
Reference-Guided De Novo Genome Assembly to Dissect a QTL Region for Submergence Tolerance Derived from Ciherang-Sub1

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
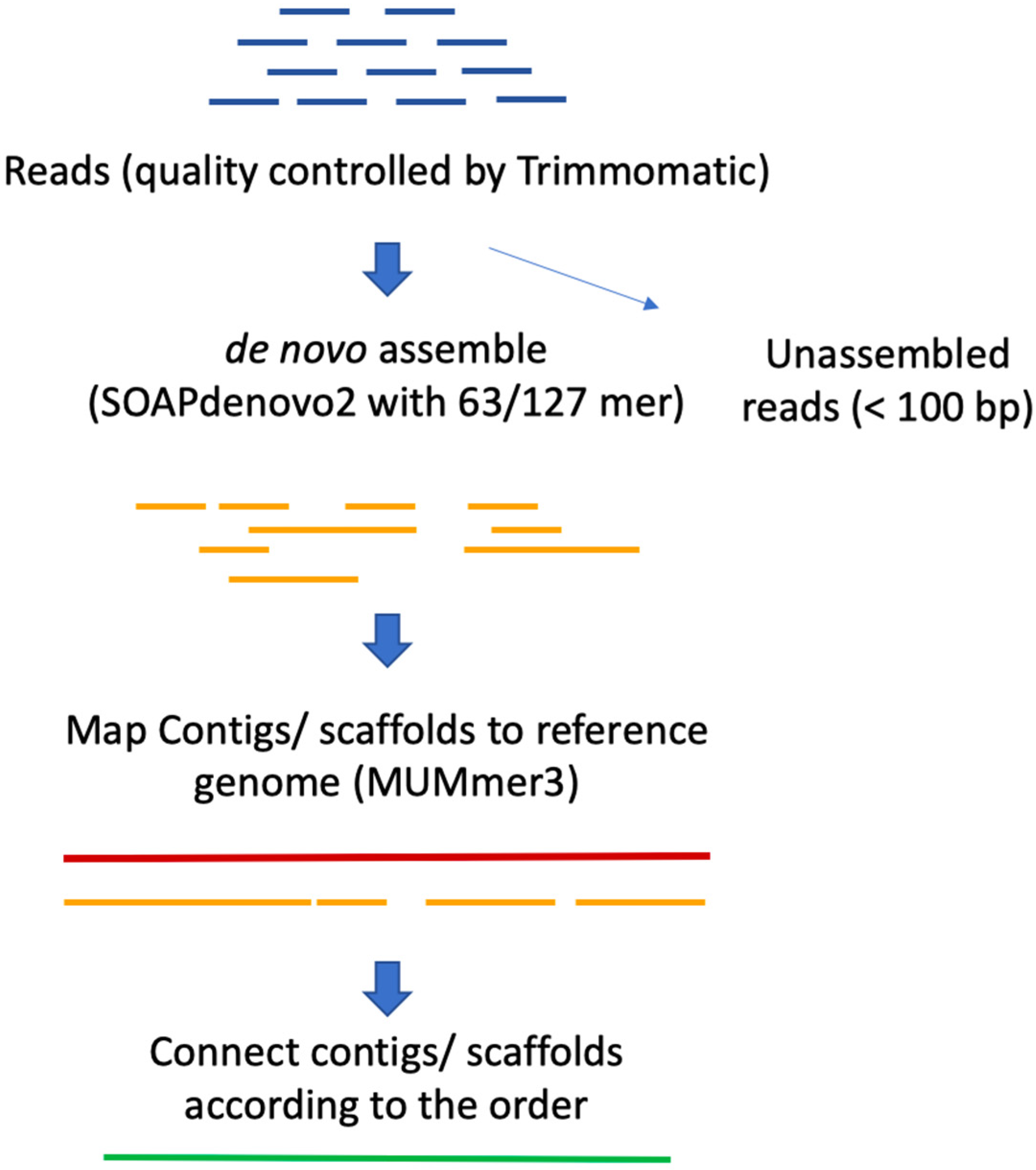
2.1. Genome Assembly
2.2. Validation of Three Assembled Genomes
2.3. Genome Profile of Ciherang-Sub1
2.4. Dissection of qSub8.1 Region
3. Materials and Methods
3.1. Plant Materials
3.2. Whole Genome Sequencing and Assembly
3.3. Variants Calling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rice Is Life: Scientific Perspectives for the 21st Century; Toriyama, K.; Heong, K.L.; Hardy, B. (Eds.) International Rice Research Institute: Los Baños, Philippines; Tsukuba, Japan, 2005. [Google Scholar]
- Fukao, T.; Bailey-Serres, J. Plant responses to hypoxia—Is survival a balancing act? Trends Plant Sci. 2004, 9, 449–456. [Google Scholar] [CrossRef]
- Fannin, B. Texas Agricultural Losses from Hurricane Harvey Estimated at more than $200 Million. AgriLife TODAY 2017. Available online: https://agrilifetoday.tamu.edu/2017/10/27/texas-agricultural-losses-hurricane-harvey-estimated-200-million/ (accessed on 8 December 2021).
