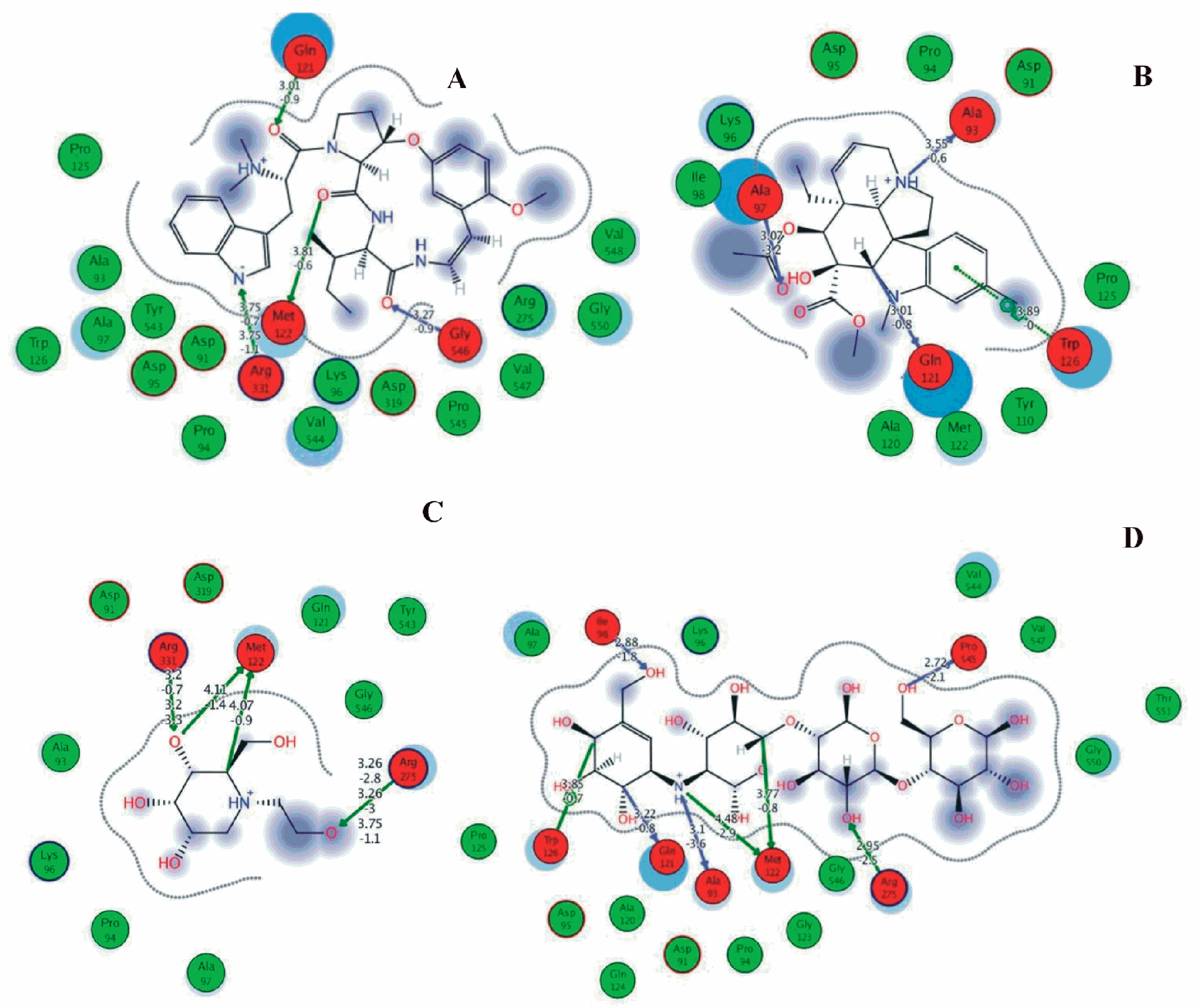
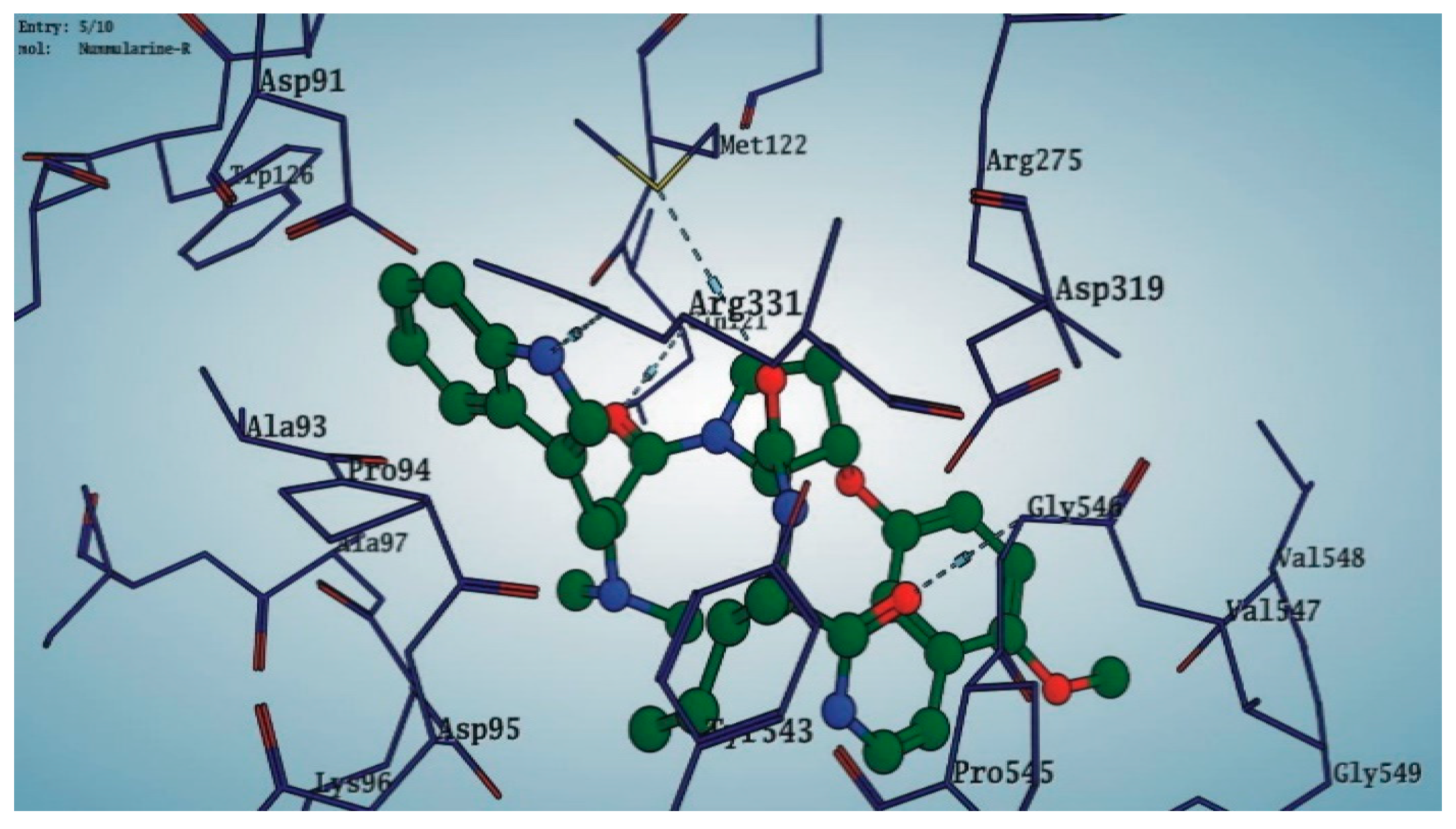
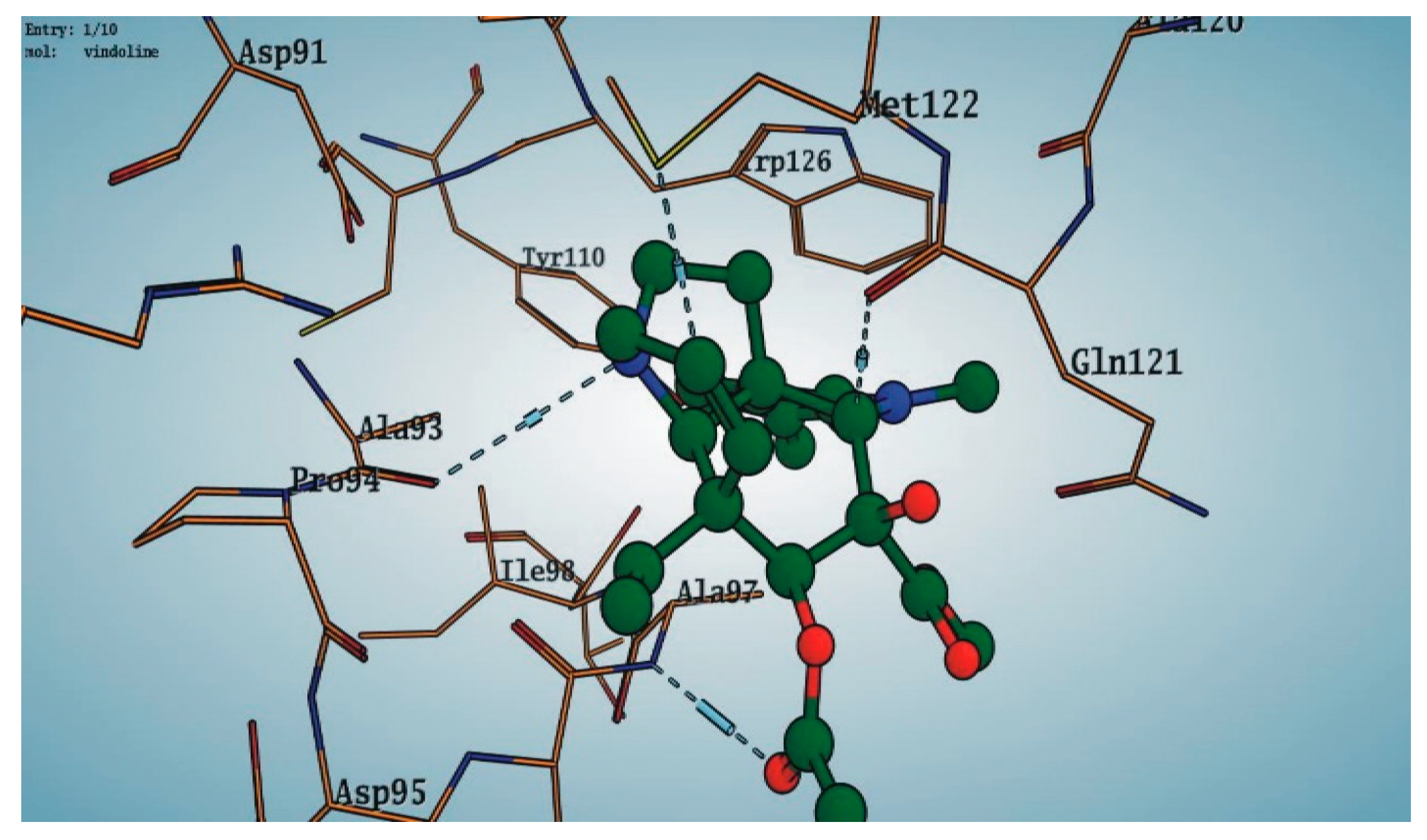
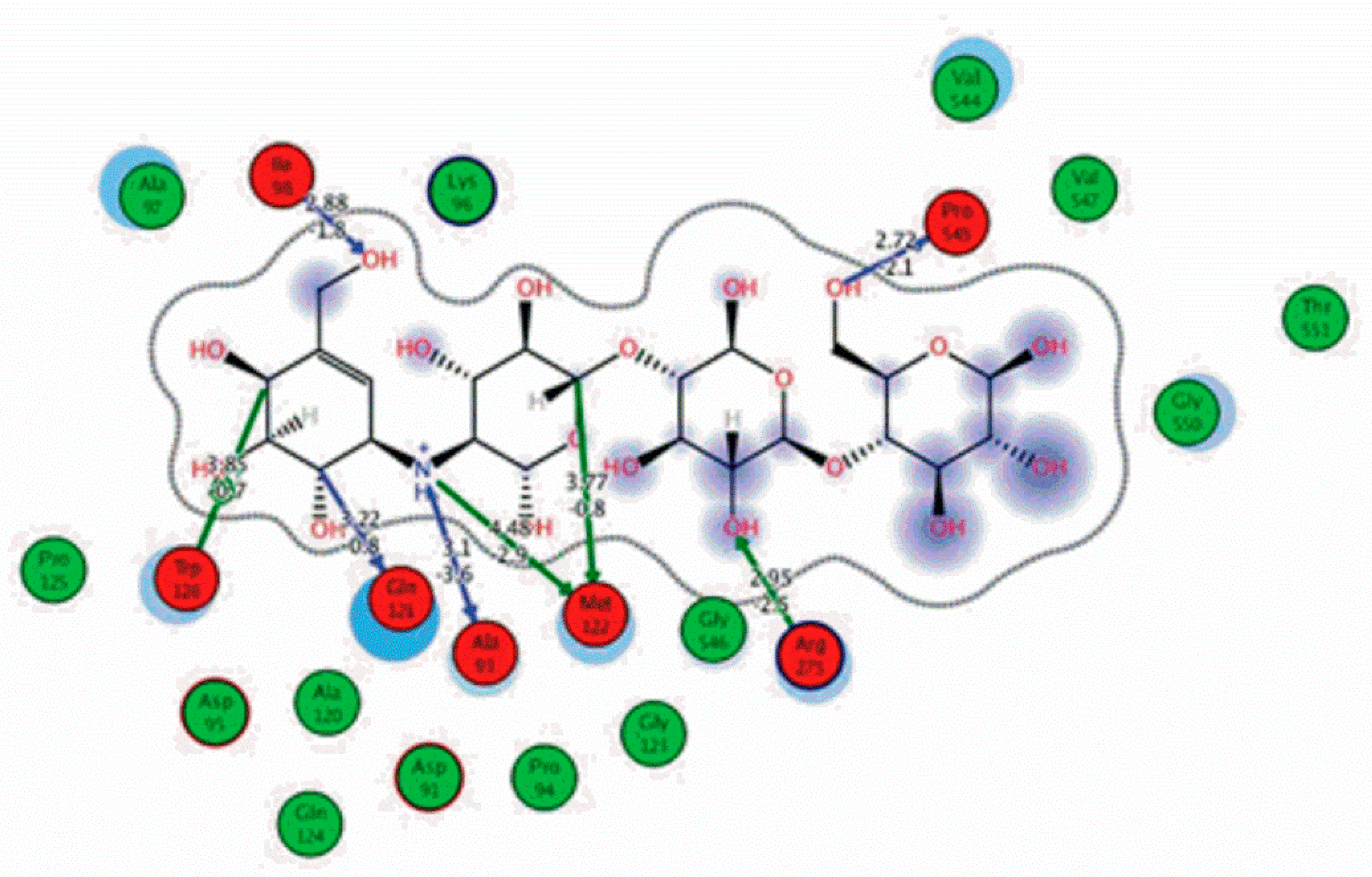
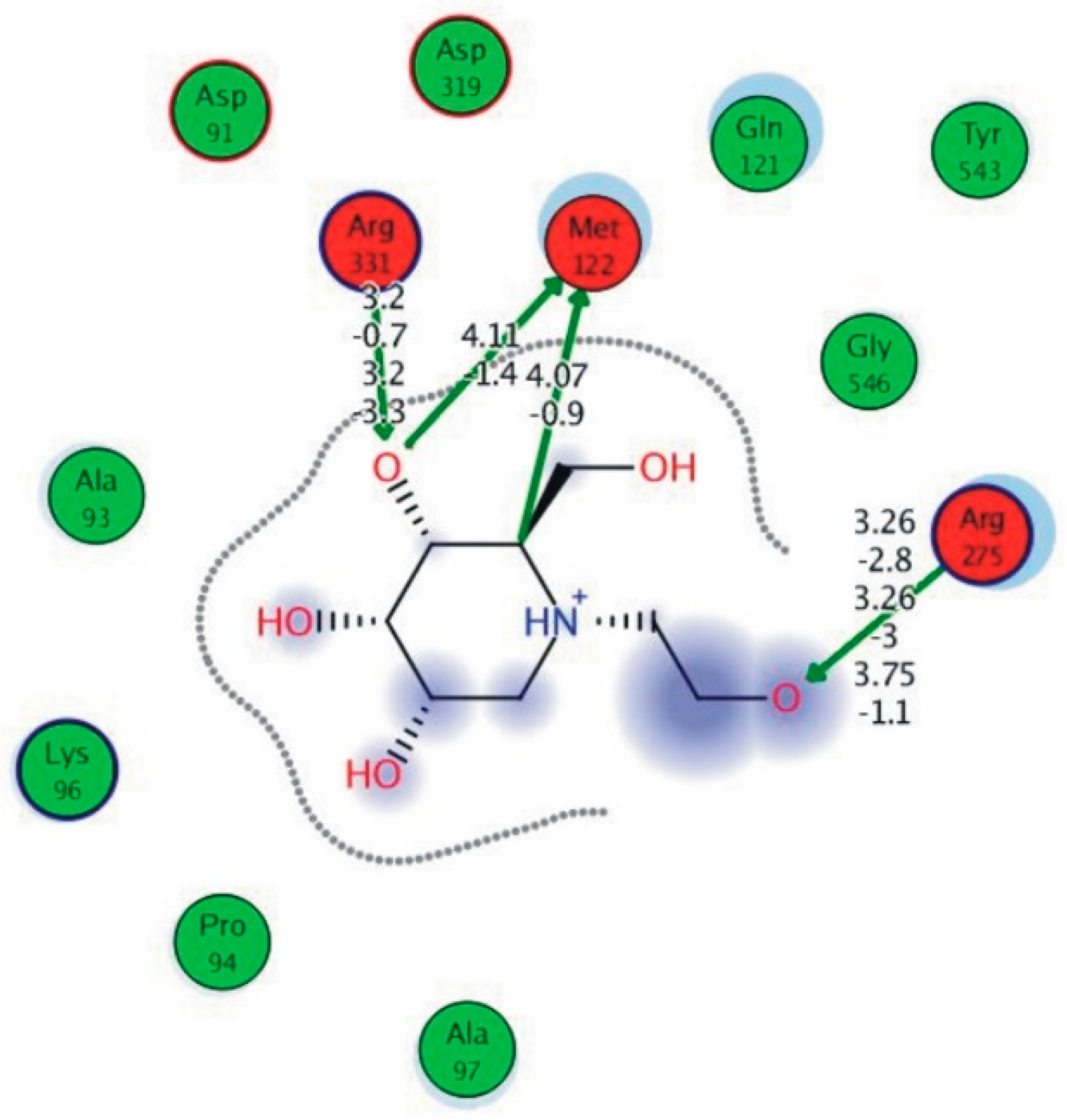
3.6. Preparation of Protein and Molecular Docking
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bharatham, K.; Bharatham, N.; Hun, K.; Woo, K. Binding mode analyses and pharmacophore model development for sulfonamide chalcone derivatives, a new class of a glucosidase inhibitors. J. Mol. Graph. Model. 2008, 26, 1202–1212. [Google Scholar] [CrossRef] [PubMed]
- Alongi, M.; Verardo, G.; Gorassini, A.; Anese, M. Effect of pasteurization on in vitro α-glucosidase inhibitory activity of apple juice. LWT 2018, 98, 366–371. [Google Scholar] [CrossRef]
- Krasikov, V.V.; Karelov, D.V.; Firsov, L.M. α-glucosidases. Biochemistry (Moscow) 2001, 66, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Bruni, C.B.; Sica, V.; Auricchio, F.; Covelli, I. Further kinetic and structural characterization of the lysosomal α-D-glucoside glucohydrolase from cattle liver. Biochim. Biophys. Acta (BBA)-Enzymol. 1970, 212, 470–477. [Google Scholar] [CrossRef]
- Flanagan, B.P.R.; Forstner, G.G. Purification of Rat Intestinal Maltase/Glucoamylase and its Anomalous Dissociation either by Heat or by Low pH. Biochem. J. 1978, 173, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Arinaminpathy, Y.; Khurana, E.; Engelman, D.M.; Gerstein, M.B. Computational analysis of membrane proteins: The largest class of drug targets. Drug Discov. Today 2009, 14, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Biochem, E.J.; Universitet, K. Amp hip hilic Pig Intestinal Microvillus Malt ase/Glucoam ylase Structure and Specificity. Eur. J. Biochem. 