The Hedgehog Signaling Pathway Emerges as a Pathogenic Target


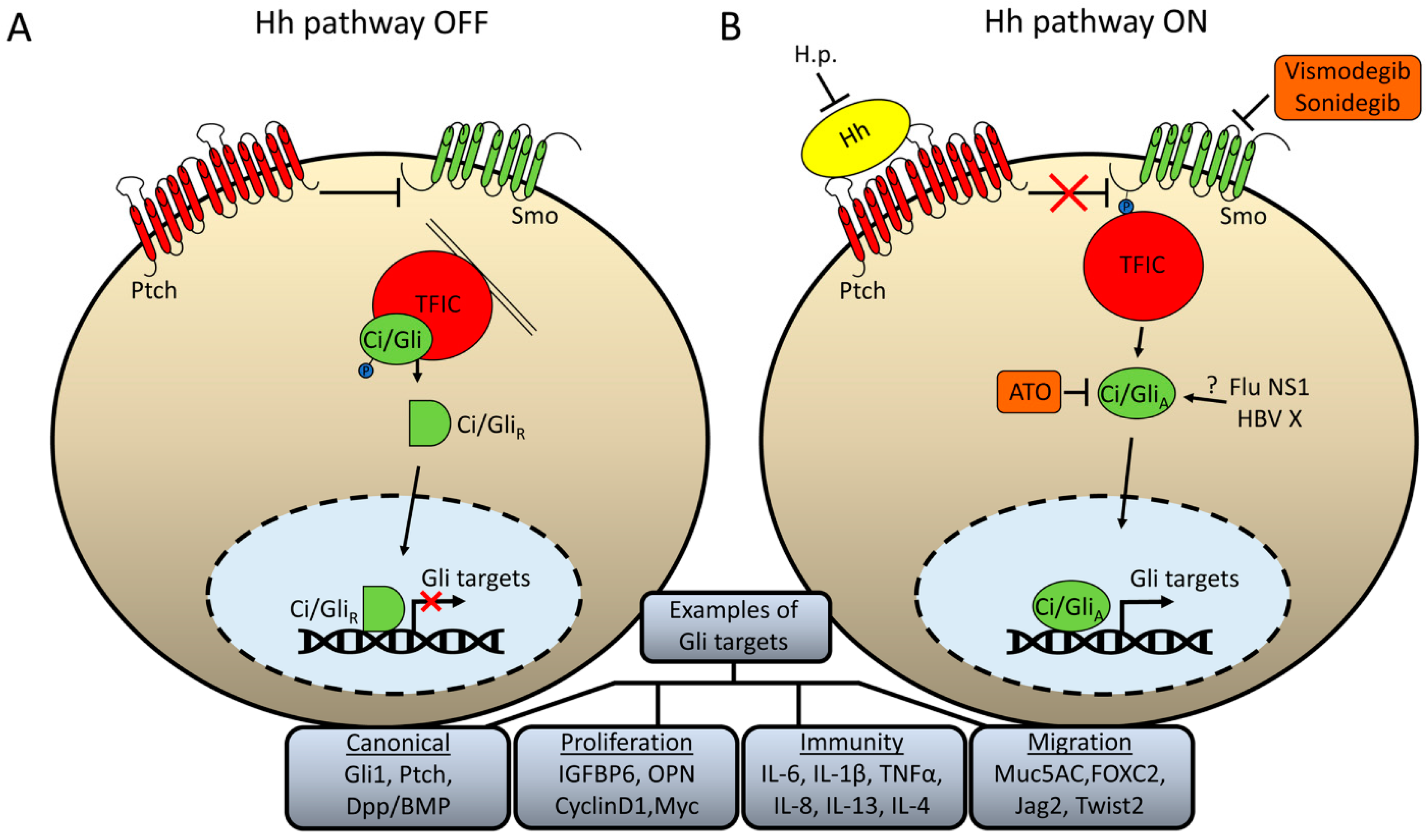{kind=link}
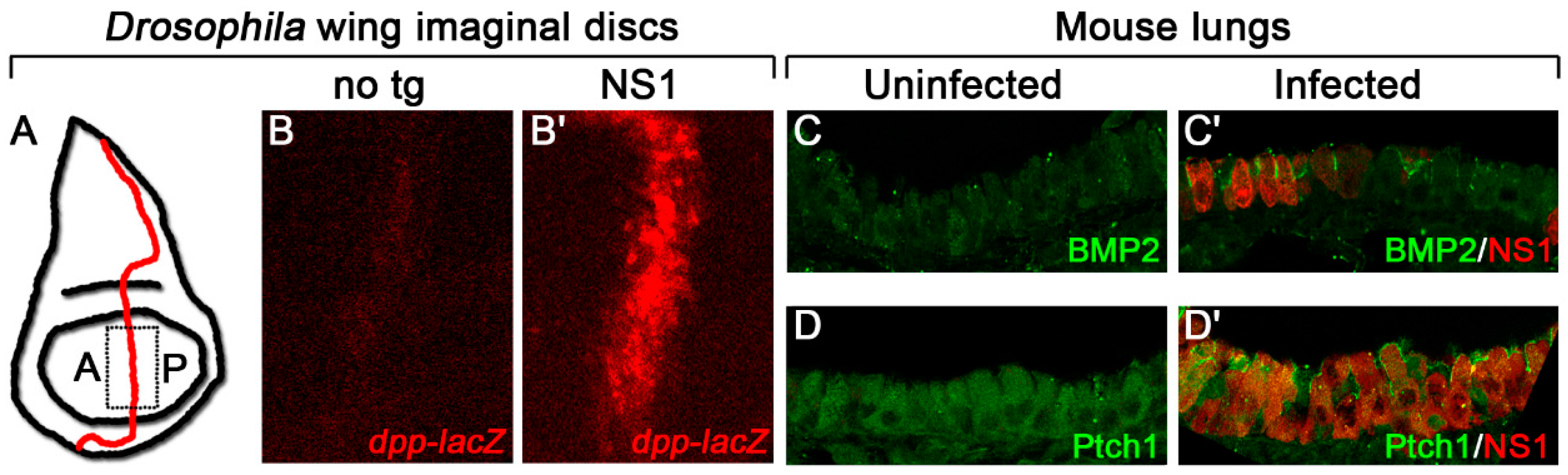{kind=link}
