A Dynamic Core in Human NQO1 Controls the Functional and Stability Effects of Ligand Binding and Their Communication across the Enzyme Dimer




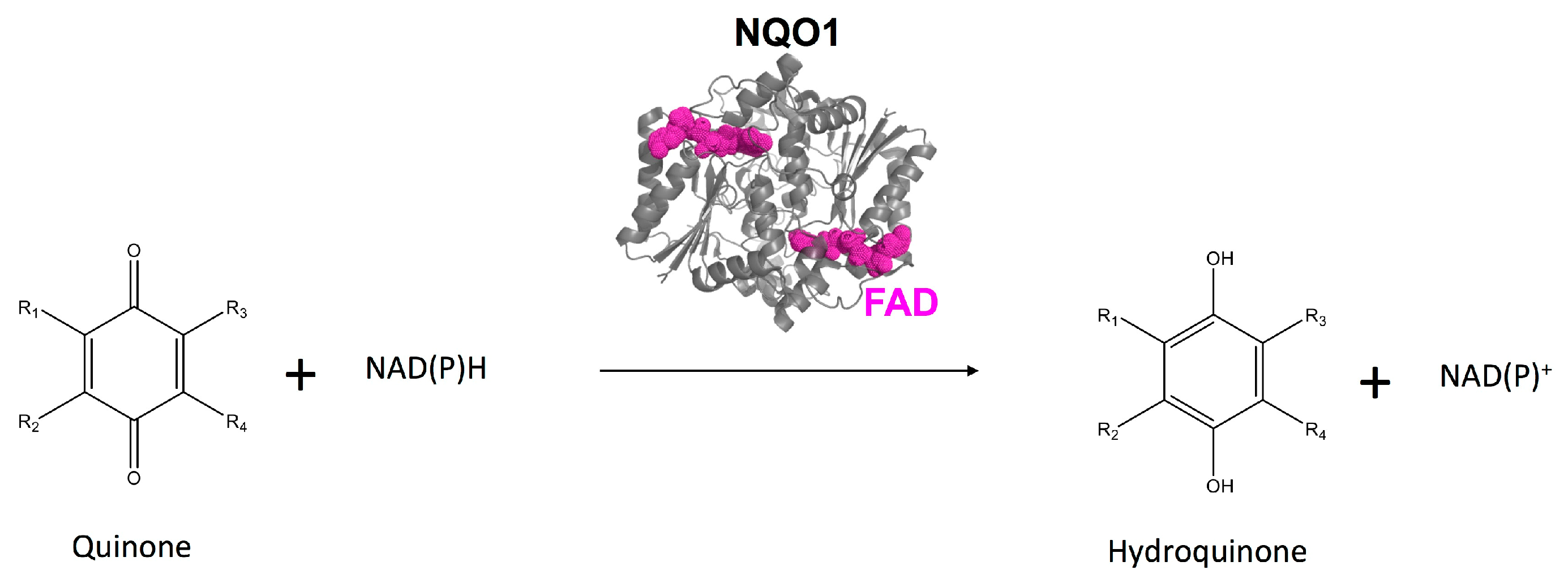{kind=link}
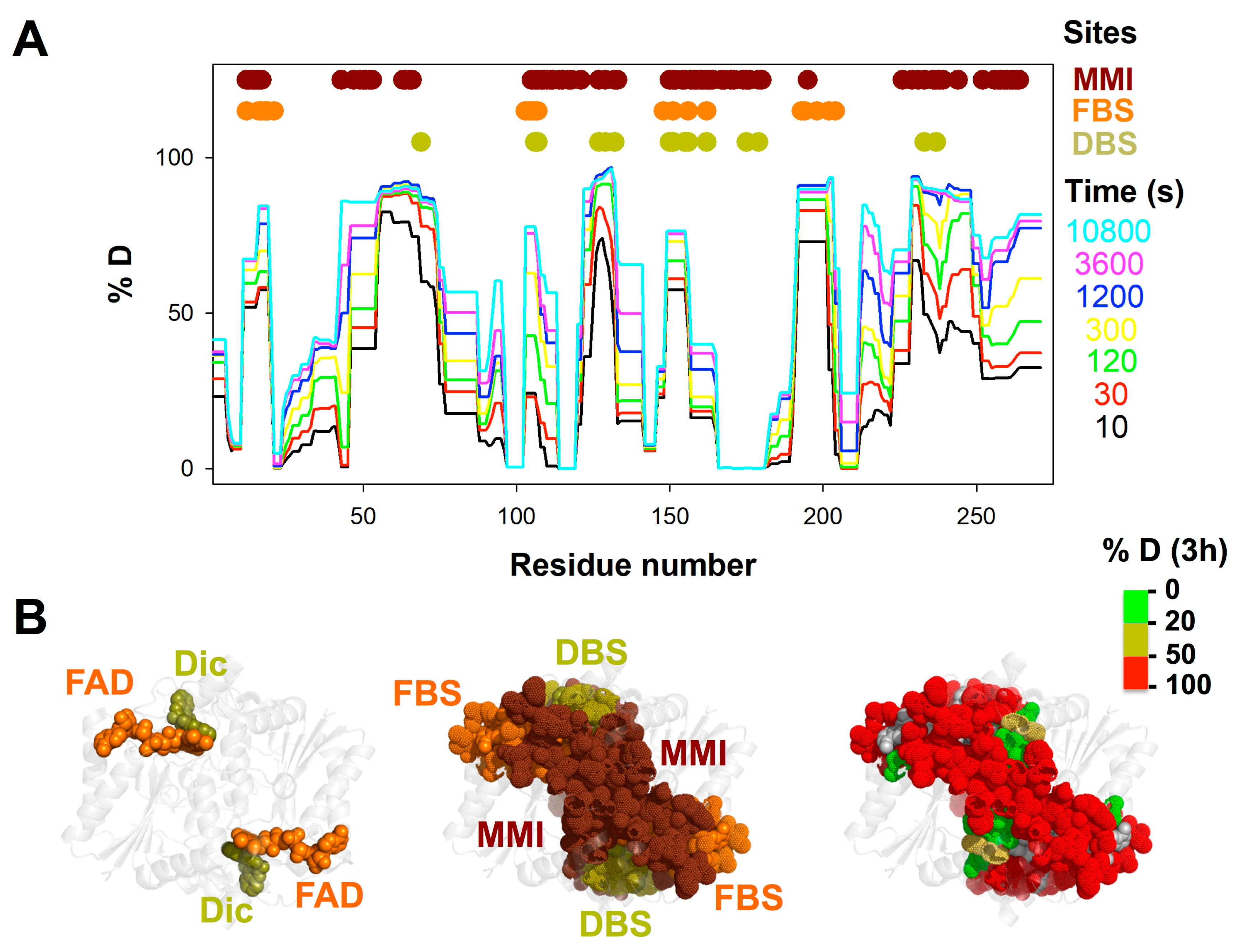{kind=link}
{kind=link}
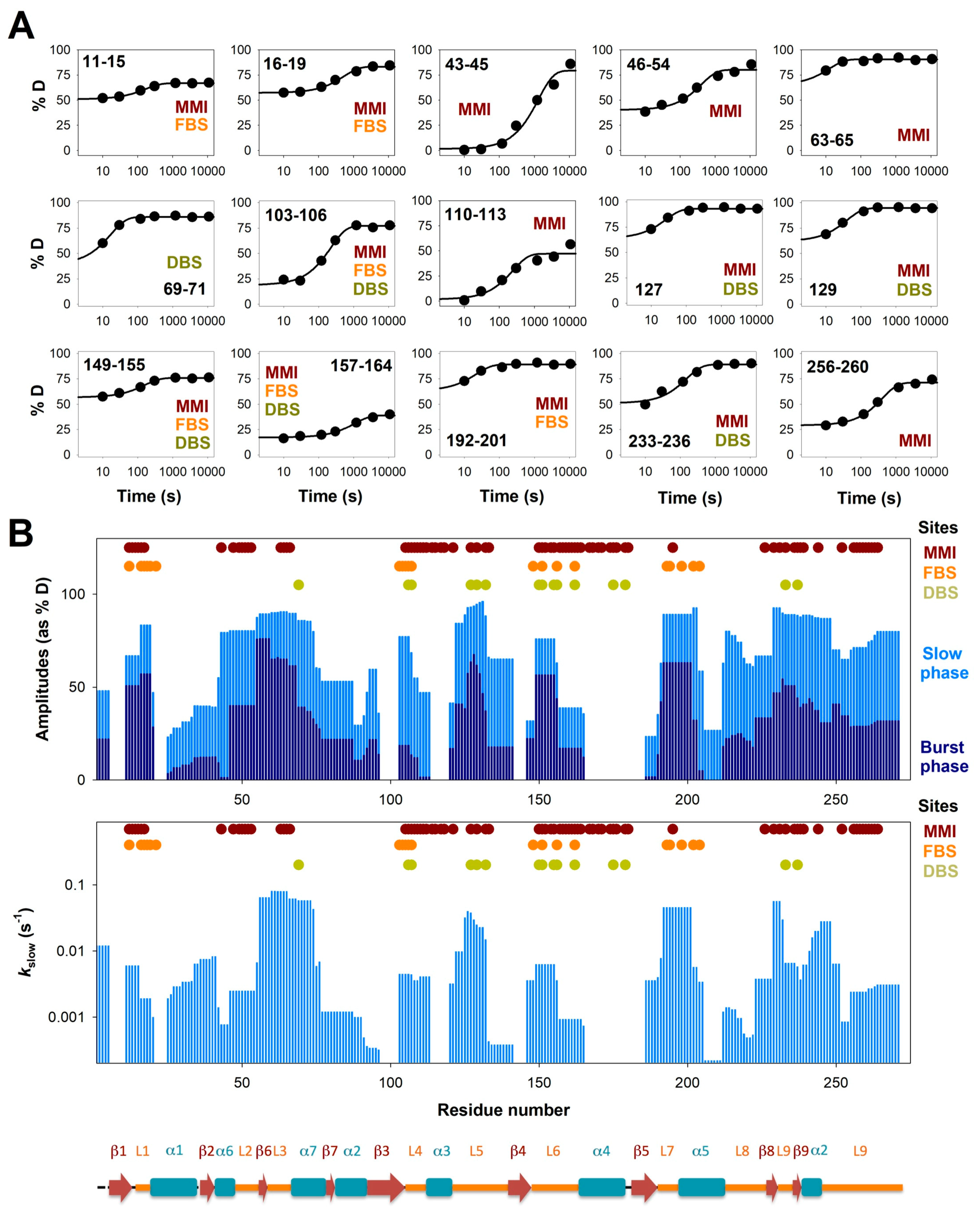{kind=link}
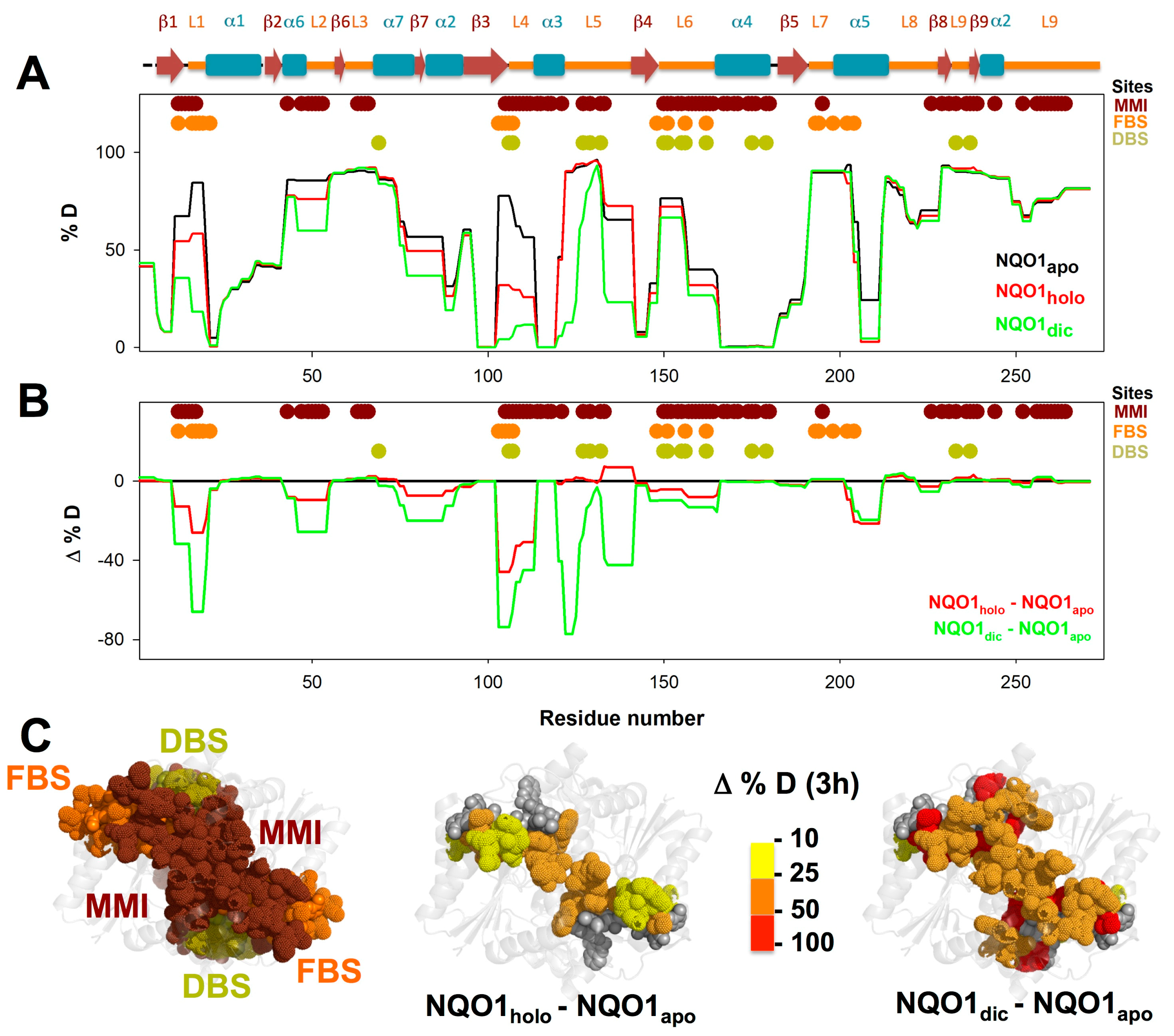{kind=link}
{kind=link}
{kind=link}
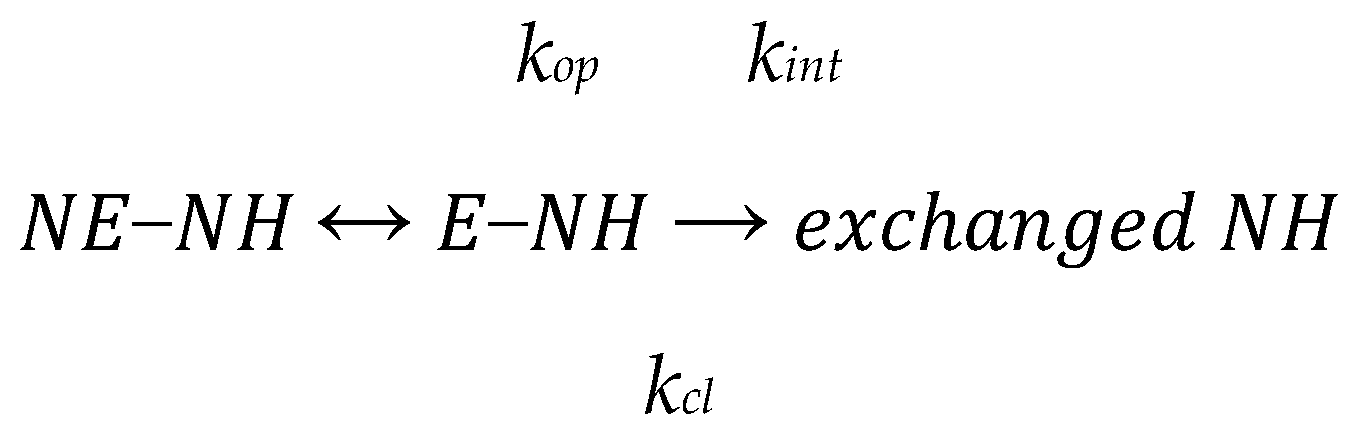{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. Hydrogen/Deuterium Exchange Mass Spectrometry (HDXMS)
3. Results and Discussion
3.1. A Stable Folded Core in NQO1apo with Highly Dynamic Functional Sites
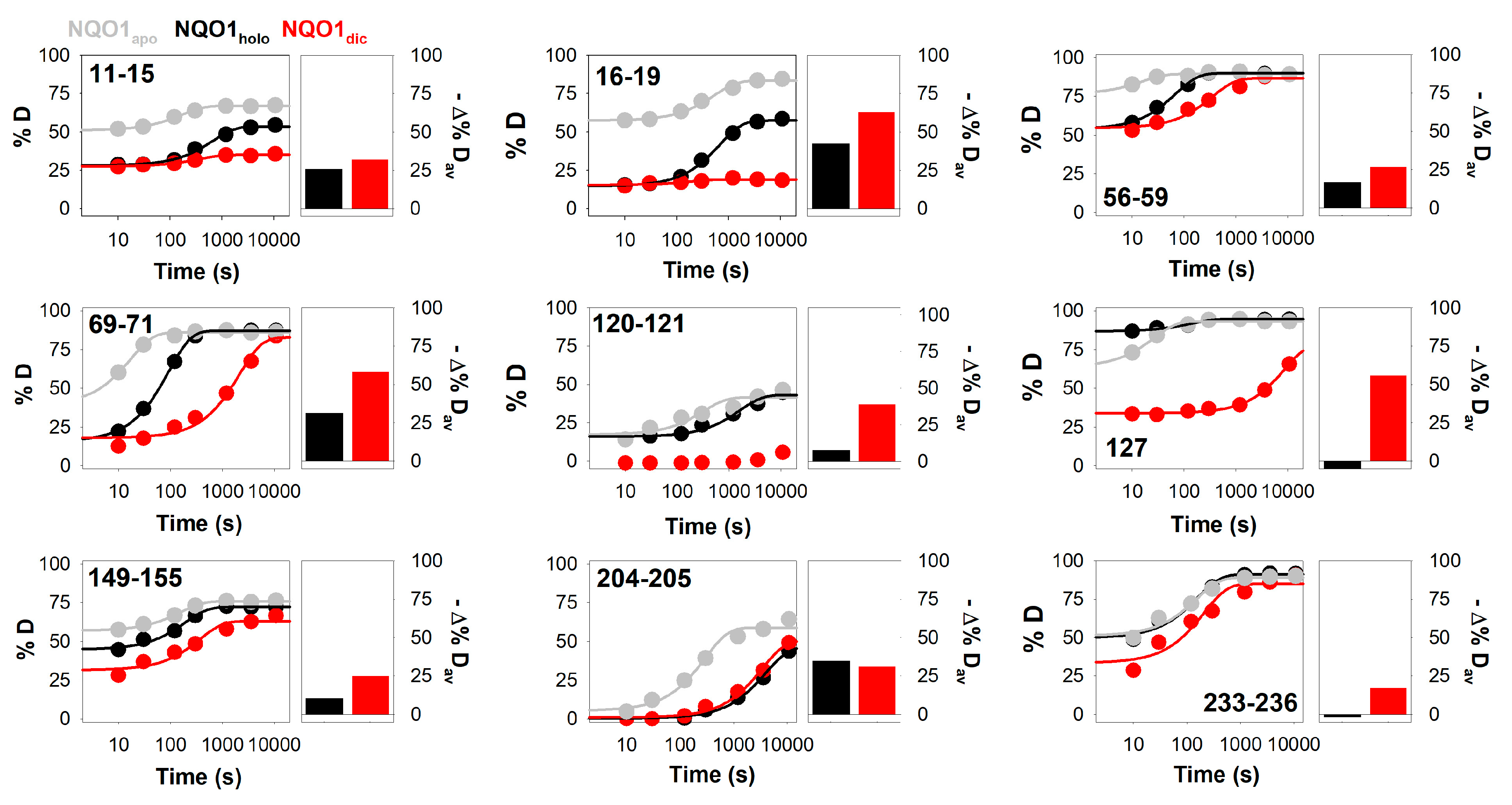
3.2. Complex HDX Kinetics
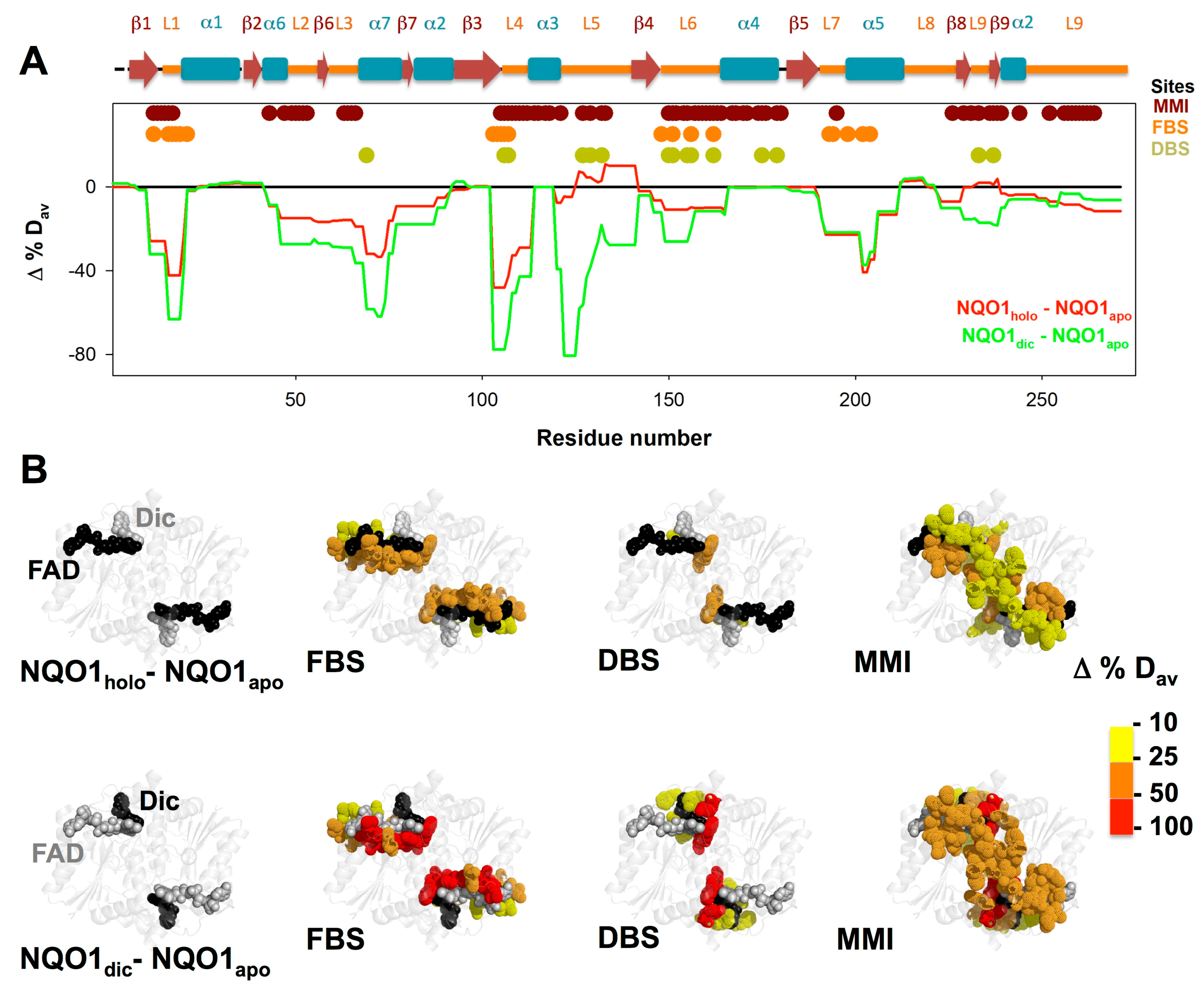
3.3. FAD and Dicoumarol Binding Cause Large-Scale Changes in Protein Structural Dynamics
3.4. Insights into Cooperative Effects upon FAD and Dicoumarol Binding from Analysis of Structural Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beaver, S.K.; Mesa-Torres, N.; Pey, A.L.; Timson, D.J. NQO1: A target for the treatment of cancer and neurological diseases, and a model to understand loss of function disease mechanisms. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.; Siegel, D. Functions of NQO1 in cellular protection and CoQ10 metabolism and its potential role as a redox sensitive molecular switch. Front. Physiol. 2017, 8, 595. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.; Siegel, D. NQO1 in protection against oxidative stress. Curr. Opin. Toxicol. 2018, 7, 67–72. [Google Scholar] [CrossRef]