- Xu, K.; Xu, X.; Fukao, T.; Canlas, P.; Maghirang-Rodriguez, R.; Heuer, S.; Ismail, A.M.; Bailey-Serres, J.; Ronald, P.C.; Mackill, D.J. Sub1A is an ethylene-response-factor-like gene that confers submergence tolerance to rice. Nature 2006, 442, 705–708. [Google Scholar] [CrossRef] [Green Version]
- Neeraja, C.N.; Maghirang-Rodriguez, R.; Pamplona, A.; Heuer, S.; Collard, B.C.Y.; Septiningsih, E.M.; Vergara, G.; Sanchez, D.; Xu, K.; Ismail, A.M.; et al. A marker-assisted backcross approach for developing submergence-tolerant rice cultivars. Theor. Appl. Genet. 2007, 115, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Septiningsih, E.M.; Pamplona, A.M.; Sanchez, D.L.; Neeraja, C.N.; Vergara, G.V.; Heuer, S.; Ismail, A.M.; Mackill, D.J. Development of submergence-tolerant rice cultivars: The Sub1 locus and beyond. Ann. Bot. 2009, 103, 151–160. [Google Scholar] [CrossRef] [Green Version]
- Septiningsih, E.M.; Mackill, D.J. Genetics and breeding of flooding tolerance in rice. In Rice Genomics, Genetics and Breeding; Springer: Berlin/Heidelberg, Germany, 2018; pp. 275–295. [Google Scholar]
- Septiningsih, E.M.; Hidayatun, N.; Sanchez, D.L.; Nugraha, Y.; Carandang, J.; Pamplona, A.M.; Collard, B.C.; Ismail, A.M.; Mackill, D.J. Accelerating the development of new submergence tolerant rice varieties: The case of Ciherang-Sub1 and PSB Rc18-Sub1. Euphytica 2015, 202, 259–268. [Google Scholar] [CrossRef]
- Iftekharuddaula, K.M.; Ahmed, H.U.; Ghosal, S.; Amin, A.; Moni, Z.R.; Ray, B.P.; Barman, H.N.; Siddique, M.A.; Collard, B.C.; Septiningsih, E.M. Development of early maturing submergence-tolerant rice varieties for Bangladesh. Field Crops Res. 2016, 190, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Iftekharuddaula, K.M.; Newaz, M.A.; Salam, M.A.; Ahmed, H.U.; Mahbub, M.a.A.; Septiningsih, E.M.; Collard, B.C.Y.; Sanchez, D.L.; Pamplona, A.M.; Mackill, D.J. Rapid and high-precision marker assisted backcrossing to introgress the SUB1 QTL into BR11, the rainfed lowland rice mega variety of Bangladesh. Euphytica 2010, 178, 83–97. [Google Scholar] [CrossRef]
- Singh, R.; Singh, Y.; Xalaxo, S.; Verulkar, S.; Yadav, N.; Singh, S.; Singh, N.; Prasad, K.; Kondayya, K.; Rao, P.R. From QTL to variety-harnessing the benefits of QTLs for drought, flood and salt tolerance in mega rice varieties of India through a multi-institutional network. Plant Sci. 2016, 242, 278–287. [Google Scholar] [CrossRef]
- Septiningsih, E.M.; Collard, B.C.; Heuer, S.; Bailey-Serres, J.; Ismail, A.M.; Mackill, D.J. Applying genomics tools for breeding submergence tolerance in rice. Transl. Genom. Crop Breed. 2013, 2, 9–30. [Google Scholar]
- Fukao, T.; Xu, K.; Ronald, P.C.; Bailey-Serres, J. A variable cluster of ethylene response factor–like genes regulates metabolic and developmental acclimation responses to submergence in rice. Plant Cell 2006, 18, 2021–2034. [Google Scholar] [CrossRef] [Green Version]
- Fukao, T.; Harris, T.; Bailey-Serres, J. Evolutionary analysis of the Sub1 gene cluster that confers submergence tolerance to domesticated rice. Ann. Bot. 2009, 103, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Niroula, R.K.; Pucciariello, C.; Ho, V.T.; Novi, G.; Fukao, T.; Perata, P. SUB1A-dependent and -independent mechanisms are involved in the flooding tolerance of wild rice species. Plant J. 2012, 72, 282–293. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Dang, T.T.; Vergara, G.V.; Pandey, D.M.; Sanchez, D.; Neeraja, C.; Septiningsih, E.M.; Mendioro, M.; Tecson-Mendoza, E.M.; Ismail, A.M. Molecular marker survey and expression analyses of the rice submergence-tolerance gene SUB1A. Theor. Appl. Genet. 2010, 121, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Iftekharuddaula, K.M.; Ghosal, S.; Gonzaga, Z.J.; Amin, A.; Barman, H.N.; Yasmeen, R.; Haque, M.M.; Carandang, J.; Collard, B.C.; Septiningsih, E.M. Allelic diversity of newly characterized submergence-tolerant rice (Oryza sativa L.) germplasm from Bangladesh. Genet. Resour. Crop Evol. 2016, 63, 859–867. [Google Scholar] [CrossRef]
- Li, Z.-X.; Septiningsih, E.; Quilloy-Mercado, S.; McNally, K.; Mackill, D. Identification of SUB1A alleles from wild rice Oryza rufipogon Griff. Genet. Resour. Crop Evol. 2011, 58, 1237–1242. [Google Scholar] [CrossRef]
- Gonzaga, Z.J.C.; Carandang, J.; Sanchez, D.L.; Mackill, D.J.; Septiningsih, E.M. Mapping additional QTLs from FR13A to increase submergence tolerance in rice beyond SUB1. Euphytica 2016, 209, 627–636. [Google Scholar] [CrossRef]
- Gonzaga, Z.J.C.; Carandang, J.; Singh, A.; Collard, B.C.Y.; Thomson, M.J.; Septiningsih, E.M. Mapping QTLs for submergence tolerance in rice using a population fixed for SUB1A tolerant allele. Mol. Breed. 2017, 37, 47. [Google Scholar] [CrossRef]
- Septiningsih, E.M.; Sanchez, D.L.; Singh, N.; Sendon, P.M.; Pamplona, A.M.; Heuer, S.; Mackill, D.J. Identifying novel QTLs for submergence tolerance in rice cultivars IR72 and Madabaru. Appl. Genet 2012, 124, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Singh, Y.; Mahato, A.K.; Jayaswal, P.K.; Singh, S.; Singh, R.; Yadav, N.; Singh, A.; Singh, P.; Singh, R. Allelic sequence variation in the Sub1A, Sub1B and Sub1C genes among diverse rice cultivars and its association with submergence tolerance. Sci. Rep. 2020, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Fukao, T.; Yeung, E.; Bailey-Serres, J. The submergence tolerance regulator SUB1A mediates crosstalk between submergence and drought tolerance in rice. Plant Cell 2011, 23, 412–427. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, B.; Shrestha, S.M.; Singh, U.S.; Manandhar, H.K.; Zaidi, N.W.; Thapa, R.B. Submergence MEDIATES Leaf Blast Resistance in Sub1 and Non-Sub1 Rice Genotypes. Glob. J. Biol. Agric. Health Sci. 2015, 4, 231–237. [Google Scholar]
- Chaudhary, B.; Shrestha, S.M.; Singh, U.S.; Manandhar, H.K.; Zaidi, N.W.; Thapa, R.B.; Dangal, N.K. Evaluation of Sub1 and Non-Sub1 Rice for Resistance to Bacterial Blight Using Submerged and Non-submerged Seedlings. Agric. Biol. Sci. J. 2015, 1, 229–234. [Google Scholar]
- Wolf, J.B.; Ellegren, H. Making sense of genomic islands of differentiation in light of speciation. Nat. Rev. Genet 2017, 18, 87–100. [Google Scholar] [CrossRef]
- Sandhu, N.; Dixit, S.; Swamy, B.P.M.; Raman, A.; Kumar, S.; Singh, S.P.; Yadaw, R.B.; Singh, O.N.; Reddy, J.N.; Anandan, A.; et al. Marker Assisted Breeding to Develop Multiple Stress Tolerant Varieties for Flood and Drought Prone Areas. Rice 2019, 12, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledo, A.M.U.; Ignacio, J.C.I.; Casal, C.; Gonzaga, Z.J.; Mendioro, M.S.; Septiningsih, E.M. Development of Improved Ciherang-Sub1 Having Tolerance to Anaerobic Germination Conditions. Plant Breed. Biotechnol. 2015, 3, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Carandang, J.; Gonzaga, Z.J.C.; Collard, B.C.Y.; Ismail, A.M.; Septiningsih, E.M. Identification of QTLs for yield and agronomic traits in rice under stagnant flooding conditions. Rice 2017, 10, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pramudyawardani, E.F.; Aswidinnoor, H.; Purwoko, B.S.; Suwarno, W.B.; Islam, M.; Verdeprado, H.; Collard, B.C. Genetic analysis and QTL mapping for agronomic and yield-related traits in Ciherang-Sub1 rice backcross populations. Plant Breed. Biotechnol. 2018, 6, 177–192. [Google Scholar] [CrossRef]