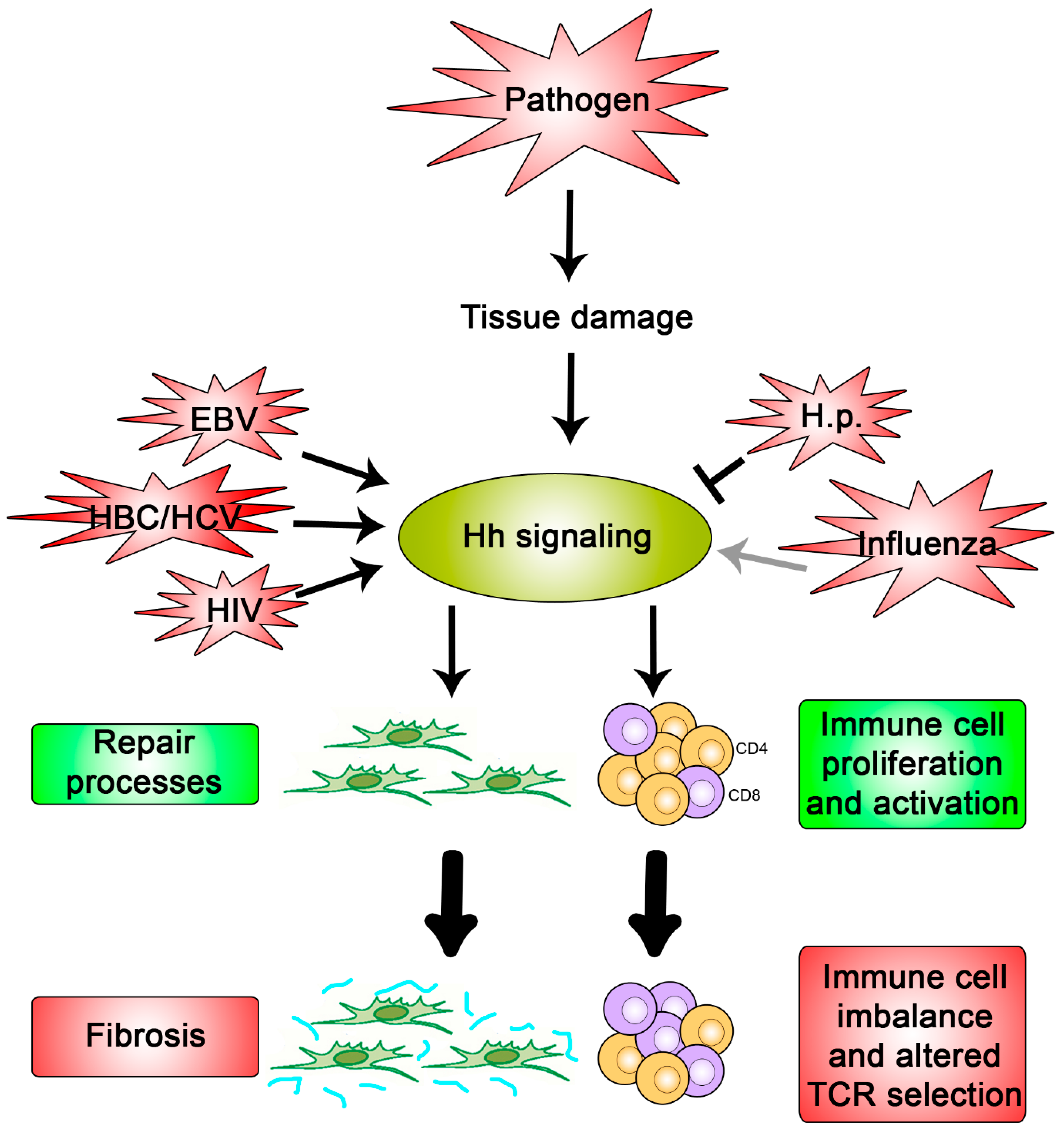{kind=link}
Abstract
:1. Introduction: Basics of the Hedgehog Signaling Pathway and Its Evolutionary Conservation