- Pey, A.L.; Megarity, C.F.; Medina-Carmona, E.; Timson, D.J. Natural small molecules as stabilizers and activators of cancer-associated NQO1 polymorphisms. Curr. Drug Targets 2016, 17, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Pey, A.L.; Megarity, C.F.; Timson, D.J. NAD (P) H quinone oxidoreductase (NQO1): An enzyme which needs just enough mobility, in just the right places. Biosci. Rep. 2019, 39, BSR20180459. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Talalay, P. NAD (P) H: Quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch. Biochem. Biophys. 2010, 501, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Gustafson, D.L.; Dehn, D.L.; Han, J.Y.; Boonchoong, P.; Berliner, L.J.; Ross, D. NAD (P) H: Quinone oxidoreductase 1: Role as a superoxide scavenger. Mol. Pharmacol. 2004, 65, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Dehn, D.D.; Bokatzian, S.S.; Quinn, K.; Backos, D.S.; DiFrancesco, A.; Bernier, M.; Reisdorph, N.; deCabo, R.; Ross, D. Redox modulation of NQO1. PLoS ONE 2018, 13, e0190717. [Google Scholar] [CrossRef] [PubMed]
- Medina-Carmona, E.; Neira, J.L.; Salido, E.; Fuchs, J.E.; Palomino-Morales, R.; Timson, D.J.; Pey, A.L. Site-to-site interdomain communication may mediate different loss-of-function mechanisms in a cancer-associated NQO1 polymorphism. Sci. Rep. 2017, 7, 44352. [Google Scholar] [CrossRef] [PubMed]
- Lienhart, W.D.; Gudipati, V.; Uhl, M.K.; Binter, A.; Pulido, S.A.; Saf, R.; Zangger, K.; Gruber, K.; Macheroux, P. Collapse of the native structure caused by a single amino acid exchange in human NAD (P) H: Quinone oxidoreductase1. FEBS J. 2014, 281, 4691–4704. [Google Scholar] [CrossRef] [PubMed]