- Liang, Y.; Biswas, S.; Kim, B.; Bailey-Serres, J.; Septiningsih, E.M. Improved Transformation and Regeneration of Indica Rice: Disruption of SUB1A as a Test Case via CRISPR-Cas9. Int. J. Mol. Sci. 2021, 22, 6989. [Google Scholar] [CrossRef]
- Thistlethwaite, F.R.; Gamal El-Dien, O.; Ratcliffe, B.; Klapste, J.; Porth, I.; Chen, C.; Stoehr, M.U.; Ingvarsson, P.K.; El-Kassaby, Y.A. Linkage disequilibrium vs. pedigree: Genomic selection prediction accuracy in conifer species. PLoS ONE 2020, 15, e0232201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, L.-L.; Sun, S.; Kudrna, D.; Copetti, D.; Li, W.; Mu, T.; Jiao, W.-B.; Xing, F.; Lee, S.; et al. Building two indica rice reference genomes with PacBio long-read and Illumina paired-end sequencing data. Sci. Data 2016, 3, 160076. [Google Scholar] [CrossRef] [PubMed]
- Blair, M.W.; Cortes, A.J.; Farmer, A.D.; Huang, W.; Ambachew, D.; Penmetsa, R.V.; Carrasquilla-Garcia, N.; Assefa, T.; Cannon, S.B. Uneven recombination rate and linkage disequilibrium across a reference SNP map for common bean (Phaseolus vulgaris L.). PLoS ONE 2018, 13, e0189597. [Google Scholar] [CrossRef] [Green Version]
- Coulton, A.; Burridge, A.J.; Edwards, K.J. Examining the Effects of Temperature on Recombination in Wheat. Front. Plant Sci. 2020, 11, 230. [Google Scholar] [CrossRef] [Green Version]
- Salina, E.A.; Adonina, I.G.; Badaeva, E.D.; Kroupin, P.Y.; Stasyuk, A.I.; Leonova, I.N.; Shishkina, A.A.; Divashuk, M.G.; Starikova, E.V.; Khuat, T.M.L.; et al. A Thinopyrum intermedium chromosome in bread wheat cultivars as a source of genes conferring resistance to fungal diseases. Euphytica 2015, 204, 91–101. [Google Scholar] [CrossRef]
- Nagy, E.D.; Chu, Y.; Guo, Y.; Khanal, S.; Tang, S.; Li, Y.; Dong, W.B.; Timper, P.; Taylor, C.; Ozias-Akins, P.; et al. Recombination is suppressed in an alien introgression in peanut harboring Rma, a dominant root-knot nematode resistance gene. Mol. Breed. 2010, 26, 357–370. [Google Scholar] [CrossRef]
- Ravinet, M.; Faria, R.; Butlin, R.K.; Galindo, J.; Bierne, N.; Rafajlovic, M.; Noor, M.A.F.; Mehlig, B.; Westram, A.M. Interpreting the genomic landscape of speciation: A road map for finding barriers to gene flow. J. Evol. Biol. 2017, 30, 1450–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadikhah, A.; Mirarab, M.; Pahlevani, M.H.; Nayyeripasand, L. Marker-Assisted Backcrossing to Develop an Elite Cytoplasmic Male Sterility Line in Rice. Plant Genome 2015, 8, 1–12. [Google Scholar] [CrossRef]
- Suprihatno, B.; Daradjat, A.A.; Baehaki, S.; Widiarta, I.; Setyono, A.; Indrasari, S.D.; Lesmana, O.S.; Sembiring, H. Deskripsi Varietas Padi (Description of Rice Varieties); Balai Besar Penelitian Tanaman Padi (Indonesian Center for Rice Research): Subang, West Java, Indonesia, 2009. [Google Scholar]
- Marathi, B.; Jena, K.K. Floral traits to enhance outcrossing for higher hybrid seed production in rice: Present status and future prospects. Euphytica 2014, 201, 1–14. [Google Scholar] [CrossRef]