2. Hh Signaling as a Target of Pathogens
3. Why Might Hh Signaling Be a Frequent Target for Pathogens?
3.1. Hh Signaling Modulates Wound Repair and Tissue Fibrosis
3.2. Hh Signaling Involved in Immune Pathways and Immune Diseases
4. Therapeutic Strategies
5. Summary
Acknowledgments
Conflicts of Interest
References
- Pak, E.; Segal, R.A. Hedgehog Signal Transduction: Key Players, Oncogenic Drivers, and Cancer Therapy. Dev. Cell 2016, 38, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.T.; Zhao, Z.; Ingham, P.W. Hedgehog signalling. Development 2016, 143, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhang, Y.; Sun, B.; McMahon, A.P.; Wang, Y. Hedgehog Signaling: From Basic Biology to Cancer Therapy. Cell Chem. Biol. 2017, 24, 252–280. [Google Scholar] [CrossRef] [PubMed]
- Nusslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Anderson, K.V. Signaling from Smo to Ci/Gli: Conservation and divergence of Hedgehog pathways from Drosophila to vertebrates. Development 2006, 133, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Ohlmeyer, J.T.; Kalderon, D. Hedgehog stimulates maturation of Cubitus interruptus into a labile transcriptional activator. Nature 1998, 396, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Kalderon, D. Hedgehog activates fused through phosphorylation to elicit a full spectrum of pathway responses. Dev. Cell 2011, 20, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Alves, G.; Limbourg-Bouchon, B.; Tricoire, H.; Brissard-Zahraoui, J.; Lamour-Isnard, C.; Busson, D. Modulation of Hedgehog target gene expression by the Fused serine-threonine kinase in wing imaginal discs. Mech. Dev. 1998, 78, 17–31. [Google Scholar] [CrossRef]
- Wang, C.; Pan, Y.; Wang, B. Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development 2010, 137, 2001–2009. [Google Scholar] [CrossRef] [PubMed]
- Humke, E.W.; Dorn, K.V.; Milenkovic, L.; Scott, M.P.; Rohatgi, R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010, 24, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Tukachinsky, H.; Lopez, L.V.; Salic, A. A mechanism for vertebrate Hedgehog signaling: Recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell Biol. 2010, 191, 415–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Hui, C.C. Hedgehog signaling in development and cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Liu, A.; Rakeman, A.S.; Murcia, N.S.; Niswander, L.; Anderson, K.V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 2003, 426, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Aberger, F.; Ruiz, I.A.A. Context-dependent signal integration by the GLI code: The oncogenic load, pathways, modifiers and implications for cancer therapy. Semin. Cell Dev. Biol. 2014, 33, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Winklmayr, M.; Schmid, C.; Laner-Plamberger, S.; Kaser, A.; Aberger, F.; Eichberger, T.; Frischauf, A.-M. Non-consensus GLI binding sites in Hedgehog target gene regulation. BMC Mol. Biol. 2010, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D. Hedgehog signalling: Emerging evidence for non-canonical pathways. Cell. Signal. 2009, 21, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Outram, S.V.; Varas, A.; Pepicelli, C.V.; Crompton, T. Hedgehog signaling regulates differentiation from double-negative to double-positive thymocyte. Immunity 2000, 13, 187–197. [Google Scholar] [CrossRef]
- Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N.P.; Guo, G.-R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J.F.; et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat. Med. 2007, 13, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Sacedon, R.; Diez, B.; Nunez, V.; Hernandez-Lopez, C.; Gutierrez-Frias, C.; Cejalvo, T.; Outram, S.V.; Crompton, T.; Zapata, A.G.; Vicente, A.; et al. Sonic hedgehog is produced by follicular dendritic cells and protects germinal center B cells from apoptosis. J. Immunol. 2005, 174, 1456–1461. [Google Scholar] [CrossRef] [PubMed]
- Le, H.; Kleinerman, R.; Lerman, O.Z.; Brown, D.; Galiano, R.; Gurtner, G.C.; Warren, S.M.; Levine, J.P.; Saadeh, P.B. Hedgehog signaling is essential for normal wound healing. Wound Repair Regener. 2008, 16, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.A.; Hoyne, G.F.; Ahmad, S.A.; Jarman, E.; Wallace, W.A.; Harrison, D.J.; Haslett, C.; Lamb, J.R.; Howie, S.E. Expression of the developmental Sonic hedgehog (Shh) signalling pathway is up-regulated in chronic lung fibrosis and the Shh receptor patched 1 is present in circulating T lymphocytes. J. Pathol. 2003, 199, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.M.; Williams, J.; van den Brink, G.R.; Lauwers, G.Y.; Roberts, D.J. Hh pathway expression in human gut tissues and in inflammatory gut diseases. Lab. Investig. 2004, 84, 1631–1642. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, G.; Murdoch, B.; Wu, D.; Baker, D.P.; Williams, K.P.; Chadwick, K.; Ling, L.E.; Karanu, F.N.; Bhatia, M. Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nat. Immunol. 2001, 2, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.I.; Outram, S.V.; Saldana, J.I.; Furmanski, A.L.; Dessens, J.T.; Crompton, T. Regulation of murine normal and stress-induced erythropoiesis by Desert Hedgehog. Blood 2012, 119, 4741–4751. [Google Scholar] [CrossRef] [PubMed]
- Petrova, R.; Joyner, A.L. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef] [PubMed]