- Faig, M.; Bianchet, M.A.; Talalay, P.; Chen, S.; Winski, S.; Ross, D.; Amzel, L.M. Structures of recombinant human and mouse NAD (P) H: Quinone oxidoreductases: Species comparison and structural changes with substrate binding and release. Proc. Natl. Acad. Sci. USA 2000, 97, 3177–3182. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Bianchet, M.A.; Talalay, P.; Amzel, L.M. The three-dimensional structure of NAD (P) H: Quinone reductase, a flavoprotein involved in cancer chemoprotection and chemotherapy: Mechanism of the two-electron reduction. Proc. Natl. Acad. Sci. USA 1995, 92, 8846–8850. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Deng, P.S.; Bailey, J.M.; Swiderek, K.M. A two-domain structure for the two subunits of NAD (P) H: Quinone acceptor oxidoreductase. Protein Sci. 1994, 3, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Medina-Carmona, E.; Palomino-Morales, R.J.; Fuchs, J.E.; Padín-Gonzalez, E.; Mesa-Torres, N.; Salido, E.; Timson, D.J.; Pey, A.L. Conformational dynamics is key to understanding loss-of-function of NQO1 cancer-associated polymorphisms and its correction by pharmacological ligands. Sci. Rep. 2016, 6, 20331. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Dym, O.; Tsvetkov, P.; Adler, J.; Shaul, Y. The crystal structure of NAD (P) H quinone oxidoreductase 1 in complex with its potent inhibitor dicoumarol. Biochemistry 2006, 45, 6372–6378. [Google Scholar] [CrossRef] [PubMed]