1982, 126, 559–568. [Google Scholar]
- Sun, Z.; Duke, H.; Henson, C.A. The Role of Pea Chloroplast a-Glucosidase in Transitory Starch Degradation. Plant Physiol. 1995, 108, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Henson, C.A.; Crops, C. Degradation of Native Starch Granules by Barley. Plant Physiol. 1990, 94, 320–327. [Google Scholar] [CrossRef]
- Ishii, T. Feruloyl oligosaccharides from cell walls of suspension-cultured spinach cells and sugar beet pulp. Plant Cell Physiol. 1994, 35, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Pervaiz, A.; Khan, R.; Anwar, F.; Mushtaq, G.; Kamal, M.A. Alkaloids: An emerging antibacterial modality against methicillin resistant Staphylococcus aureus. Curr. Pharm. Des. 2016, 22, 4420–4429. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Khan, H.; Siddiqui, Z.; Khan, M.Y.; Rehman, S.; Shahab, U.; Godovikova, T.; Silnikov, V. April. AGEs, RAGEs and s-RAGE; friend or foe for cancer. Semin. Cancer Biol. 2018, 49, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Perviz, S.; Khan, H.; Pervaiz, A. Plant Alkaloids as an Emerging Therapeutic Alternative for the Treatment of Depression. Front. Pharmacol. 2016, 7, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, H.; Mubarak, M.S.; Pervaiz, A. Plant Alkaloids as Antiplatelet Agent: Drugs of the Future in the Light of Recent Developments. Front. Pharmacol. 2016, 7, 292. [Google Scholar] [CrossRef]