- El Andaloussi, A.; Graves, S.; Meng, F.; Mandal, M.; Mashayekhi, M.; Aifantis, I. Hedgehog signaling controls thymocyte progenitor homeostasis and differentiation in the thymus. Nat. Immunol. 2006, 7, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Rowbotham, N.J.; Furmanski, A.L.; Hager-Theodorides, A.L.; Ross, S.E.; Drakopoulou, E.; Koufaris, C.; Outram, S.V.; Crompton, T. Repression of hedgehog signal transduction in T-lineage cells increases TCR-induced activation and proliferation. Cell Cycle 2008, 7, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Rowbotham, N.J.; Hager-Theodorides, A.L.; Cebecauer, M.; Shah, D.K.; Drakopoulou, E.; Dyson, J.; Outram, S.V.; Crompton, T. Activation of the Hedgehog signaling pathway in T-lineage cells inhibits TCR repertoire selection in the thymus and peripheral T-cell activation. Blood 2007, 109, 3757–3766. [Google Scholar] [CrossRef] [PubMed]
- Furmanski, A.L.; Saldana, J.I.; Ono, M.; Sahni, H.; Paschalidis, N.; D’Acquisto, F.; Crompton, T. Tissue-derived hedgehog proteins modulate Th differentiation and disease. J. Immunol. 2013, 190, 2641–2649. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.; Park, S.H. The epidemiology of hepatocellular cancer: From the perspectives of public health problem to tumor biology. J. Gastroenterol. 2009, 44 (Suppl. 19), 96–101. [Google Scholar] [CrossRef] [PubMed]
- De Almeida Pereira, T.; Witek, R.P.; Syn, W.K.; Choi, S.S.; Bradrick, S.; Karaca, G.F.; Agboola, K.M.; Jung, Y.; Omenetti, A.; Moylan, C.A.; et al. Viral factors induce Hedgehog pathway activation in humans with viral hepatitis, cirrhosis, and hepatocellular carcinoma. Lab. Investig. 2010, 90, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Zompetta, C.; Vescarelli, E.; Rizzello, C.; Cardi, A.; Valia, S.; Antonelli, G.; Marchese, C.; Torrisi, M.R.; Faggioni, A.; et al. HCV derived from sera of HCV-infected patients induces pro-fibrotic effects in human primary fibroblasts by activating GLI2. Sci. Rep. 2016, 6, 30649. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Cho, H.K.; Hong, S.P.; Cheong, J. Hepatitis B virus X protein stimulates the Hedgehog-Gli activation through protein stabilization and nuclear localization of Gli1 in liver cancer cells. Cancer Lett. 2011, 309, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Arzumanyan, A.; Sambandam, V.; Clayton, M.M.; Choi, S.S.; Xie, G.; Diehl, A.M.; Yu, D.Y.; Feitelson, M.A. Hedgehog signaling blockade delays hepatocarcinogenesis induced by hepatitis B virus X protein. Cancer Res. 2012, 72, 5912–5920. [Google Scholar] [CrossRef] [PubMed]
- Jo, B.B.; Jeong, M.S.; Park, S.Y.; Cheong, J.; Jang, S.B. The Binding of Hepatitis B Virus X Protein to Glioma-Associated Oncogene Homologue 1 and its Biological Characterization In vitro. Appl. Biochem. Biotechnol. 2011, 165, 109. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Bradrick, S.; Qiang, G.; Mostafavi, A.; Chaturvedi, G.; Weinman, S.A.; Diehl, A.M.; Jhaveri, R. Up-regulation of Hedgehog pathway is associated with cellular permissiveness for hepatitis C virus replication. Hepatology 2011, 54, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Port, R.J.; Pinheiro-Maia, S.; Hu, C.; Arrand, J.R.; Wei, W.; Young, L.S.; Dawson, C.W. Epstein-Barr virus induction of the Hedgehog signalling pathway imposes a stem cell phenotype on human epithelial cells. J. Pathol. 2013, 231, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.D.; Banerjee, S. Epstein-Barr virus latent membrane protein 2A mediated activation of Sonic Hedgehog pathway induces HLA class Ia downregulation in gastric cancer cells. Virology 2015, 484, 22–32. [Google Scholar]
- Lan, X.; Wen, H.; Cheng, K.; Plagov, A.; Shoshtari, S.S.M.; Malhotra, A.; Singhal, P.C. Hedgehog pathway plays a vital role in HIV-induced epithelial-mesenchymal transition of podocyte. Exp. Cell Res. 2017, 352, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Fuccio, L.; Eusebi, L.H.; Bazzoli, F. Gastric cancer, Helicobacter pylori infection and other risk factors. World J. Gastrointest. Oncol. 2010, 2, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Suerbaum, S.; Michetti, P. Helicobacter pylori infection. N. Engl. J. Med. 2002, 347, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.A.; Donnelly, J.M.; Engevik, A.C.; Xiao, C.; Yang, L.; Kenny, S.; Varro, A.; Hollande, F.; Samuelson, L.C.; Zavros, Y. Gastric Sonic Hedgehog acts as a macrophage chemoattractant during the immune response to Helicobacter pylori. Gastroenterology 2012, 142, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.A.; Feng, R.; Aihara, E.; Engevik, A.C.; Montrose, M.H.; Ottemann, K.M.; Zavros, Y. Helicobacter pylori-induced Sonic Hedgehog expression is regulated by NFkappaB pathway activation: The use of a novel in vitro model to study epithelial response to infection. Helicobacter 2015, 20, 19–28. [Google Scholar] [CrossRef] [PubMed]
- El-Zaatari, M.; Tobias, A.; Grabowska, A.M.; Kumari, R.; Scotting, P.J.; Kaye, P.; Atherton, J.; Clarke, P.A.; Powe, D.G.; Watson, S.A. De-regulation of the sonic hedgehog pathway in the InsGas mouse model of gastric carcinogenesis. Br. J. Cancer 2007, 96, 1855–1861. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Minegishi, Y.; Nomoto, Y.; Ota, T.; Masaoka, T.; van den Brink, G.R.; Hibi, T. Down-regulation of a morphogen (sonic hedgehog) gradient in the gastric epithelium of Helicobacter pylori-infected Mongolian gerbils. J. Pathol. 2005, 206, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Stasikowska-Kanicka, O.; Wagrowska-Danilewicz, M.; Bialek, I.; Danilewicz, M. The immunoexpression of Shh, Smo and Gli2 in Helicobacter pylori positive and negative gastric biopsies. Pol. J. Pathol. 2012, 63, 25–30. [Google Scholar] [PubMed]