- Moscovitz, O.; Tsvetkov, P.; Hazan, N.; Michaelevski, I.; Keisar, H.; Ben-Nissan, G.; Shaul, Y.; Sharon, M. A mutually inhibitory feedback loop between the 20S proteasome and its regulator, NQO1. Mol. Cell 2012, 47, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Tsvetkov, P.; Kahana, C.; Shaul, Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005, 19, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.T.; Kim, J.W.; Kim, J.M.; Kim, S.J.; Lee, J.S.; Hong, S.S.; Goodwin, J.; Ruthenborg, R.J.; Jung, M.G.; Lee, H.J.; et al. NQO1 inhibits proteasome-mediated degradation of HIF-1alpha. Nat. Commun. 2016, 7, 13593. [Google Scholar] [CrossRef] [PubMed]
- Lata, S.; Ali, A.; Sood, V.; Raja, R.; Banerjea, A.C. HIV-1 Rev downregulates Tat expression and viral replication via modulation of NAD (P) H: Quinine oxidoreductase 1 (NQO1). Nat. Commun. 2015, 6, 7244. [Google Scholar] [CrossRef] [PubMed]
- DiFrancesco, A.; DiGermanio, C.; Panda, A.C.; Huynh, P.; Peaden, R.; Navas-Enamorado, I.; Bastian, P.; Lehrmann, E.; Diaz-Ruiz, A.; Ross, D.; et al. Novel RNA-binding activity of NQO1 promotes SERPINA1 mRNA translation. Free Radic. Biol. Med. 2016, 99, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Betancor-Fernandez, I.; Timson, D.J.; Salido, E.; Pey, A.L. Natural (and unnatural) small molecules as pharmacological chaperones and inhibitors in cancer. Handb. Exp. Pharmacol. 2018, 45, 345–383. [Google Scholar]
- Pey, A.L.; Megarity, C.F.; Timson, D.J. FAD binding overcomes defects in activity and stability displayed by cancer-associated variants of human NQO1. Biochim. Biophys. Acta 2014, 1842, 2163–2173. [Google Scholar] [CrossRef] [PubMed]