- Fujii, M.; Takei, I.; Umezawa, K. Antidiabetic effect of orally administered conophylline-containing plant extract on streptozotocin-treated and Goto-Kakizaki rats. Biomed Pharm. 2009, 63, 710–716. [Google Scholar] [CrossRef]
- Sangeetha, M.K.; Priya, C.D.M.; Vasanthi, H.R. Phytomedicine Anti-diabetic property of Tinospora cordifolia and its active compound is mediated through the expression of Glut-4 in L6 myotubes. Eur. J. Integr. Med. 2013, 20, 246–248. [Google Scholar] [CrossRef]
- Patel, M.B.; Mishra, S. Phytomedicine Hypoglycemic activity of alkaloidal fraction of Tinospora cordifolia. Eur. J. Integr. Med. 2011, 18, 1045–1052. [Google Scholar] [CrossRef]
- Choudhary, M.I.; Adhikari, A.; Rasheed, S.; Marasini, B.P.; Hussain, N.; Kaleem, W.A. Cyclopeptide alkaloids of Ziziphus oxyphylla Edgw as novel inhibitors of α-glucosidase enzyme and protein glycation. Phytochem. Lett. 2011, 4, 404–406. [Google Scholar] [CrossRef]
- Don, C.L.G. Antidiabetic and Antioxidant Properties of Alkaloids from Catharanthus roseus (L.) G. Don. Molecules 2013, 18, 9770–9784. [Google Scholar] [CrossRef]
- Al, M.G.E.T.; Gulfraz, M.; Mehmood, S.; Ahmad, A.; Fatima, N.; Praveen, Z.; Williamson, E.M. Comparison of the Antidiabetic Activity of Berberis lyceum Root Extract and Berberine in Alloxan-induced Diabetic Rats. Phytother. Res. 2008, 22, 1208–1212. [Google Scholar] [CrossRef]