- Nishizawa, T.; Suzuki, H.; Nakagawa, I.; Minegishi, Y.; Masaoka, T.; Iwasaki, E.; Hibi, T. Early Helicobacter pylori eradication restores sonic hedgehog expression in the gastric mucosa of Mongolian gerbils. Digestion 2009, 79, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Shiotani, A.; Uedo, N.; Iishi, H.; Tatsuta, M.; Ishiguro, S.; Nakae, Y.; Kamada, T.; Haruma, K.; Merchant, J.L. Re-expression of sonic hedgehog and reduction of CDX2 after Helicobacter pylori eradication prior to incomplete intestinal metaplasia. Int. J. Cancer 2007, 121, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, T.; Suzuki, H.; Masaoka, T.; Minegishi, Y.; Iwasahi, E.; Hibi, T. Helicobacter pylori eradication restored sonic hedgehog expression in the stomach. Hepato Gastroenterol. 2007, 54, 697–700. [Google Scholar]
- Jung, D.H.; Kim, J.H.; Chung, H.S.; Park, J.C.; Shin, S.K.; Lee, S.K.; Lee, Y.C. Helicobacter pylori Eradication on the Prevention of Metachronous Lesions after Endoscopic Resection of Gastric Neoplasm: A Meta-Analysis. PLoS ONE 2015, 10, e0124725. [Google Scholar] [CrossRef] [PubMed]
- Mutoh, H.; Hayakawa, H.; Sashikawa, M.; Sakamoto, H.; Sugano, K. Direct repression of Sonic Hedgehog expression in the stomach by Cdx2 leads to intestinal transformation. Biochem. J. 2010, 427, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Barros, R.; Marcos, N.; Reis, C.A.; De Luca, A.; David, L.; Almeida, R. CDX2 expression is induced by Helicobacter pylori in AGS cells. Scand. J. Gastroenterol. 2009, 44, 124–125. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Yamauchi, K.; Matsumoto, T.; Sano, K.; Yamaoka, Y.; Ota, H. Quantitative analysis of the effect of Helicobacter pylori on the expressions of SOX2, CDX2, MUC2, MUC5AC, MUC6, TFF1, TFF2, and TFF3 mRNAs in human gastric carcinoma cells. Scand. J. Gastroenterol. 2008, 43, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Smelkinson, M.G.; Guichard, A.; Teijaro, J.R.; Malur, M.; Loureiro, M.E.; Jain, P.; Ganesan, S.; Zúñiga, E.I.; Krug, R.M.; Oldstone, M.B.; et al. Influenza NS1 directly modulates Hedgehog signaling during infection. PLoS Pathog. 2017, 13, e1006588. [Google Scholar] [CrossRef] [PubMed]
- Bier, E.; Guichard, A. Deconstructing host-pathogen interactions in Drosophila. Dis. Model. Mech. 2012, 5, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Bier, E. Drosophila, the golden bug, emerges as a tool for human genetics. Nat. Rev. Genet. 2005, 6, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Reiter, L.T.; Potocki, L.; Chien, S.; Gribskov, M.; Bier, E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001, 11, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.T.; Allen, A.L.; Bardin, J.E.; Christian, M.N.; Daimon, K.; Dozier, K.D.; Hansen, C.L.; Holcomb, L.M. Ahlander, J. Drosophila as a genetic model for studying pathogenic human viruses. Virology 2012, 423, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.A.; Kim, B.; Bhin, J.; Kim, D.H.; You, H.; Kim, E.K.; Kim, S.H.; Ryu, J.H.; Hwang, D.; Lee, W.J. Bacterial uracil modulates Drosophila DUOX-dependent gut immunity via Hedgehog-induced signaling endosomes. Cell Host Microbe 2015, 17, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Biehs, B.; Sturtevant, M.A.; Bier, E. Boundaries in the Drosophila wing imaginal disc organize vein-specific genetic programs. Development 1998, 125, 4245–4257. [Google Scholar] [PubMed]
- St Johnston, R.D.; Hoffmann, F.M.; Blackman, R.K.; Segal, D.; Grimaila, R.; Padgett, R.W.; Irick, H.A.; Gelbart, W.M. Molecular organization of the decapentaplegic gene in Drosophila melanogaster. Genes Dev. 1990, 4, 1114–1127. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A.; Krug, R.M. Othromyxoviridae: The viruses and their replication. In Fundamental Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001. [Google Scholar]
- Hale, B.G.; Randall, R.E.; Ortin, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef] [PubMed]
- Marc, D. Influenza virus non-structural protein NS1: Interferon antagonism and beyond. J. Gen. Virol. 2014, 95, 2594–2611. [Google Scholar] [CrossRef] [PubMed]
- El-Zaatari, M.; Kao, J.Y.; Tessier, A.; Bai, L.; Hayes, M.M.; Fontaine, C.; Eaton, K.A.; Merchant, J.L. Gli1 deletion prevents Helicobacter-induced gastric metaplasia and expansion of myeloid cell subsets. PLoS ONE 2013, 8, e58935. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.L.; Li, C.; Ping, J.; Zhou, Y.; Li, Y.; Hao, P. The domain landscape of virus-host interactomes. Biomed. Res. Int. 2014, 2014, 867235. [Google Scholar] [CrossRef] [PubMed]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Pogach, M.S.; Cao, Y.; Millien, G.; Ramirez, M.I.; Williams, M.C. Key developmental regulators change during hyperoxia-induced injury and recovery in adult mouse lung. J. Cell. Biochem. 