- Claveria-Gimeno, R.; Velazquez-Campoy, A.; Pey, A.L. Thermodynamics of cooperative binding of FAD to human NQO1: Implications to understanding cofactor-dependent function and stability of the flavoproteome. Arch. Biochem. Biophys. 2017, 636, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Anwar, A.; Winski, S.L.; Kepa, J.K.; Zolman, K.L.; Ross, D. Rapid polyubiquitination and proteasomal degradation of a mutant form of NAD (P) H: Quinone oxidoreductase 1. Mol. Pharmacol. 2001, 59, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Limon, A.; Alriquet, M.; Lang, W.H.; Calloni, G.; Wittig, I.; Vabulas, R.M. Recognition of enzymes lacking bound cofactor by protein quality control. Proc. Natl. Acad. Sci. USA 2016, 113, 12156–12161. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; SuKang, S.; Wang, Z.H.; Liu, X.; Day, J.X.; Wu, Z.; Peng, J.; Xiang, D.; Springer, W.; Ye, K. Akt Phosphorylates NQO1 and Triggers its Degradation, Abolishing Its Antioxidative Activities in Parkinson’s Disease. J. Neurosci. 2019, 39, 7291–7305. [Google Scholar] [CrossRef] [PubMed]
- Nolan, K.A.; Scott, K.A.; Barnes, J.; Doncaster, J.; Whitehead, R.C.; Stratford, I.J. Pharmacological inhibitors of NAD (P) H quinone oxidoreductase, NQO1: Structure/activity relationships and functional activity in tumour cells. Biochem. Pharmacol. 2010, 80, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Nolan, K.A.; Zhao, H.; Faulder, P.F.; Frenkel, A.D.; Timson, D.J.; Siegel, D.; Ross, D.; Burke, T.R., Jr.; Stratford, I.J.; Bryce, R.A. Coumarin-based inhibitors of human NAD (P) H: Quinone oxidoreductase-1. Identification, structure–activity, off-target effects and in vitro human pancreatic cancer toxicity. J. Med. Chem. 2007, 50, 6316–6325. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.A.; Barnes, J.; Whitehead, R.C.; Stratford, I.J.; Nolan, K.A. Inhibitors of NQO1: Identification of compounds more potent than dicoumarol without associated off-target effects. Biochem. Pharmacol. 2011, 81, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Medina-Carmona, E.; Rizzuti, B.; Martin-Escolano, R.; Pacheco-Garcia, J.L.; Mesa-Torres, N.; Neira, J.L.; Guzzi, R.; Pey, A.L. Phosphorylation compromises FAD binding and intracellular stability of wild-type and cancer-associated NQO1: Insights into flavo-proteome stability. Int. J. Biol. Macromol. 2019, 125, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- Mesa-Torres, N.; Betancor-Fernández, I.; Oppici, E.; Cellini, B.; Salido, E.; Pey, A.L. Evolutionary divergent suppressor mutations in conformational diseases. Genes 2018, 9, 352. [Google Scholar] [CrossRef] [PubMed]
- Medina-Carmona, E.; Betancor-Fernández, I.; Santos, J.; Mesa-Torres, N.; Grottelli, S.; Batlle, C.; Naganathan, A.N.; Oppici, O.; Cellini, B.; Ventura, S.; et al. Insight into the specificity and severity of pathogenic mechanisms associated with missense mutations through experimental and structural perturbation analyses. Hum. Mol. Genet. 2019, 28, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Pey, A.L. Biophysical and functional perturbation analyses at cancer-associated P187 and K240 sites of the multifunctional NADP (H): Quinone oxidoreductase 1. Int. J. Biol. Macromol. 2018, 118, 1912–1923. [Google Scholar] [CrossRef] [PubMed]