- Jung, H.A.; Yoon, N.Y.; Bae, H.J.; Min, B.-S.; Choi, J.S. Inhibitory activities of the alkaloids from Coptidis Rhizoma against aldose reductase. Arch. Pharm. Res. 2008, 31, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Park, M.; Lee, H.C.; Kang, Y.; Kang, E.S.; Kim, K. Antidiabetic Agents from Medicinal Plants. Curr. Med. Chem. 2006, 13, 1203–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.; Igoli, J.; Clements, C.; Semaan, D.; Alamzeb, M.; Mamoon-Ur-Rashid Shah, S.Q.; Ferro, V.A.; Gray, A.I.; Khan, M.R. Antidiabetic and antimicrobial activities of fractions and compounds isolated from Berberis brevissima Jafri and Berberis parkeriana Schneid. Bangladesh J. Pharm. 2013, 8, 336–342. [Google Scholar] [CrossRef] [Green Version]
- Adebajo, A.C.; Ayoola, O.F.; Iwalewa, E.O.; Akindahunsi, A.A.; Omisore, N.O.A.; Adewunmi, C.O.; Adenowo, T.K. Anti-trichomonal, biochemical and toxicological activities of methanolic extract and some carbazole alkaloids isolated from the leaves of Murraya koenigii growing in Nigeria. Phytomedicine 2006, 13, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.B.; Xin, X.L.; Yang, Y.; Lee, S.S.; Aisa, H.A. Highly conjugated norditerpenoid and pyrroloquinoline alkaloids with potent ptp1b inhibitory activity from nigella glandulifera. J. Nat. Prod. 2014, 77, 807–812. [Google Scholar] [CrossRef]
- Costantino, L.; Raimondi, L.; Pirisino, R.; Brunetti, T.; Pessotto, P.; Giannessi, F.; Lins, A.P.; Barlocco, D.; Antolini, L.; El-Abady, S.A. Isolation and pharmacological activities of the Tecoma stans alkaloids. Farmaco 2003, 58, 781–785. [Google Scholar] [CrossRef]