2007, 100, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- Moshai, E.F.; Wemeau-Stervinou, L.; Cigna, N.; Brayer, S.; Somme, J.M.; Crestani, B.; Mailleux, A.A. Targeting the hedgehog-glioma-associated oncogene homolog pathway inhibits bleomycin-induced lung fibrosis in mice. Am. J. Respir. Cell Mol. Biol. 2014, 51, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.E.; Ahmad, S.A.; Fitch, P.M.; Lamb, J.R.; Howie, S.E. FITC-induced murine pulmonary inflammation: CC10 up-regulation and concurrent Shh expression. Cell Biol. Int. 2005, 29, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Asai, J.; Takenaka, H.; Kusano, K.F.; Ii, M.; Luedemann, C.; Curry, C.; Eaton, E.; Iwakura, A.; Tsutsumi, Y.; Hamada, H.; et al. Topical sonic hedgehog gene therapy accelerates wound healing in diabetes by enhancing endothelial progenitor cell-mediated microvascular remodeling. Circulation 2006, 113, 2413–2424. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.D.; Hu, T.P.; Wang, L.; Chen, M.S.; Liu, S.M.; Chen, A.F. Sonic hedgehog improves delayed wound healing via enhancing cutaneous nitric oxide function in diabetes. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E525–E531. [Google Scholar] [CrossRef] [PubMed]
- Mathew, E.; Collins, M.A.; Fernandez-Barrena, M.G.; Holtz, A.M.; Yan, W.; Hogan, J.O.; Tata, Z.; Allen, B.L.; Fernandez-Zapico, M.E.; Di Magliano, M.P. The transcription factor GLI1 modulates the inflammatory response during pancreatic tissue remodeling. J. Biol. Chem. 2014, 289, 27727–27743. [Google Scholar] [CrossRef] [PubMed]
- Mills, L.D.; Zhang, Y.; Marler, R.J.; Herreros-Villanueva, M.; Zhang, L.; Almada, L.L.; Couch, F.; Wetmore, C.; Di Magliano, M.P.; Fernandez-Zapico, M.E. Loss of the transcription factor GLI1 identifies a signaling network in the tumor microenvironment mediating KRAS oncogene-induced transformation. J. Biol. Chem. 2013, 288, 11786–11794. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Lin, X.; Lu, H.; Chen, B.; Bai, Y. An overview of hedgehog signaling in fibrosis. Mol. Pharmacol. 2015, 87, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Bolanos, A.L.; Milla, C.M.; Lira, J.C.; Ramirez, R.; Checa, M.; Barrera, L.; García-Alvarez, J.; Carbajal, V.; Becerril, C.; Gaxiola, M.; et al. Role of Sonic Hedgehog in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L978–L990. [Google Scholar] [CrossRef] [PubMed]
- Cigna, N.; Moshai, E.F.; Brayer, S.; Marchal-Somme, J.; Wemeau-Stervinou, L.; Fabre, A.; Mal, H.; Lesèche, G.; Dehoux, M.; Soler, P.; et al. The hedgehog system machinery controls transforming growth factor-beta-dependent myofibroblastic differentiation in humans: Involvement in idiopathic pulmonary fibrosis. Am. J. Pathol. 2012, 181, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Lowrey, J.A.; Stewart, G.A.; Lindey, S.; Hoyne, G.F.; Dallman, M.J.; Howie, S.E.; Lamb, J.R. Sonic hedgehog promotes cell cycle progression in activated peripheral CD4(+) T lymphocytes. J. Immunol. 2002, 169, 1869–1875. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.A.; Lowrey, J.A.; Wakelin, S.J.; Fitch, P.M.; Lindey, S.; Dallman, M.J.; Lamb, J.R.; Howie, S.E. Sonic hedgehog signaling modulates activation of and cytokine production by human peripheral CD4+ T cells. J. Immunol. 2002, 169, 5451–5457. [Google Scholar] [CrossRef] [PubMed]
- Fitch, P.M.; Howie, S.E.; Wallace, W.A. Oxidative damage and TGF-beta differentially induce lung epithelial cell sonic hedgehog and tenascin-C expression: Implications for the regulation of lung remodelling in idiopathic interstitial lung disease. Int. J. Exp. Pathol. 2011, 92, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Coon, D.R.; Roberts, D.J.; Loscertales, M.; Kradin, R. Differential epithelial expression of SHH and FOXF1 in usual and nonspecific interstitial pneumonia. Exp. Mol. Pathol. 2006, 80, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Kugler, M.C.; Loomis, C.A.; Samdani, R.; Zhao, Z.; Chen, G.J.; Brandt, J.P.; Brownell, I.; Joyner, A.L.; Rom, W.N.; et al. Hedgehog signaling in neonatal and adult lung. Am. J. Respir. Cell Mol. Biol. 2013, 48, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Diehl, A.M. Non-alcoholic steatohepatitis pathogenesis: Role of repair in regulating the disease progression. Dig. Dis. 2010, 28, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Diehl, A.M. Hedgehog signaling in cholangiocytes. Curr. Opin. Gastroenterol. 2011, 27, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, B.; Syn, W.K.; Delgado, I.; Karaca, G.F.; Jung, Y.; Wang, J.; Zubiaga, A.M.; Fresnedo, O.; Omenetti, A.; Zdanowicz, M.; et al. Hedgehog signaling is critical for normal liver regeneration after partial hepatectomy in mice. Hepatology 2010, 51, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Syn, W.K.; Choi, S.S.; Liaskou, E.; Karaca, G.F.; Agboola, K.M.; Oo, Y.H.; Mi, Z.; Pereira, T.A.; Zdanowicz, M.; Malladi, P.; Chen, Y. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology 2011, 53, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Harris, L.G.; Metge, B.J.; Liu, S.; Riker, A.I.; Samant, R.S.; Shevde, L.A. The hedgehog pathway transcription factor GLI1 promotes malignant behavior of cancer cells by up-regulating osteopontin. J. Biol. Chem. 2009, 284, 22888–22897. [Google Scholar] [CrossRef] [PubMed]