- Munoz, I.G.; Morel, B.; Medina-Carmona, E.; Pey, A.L. A mechanism for cancer-associated inactivation of NQO1 due to P187S and its reactivation by the consensus mutation H80R. FEBS Lett. 2017, 591, 2826–2835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lienhart, W.D.; Strandback, E.; Gudipati, V.; Koch, K.; Binter, A.; Uhl, M.K.; Rantasa, D.M.; Bourgeois, B.; Madl, T.; Zangger, K.; et al. Catalytic competence, structure and stability of the cancer-associated R139W variant of the human NAD (P) H: Quinone oxidoreductase 1 (NQO 1). FEBS J. 2017, 284, 1233–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina-Carmona, E.; Fuchs, J.E.; Gavira, J.A.; Mesa-Torres, N.; Neira, J.L.; Salido, E.; Palomino-Morales, R.; Burgos, M.; Timson, D.J.; Pey, A.L. Enhanced vulnerability of human proteins towards disease-associated inactivation through divergent evolution. Hum. Mol. Genet. 2017, 26, 3531–3544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Smith, D.L. Determination of amide hydrogen exchange by mass spectrometry: A new tool for protein structure elucidation. Protein Sci. 1993, 2, 522–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trcka, F.; Durech, M.; Vankova, P.; Chmelik, J.; Martinkova, V.; Hausner, J.; Kadek, A.; Marcoux, J.; Klumpler, T.; Vojtesek, B.; et al. Human stress-inducible Hsp70 has a high propensity to form ATP-dependent antiparallel dimers that are differentially regulated by Cochaperone binding. Mol. Cell Proteom. 2019, 18, 320–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y. Hydrogen Exchange Experiments: Detection and Characterization of Protein Folding Intermediates. In Protein Folding, Misfolding and Aggregation; Muñoz, V., Ed.; Royal Society of Chemistry: Cambridge, UK, 2008; pp. 70–83. [Google Scholar]