- Goodford, P.J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef]
- Seeliger, D.; Groot, B.L.; De Pymol, V. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Rauf, M.A.; Zubair, S.; Azhar, A. Ligand docking and binding site analysis with pymol and autodock/vina. Int. J. Basic Appl. Sci. 2015, 4, 168–177. [Google Scholar] [CrossRef]
- Blonde, L. Benefits and Risks for Intensive Glycemic Control in Patients With Diabetes Mellitus. Am. J. Med. Sci. 2012, 343, 17–20. [Google Scholar] [CrossRef]
- Zafar, M.; Khan, H.; Rauf, A.; Khan, A.; Lodhi, M.A. In Silico study of alkaloids as α-glucosidase inhibitors: Hope for the Discovery of effective lead compounds. Front. Endocrinol. 2016, 7, 153. [Google Scholar] [CrossRef] [PubMed]
- Bolen, S.; Feldman, L.; Vassy, J.; Wilson, L.; Yeh, H. Review Annals of Internal Medicine Systematic Review: Comparative Effectiveness and Safety of Oral Medications for Type 2 Diabetes Mellitus. Ann. Intern. Med. 2019, 147, 386–399. [Google Scholar] [CrossRef] [PubMed]
- Benalla, W.; Bellahcen, S.; Bnouham, M. Antidiabetic Medicinal Plants as a Source of Alpha Glucosidase Inhibitors. Curr. Diabetes Rev. 2010, 6, 247–254. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plant | Molecular Structures | Activity | Docking Score | Ref. |
|---|---|---|---|---|
| Ervatamia microphylla (leaves) |  Conophyline 1 | In vivo p.o. stimulate iPMSCs proliferation | −12.6274 | [1] |