- Fabian, S.L.; Penchev, R.R.; Jacques, B.S.; Rao, A.N.; Sipila, P.; West, K.A.; McMahon, A.P.; Humphreys, B.D. Hedgehog-Gli Pathway Activation during Kidney Fibrosis. Am. J. Pathol. 2012, 180, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Zhou, D.; Hao, S.; Zhou, L.; He, W.; Nie, J.; Hou, F.F.; Liu, Y. Sonic Hedgehog Signaling Mediates Epithelial-Mesenchymal Communication and Promotes Renal Fibrosis. J. Am. Soc. Nephrol. 2012, 23, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Kayed, H.; Kleeff, J.; Keleg, S.; Buchler, M.W.; Friess, H. Distribution of Indian hedgehog and its receptors patched and smoothened in human chronic pancreatitis. J. Endocrinol. 2003, 178, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.H.; Jung, D.E.; Park, Y.N.; Song, S.Y.; Park, S.W. Aberrant Hedgehog ligands induce progressive pancreatic fibrosis by paracrine activation of myofibroblasts and ductular cells in transgenic zebrafish. PLoS ONE 2011, 6, e27941. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, S.; Ohnishi, H.; Hama, K.; Kita, H.; Yamamoto, H.; Osawa, H.; Sato, K.; Tamada, K.; Mashima, H.; Sugano, K. Indian hedgehog promotes the migration of rat activated pancreatic stellate cells by increasing membrane type-1 matrix metalloproteinase on the plasma membrane. J. Cell. Physiol. 2008, 216, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Bier, E.; Nizet, V. Hedgehog: Linking uracil to innate defense. Cell Host Microbe 2015, 17, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.A.; Kim, B.; You, H.; Lee, W.J. Uracil-induced signaling pathways for DUOX-dependent gut immunity. Fly 2015, 9, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Crompton, T.; Outram, S.V.; Hager-Theodorides, A.L. Sonic hedgehog signalling in T-cell development and activation. Nat. Rev. Immunol. 2007, 7, 726–735. [Google Scholar] [CrossRef] [PubMed]
- De la Roche, M.; Ritter, A.T.; Angus, K.L.; Dinsmore, C.; Earnshaw, C.H.; Reiter, J.F.; Griffiths, G.M. Hedgehog signaling controls T cell killing at the immunological synapse. Science 2013, 342, 1247–1250. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, D.I.; Zlotnik, A. Control points in early T-cell development. Immunol. Today 1993, 14, 547–553. [Google Scholar] [CrossRef]
- Haks, M.C.; Oosterwegel, M.A.; Blom, B.; Spits, H.M.; Kruisbeek, A.M. Cell-fate decisions in early T cell development: Regulation by cytokine receptors and the pre-TCR. Semin. Immunol. 1999, 11, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Borowski, C.; Martin, C.; Gounari, F.; Haughn, L.; Aifantis, I.; Grassi, F.; von Boehmer, H. On the brink of becoming a T cell. Curr. Opin. Immunol. 2002, 14, 200–206. [Google Scholar] [CrossRef]
- Shah, D.K.; Hager-Theodorides, A.L.; Outram, S.V.; Ross, S.E.; Varas, A.; Crompton, T. Reduced thymocyte development in sonic hedgehog knockout embryos. J. Immunol. 2004, 172, 2296–2306. [Google Scholar] [CrossRef] [PubMed]
- Hager-Theodorides, A.L.; Dessens, J.T.; Outram, S.V.; Crompton, T. The transcription factor Gli3 regulates differentiation of fetal CD4–CD8–double-negative thymocytes. Blood 2005, 106, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Outram, S.V.; Hager-Theodorides, A.L.; Shah, D.K.; Rowbotham, N.J.; Drakopoulou, E.; Ross, S.E.; Lanske, B.; Dessens, J.T.; Crompton, T. Indian hedgehog (Ihh) both promotes and restricts thymocyte differentiation. Blood 2009, 113, 2217–2228. [Google Scholar] [CrossRef] [PubMed]
- Wakelin, S.J.; Forsythe, J.L.; Garden, O.J.; Howie, S.E. Commercially available recombinant sonic hedgehog up-regulates Ptc and modulates the cytokine and chemokine expression of human macrophages: An effect mediated by endotoxin contamination? Immunobiology 2008, 213, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Marwaha, S.; Schumacher, M.A.; Zavros, Y.; Eghbalnia, H.R. Crosstalks between cytokines and Sonic Hedgehog in Helicobacter pylori infection: A mathematical model. PLoS ONE 2014, 9, e111338. [Google Scholar] [CrossRef] [PubMed]
- Chiaramonte, M.G.; Schopf, L.R.; Neben, T.Y.; Cheever, A.W.; Donaldson, D.D.; Wynn, T.A. IL-13 is a key regulatory cytokine for Th2 cell-mediated pulmonary granuloma formation and IgE responses induced by Schistosoma mansoni eggs. J. Immunol. 1999, 162, 920–930. [Google Scholar] [PubMed]
- Fichtner-Feigl, S.; Strober, W.; Kawakami, K.; Puri, R.K.; Kitani, A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat. Med. 2006, 12, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Syn, W.K.; Witek, R.P.; Curbishley, S.M.; Jung, Y.; Choi, S.S.; Enrich, B.; Omenetti, A.; Agboola, K.M.; Fearing, C.M.; Tilg, H. Role for hedgehog pathway in regulating growth and function of invariant NKT cells. Eur. J. Immunol. 2009, 39, 1879–1892. [Google Scholar] [CrossRef] [PubMed]
- Standing, A.S.; Yanez, D.C.; Ross, R.; Crompton, T.; Furmanski, A.L. Frontline Science: Shh production and Gli signaling is activated in vivo in lung, enhancing the Th2 response during a murine model of allergic asthma. J. Leukoc. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Sun, D.; Li, H.; Li, X.; Pan, W.; Yan, C.; Tang, R.; Liu, X. The Effect of SHH-Gli Signaling Pathway on the Synovial Fibroblast Proliferation in Rheumatoid Arthritis. Inflammation 2016, 39, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Samanta, J.; Grund, E.M.; Silva, H.M.; Lafaille, J.J.; Fishell, G.; Salzer, J.L. Inhibition of Gli1 mobilizes endogenous neural stem cells for remyelination. Nature 2015, 526, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Seifert, T.; Bauer, J.; Weissert, R.; Fazekas, F.; Storch, M.K. Differential expression of sonic hedgehog immunoreactivity during lesion evolution in autoimmune encephalomyelitis. J. Neuropathol. Exp. Neurol. 2005, 64, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Kobune, M.; Takimoto, R.; Murase, K.; Iyama, S.; Sato, T.; Kikuchi, S.; Kawano, Y.; Miyanishi, K.; Sato, Y.; Niitsu, Y.; et al. Drug resistance is dramatically restored by hedgehog inhibitors in CD34+ leukemic cells. Cancer Sci. 2009, 100, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Dagklis, A.; Pauwels, D.; Lahortiga, I.; Geerdens, E.; Bittoun, E.; Cauwelier, B.; Tousseyn, T.; Uyttebroeck, A.; Maertens, J.; Verhoef, G.; et al. Hedgehog pathway mutations in T-cell acute lymphoblastic leukemia. Haematologica 2015, 100, e102–e105. [Google Scholar] [CrossRef] [PubMed]