- Konermann, L.; Pan, J.; Liu, Y.H. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 2011, 40, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Megarity, C.F.; Abdel-Bettley, H.; Caraher, M.C.; Scott, K.A.; RA, W.; Jowitt, T.A.; Gutierrez, A.; Bryce, R.A.; Nolan, K.A.; Stratford, I.J.; et al. Negative cooperativity in NAD (P) H quinone oxidoreductase 1 (NQO1). ChemBioChem 2019. [CrossRef] [PubMed]
- Wyman, J.; Gill, S.J. Binding and Linkage: Functional Chemistry of Biological Macromolecules; University Science Books: Mill Valley, CA, USA, 1990. [Google Scholar]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vankova, P.; Salido, E.; Timson, D.J.; Man, P.; Pey, A.L. A Dynamic Core in Human NQO1 Controls the Functional and Stability Effects of Ligand Binding and Their Communication across the Enzyme Dimer. Biomolecules 2019, 9, 728. https://doi.org/10.3390/biom9110728
Vankova P, Salido E, Timson DJ, Man P, Pey AL. A Dynamic Core in Human NQO1 Controls the Functional and Stability Effects of Ligand Binding and Their Communication across the Enzyme Dimer. Biomolecules. 2019; 9(11):728. https://doi.org/10.3390/biom9110728
Chicago/Turabian StyleVankova, Pavla, Eduardo Salido, David J. Timson, Petr Man, and Angel L. Pey. 2019. "A Dynamic Core in Human NQO1 Controls the Functional and Stability Effects of Ligand Binding and Their Communication across the Enzyme Dimer" Biomolecules 9, no. 11: 728. https://doi.org/10.3390/biom9110728
APA StyleVankova, P., Salido, E., Timson, D. J., Man, P., & Pey, A. L. (2019). A Dynamic Core in Human NQO1 Controls the Functional and Stability Effects of Ligand Binding and Their Communication across the Enzyme Dimer. Biomolecules, 9(11), 728. https://doi.org/10.3390/biom9110728