| Murraya koenigii (leaves) |  Bicyclomahanimbiline 2 | In vivo p.o. Hypoglycemic Activity | −11.7634 | [2] |
| Murraya koenigii (leaves) |  Girinimbilylacetat 3 | In vivo p.o. Hypoglycemic Activity | −9.5231 | [2] |
| Murraya koenigii (leaves) |  Girinimbine 4 | In vivo p.o. Hypoglycemic Activity | −9.9589 | [2] |
| Murraya koenigii (leaves) |  Mahanimbilylacetate 5 | In vivo p.o. Hypoglycemic Activity | −12.9971 | [2] |
| Coptis chinensis (Rhizome) |  Jatrorrhizine 6 | In vitro anti-diabetic | −9.3385 | [3,4] |
| Coptis chinensis (Rhizome) |  Magnoflorine 7 | In vitro anti-diabetic | −11.2586 | [3,4] |
| Coptis chinensis (Rhizome) |  Palmatine 8 | In vitro Anti-diabetic | −10.0536 | [3,4] |
| Catharanthus roseus (Leaves) |  Vindolicine 9 | In vitro Anti-diabetic | −9.2272 | [5] |
| Catharanthus roseus (Leaves) |  Vindoline 10 | In vitro Anti-diabetic | −13.2250 | [5] |
| Catharanthus roseus (Leaves) |  Vindolinine 11 | In vitro Anti-diabetic | −5.5275 | [5] |
| Ziziphus oxyphylla (Whole plant) |  Hemsine-A 12 | In vitro Control the postprandial hyperglycemia | −10.4509 | [6] |
| Ziziphus oxyphylla (Whole plant) |  Nummularin-C 13 | In vitro Anti-diabetic Control the postprandial hyperglycemia | −10.3726 | [6] |
| Ziziphus oxyphylla (Whole plant) |  Nummularine-R 14 | In vitro Anti-diabetic Control the postprandial hyperglycemia | −14.5691 | [6] |
| Berberis lyceum (Root) |  Berberine 15 | In vitro Anti-diabetic Hypoglycemic Activity | −10.5667 | [7] |