- Pomes, A.; Chapman, M.D.; Wunschmann, S. Indoor Allergens and Allergic Respiratory Disease. Curr. Allergy Asthma Rep. 2016, 16, 43. [Google Scholar] [CrossRef] [PubMed]
- Nhu, Q.M.; Aceves, S.S. Tissue Remodeling in Chronic Eosinophilic Esophageal Inflammation: Parallels in Asthma and Therapeutic Perspectives. Front. Med. 2017, 4, 128. [Google Scholar] [CrossRef] [PubMed]
- Potter, C.W.; Jennings, R. A definition for influenza pandemics based on historical records. J. Infect. 2011, 63, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Aditya, S.; Rattan, A. Vismodegib: A smoothened inhibitor for the treatment of advanced basal cell carcinoma. Indian Dermatol. Online J. 2013, 4, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.W. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers 2016, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Philips, G.M.; Chan, I.S.; Swiderska, M.; Schroder, V.T.; Guy, C.; Karaca, G.F.; Moylan, C.; Venkatraman, T.; Feuerlein, S.; Syn, W.K.; et al. Hedgehog signaling antagonist promotes regression of both liver fibrosis and hepatocellular carcinoma in a murine model of primary liver cancer. PLoS ONE 2011, 6, e23943. [Google Scholar] [CrossRef] [PubMed]
- Pratap, A.; Panakanti, R.; Yang, N.; Eason, J.D.; Mahato, R.I. Inhibition of endogenous hedgehog signaling protects against acute liver injury after ischemia reperfusion. Pharm. Res. 2010, 27, 2492–2504. [Google Scholar] [CrossRef] [PubMed]
- Samarzija, I.; Beard, P. Hedgehog pathway regulators influence cervical cancer cell proliferation, survival and migration. Biochem. Biophys. Res. Commun. 2012, 425, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Yu, K.; Zhang, L.; Li, Y.; Li, Q.; Yang, Z.; Shen, T.; Duan, L.; Xiong, W.; Wang, W. Synergistic inhibition of colon carcinoma cell growth by Hedgehog-Gli1 inhibitor arsenic trioxide and phosphoinositide 3-kinase inhibitor LY294002. OncoTargets Ther. 2015, 8, 877–883. [Google Scholar]
- Kerl, K.; Moreno, N.; Holsten, T.; Ahlfeld, J.; Mertins, J.; Hotfilder, M.; Kool, M.; Bartelheim, K.; Schleicher, S.; Handgretinger, R.; et al. Arsenic trioxide inhibits tumor cell growth in malignant rhabdoid tumors in vitro and in vivo by targeting overexpressed Gli1. Int. J. Cancer 2014, 135, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Nagano, S.; Nagao, H.; Ishidou, Y.; Yokouchi, M.; Abematsu, M.; Yamamoto, T.; Komiya, S.; Setoguchi, T. Arsenic trioxide prevents osteosarcoma growth by inhibition of GLI transcription via DNA damage accumulation. PLoS ONE 2013, 8, e69466. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Cao, F.; Ye, X.; Zhao, H.; Liu, X.; Li, Y.; Shi, C.; Wang, H.; Zhou, J. Arsenic trioxide inhibits the Hedgehog pathway which is aberrantly activated in acute promyelocytic leukemia. Acta Haematol. 2013, 130, 260–267. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Varona-Santos, J.; Singh, S.; Robbins, D.J.; Savaraj, N.; Nguyen, D.M. Targeting of the Hedgehog signal transduction pathway suppresses survival of malignant pleural mesothelioma cells in vitro. J. Thorac. Cardiovasc. Surg. 2014, 147, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Bansal, N.; Farley, N.J.; Wu, L.; Lewis, J.; Youssoufian, H.; Bertino, J.R. Darinaparsin inhibits prostate tumor-initiating cells and Du145 xenografts and is an inhibitor of hedgehog signaling. Mol. Cancer Ther. 2015, 14, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.B.; Singh, M.V.; Gorantla, S.; Poluektova, L.Y.; Maggirwar, S.B. Smoothened Agonist Reduces Human Immunodeficiency Virus Type-1-Induced Blood-Brain Barrier Breakdown in Humanized Mice. Sci. Rep. 2016, 6, 26876. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.B.; Singh, M.V.; Piekna-Przybylska, D.; Gorantla, S.; Poluektova, L.Y.; Maggirwar, S.B. Sonic Hedgehog mimetic prevents leukocyte infiltration into the CNS during acute HIV infection. Sci. Rep. 2017, 7, 9578. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smelkinson, M.G. The Hedgehog Signaling Pathway Emerges as a Pathogenic Target. J. Dev. Biol. 2017, 5, 14. https://doi.org/10.3390/jdb5040014
Smelkinson MG. The Hedgehog Signaling Pathway Emerges as a Pathogenic Target. Journal of Developmental Biology. 2017; 5(4):14. https://doi.org/10.3390/jdb5040014
Chicago/Turabian StyleSmelkinson, Margery G. 2017. "The Hedgehog Signaling Pathway Emerges as a Pathogenic Target" Journal of Developmental Biology 5, no. 4: 14. https://doi.org/10.3390/jdb5040014
APA StyleSmelkinson, M. G. (2017). The Hedgehog Signaling Pathway Emerges as a Pathogenic Target. Journal of Developmental Biology, 5(4), 14. https://doi.org/10.3390/jdb5040014