| Coptis japonica (Root) |  Columbamine 16 | In vitro Anti-diabetic Aldose Reductase Inhibitory Activity | −7.4609 | [3] |
| Coptis chinensis (Rhizome) |  Coptisine 17 | Anti-diabetic | −8.9123 | [3,4] |
| Coptis chinensis (Rhizome) |  Epiberberine 18 | In vitro Anti-diabetic | −12.9822 | [3,4] |
| Coptis japonica (Root) |  Glutamic acid 19 | In vitro Anti-diabetic Aldose Reductase Inhibitory Activity | −12.6023 | [3] |
| Coptis chinensis (Rhizome) |  Groenlandicine 20 | In vitro Anti-diabetic | −7.0817 | [3,4] |
| Coptis chinensis (Rhizome) |  Jateorrhizine 21 | In vitro Anti-diabetic | −11.4544 | [3,4] |
| Coptis japonica (Root) |  Dehydrocheilanthifoline 22 | In vitro Anti-diabetic Aldose Reductase Inhibitory Activity | −10.8606 | [3] |
| Tecoma stans (Leaves) |  β-hydroxyskitanthine 23 | In Vivo and In Vitro Potent stimulating effect on the basal glucose uptake rate | −10.2216 | [8] |
| Tecoma stans (Leaves) |  Boschnlakine 24 | In Vivo and In Vitro Potent stimulating effect on the basal glucose uptake rate | −7.6929 | [8] |
| Tecoma stan (Leaves) |  Tecomine 25 | In Vivo and In Vitro Potent stimulating effect on the basal glucose uptake rate | −9.1085 | [8] |
| Tecoma stans (Leaves) |  Tecostanine 26 | In Vivo and In Vitro Potent stimulating effect on the basal glucose uptake rate | −9.9845 | [8] |
| Nigella glandulifera. (Seed) |  Nigelladines A 27 | In Vitro PTP1B inhibitory activity | - | [9] |
| Nigella glandulifera. (Seed) |  Nigelladines B 28 | In Vitro PTP1B inhibitory activity | −9.7263 | [9] |
| Nigella glandulifera. (Seed) |  Nigelladines C 29 | In Vivo and In Vitro PTP1B inhibitory activity | −9.9462 | [9] |
| Nigella glandulifera. (Seed) |  Nigellaquinomine 30 | In Vitro PTP1B inhibitory activity | −10.7638 | [9] |
| Nigella glandulifera. (Seed) |  Pyrroloquinoline 31 | In Vitro PTP1B inhibitory activity | −9.4846 | [9] |
| Brassica oleracea var. capitate (Seed) |  2,3-Dicyano-5,6-diphenylpyrazine 32 | Antidiabetic activity | −9.6067 | [10] |
| 34 |  Miglitol | −15.4423 | ||
| 35 |  Acarbose | −14.7983 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, N.; Muhammad, I.; Gul-E-Nayab; Khan, H.; Aschner, M.; Filosa, R.; Daglia, M. Molecular Docking of Isolated Alkaloids for Possible α-Glucosidase Inhibition. Biomolecules 2019, 9, 544. https://doi.org/10.3390/biom9100544
Rahman N, Muhammad I, Gul-E-Nayab, Khan H, Aschner M, Filosa R, Daglia M. Molecular Docking of Isolated Alkaloids for Possible α-Glucosidase Inhibition. Biomolecules. 2019; 9(10):544. https://doi.org/10.3390/biom9100544
Chicago/Turabian StyleRahman, Noor, Ijaz Muhammad, Gul-E-Nayab, Haroon Khan, Michael Aschner, Rosanna Filosa, and Maria Daglia. 2019. "Molecular Docking of Isolated Alkaloids for Possible α-Glucosidase Inhibition" Biomolecules 9, no. 10: 544. https://doi.org/10.3390/biom9100544
APA StyleRahman, N., Muhammad, I., Gul-E-Nayab, Khan, H., Aschner, M., Filosa, R., & Daglia, M. (2019). Molecular Docking of Isolated Alkaloids for Possible α-Glucosidase Inhibition. Biomolecules, 9(10), 544. https://doi.org/10.3390/biom9100544