- Mather, K.A.; Caicedo, A.L.; Polato, N.R.; Olsen, K.M.; McCouch, S.; Purugganan, M.D. The extent of linkage disequilibrium in rice (Oryza sativa L.). Genetics 2007, 177, 2223–2232. [Google Scholar] [CrossRef] [Green Version]
- Bernier, J.; Kumar, A.; Ramaiah, V.; Spaner, D.; Atlin, G. A Large-Effect QTL for Grain Yield under Reproductive-Stage Drought Stress in Upland Rice. Crop Sci. 2007, 47, 507–516. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 30. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [Green Version]
- Song, J.M.; Lei, Y.; Shu, C.C.; Ding, Y.; Xing, F.; Liu, H.; Wang, J.; Xie, W.; Zhang, J.; Chen, L.L. Rice Information GateWay: A Comprehensive Bioinformatics Platform for Indica Rice Genomes. Mol. Plant 2018, 11, 505–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Meth. 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Genome Size (bp) | |||||
|---|---|---|---|---|---|
| Variety | Number of Reads | Total Read Length (bp) | Sequencing Depth (X) | Nipponbare Reference * | MH63 Reference * |
| Ciherang-Sub1 | 102,213,313 | 15,331,996,950 | 38 | 345,442,284 (92.6%) | 354,934,762 (91.6%) |
| Ciherang | 115,172,273 | 17,275,840,950 | 43 | 343,737,849 (92.2%) | 353,859,778 (91.3%) |
| IR64-Sub1 | 114,085,140 | 17,112,771,000 | 43 | 344,678,967 (92.4%) | 353,859,751 (91.3%) |
| Gene | Position of the Top Match in Each Genome Assembly (bp) | |||
|---|---|---|---|---|
| Nipponbare | Ciherang-Sub1 | Ciherang | IR64-Sub1 | |
| SD1 (Os01g0883800) | chr01:38382385-38385469 | chr01:42499233-42501392 | chr01:43076922-43079161 | chr01:42595591-42597830 |
| GW5 (Os05g0187500) Sub1A (DQ011598) | chr05:5365122-5366701 absent 1 | chr05:4832476-4834024 NA 2 | chr05:4873155-4874734 NA | chr05:5262475-5264054 NA |
| Sub1B (Os09g0287000) | chr09:6404482-6406039 | chr09:5266747-5267932 | chr1:4258518-4259713 | chr09:4932683-4933868 |
| Sub1C (Os09g0286600) | chr09: 6387891-6389789 | chr09: 5250965-5252048 | chr4: 13931868-13933764 | chr09: 4916471-4917910 |
| Gene | Position of the Top Match in Each Genome Assembly (bp) | |||
|---|---|---|---|---|
| MH63 | Ciherang-Sub1 | Ciherang | IR64-Sub1 | |
| SD1 (OsMH_01G0636900) | chr01:39643093-39644426 | chr01:42732034-42734193 | chr01:42311555-42313794 | chr01:42124122-42126361 |
| GW5 (OsMH_05G0081900) | chr05:5428533-5430112 | chr05:5278241-5279789 | chr05:5178118-5179697 | chr05:4864637-4866216 |
| Sub1A (DQ011598) | chr06:22422489-22419199 | chr11:17912307-17916149 | chr11:8553735-8556945 | chr11:18314723-18318566 |
| Sub1B (OsMH_09G0114700) | chr09: 7179132-7180335 | chr09: 4262487-4264027 | chr12: 7911412-7912963 | chr09: 5209175-5210715 |
| Sub1C (No annotation) | chr09: 7162325-7164223 | chr10: 10781009-10782407 | chr8: 20306150-20308046 | chr09: 5193229-5195103 |
| All SNPs | 50 kb Window Size | 100 kb Window Size | |
|---|---|---|---|
| Ciherang-like genotype | 515,698 (87%) | 4406 (59%) | 2328 (63%) |
| IR64-Sub1-like genotype | 65,426 (11%) | 1791 (24%) | 832 (22%) |
| Other | 9540 (2%) | 1256 (17%) | 564 (15%) |
| Total SNP number | 590,664 | 7453 | 3724 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, Y.; Wang, S.; Harper, C.L.; Subramanian, N.K.; Tabien, R.E.; Johnson, C.D.; Bailey-Serres, J.; Septiningsih, E.M. Reference-Guided De Novo Genome Assembly to Dissect a QTL Region for Submergence Tolerance Derived from Ciherang-Sub1. Plants 2021, 10, 2740. https://doi.org/10.3390/plants10122740
Liang Y, Wang S, Harper CL, Subramanian NK, Tabien RE, Johnson CD, Bailey-Serres J, Septiningsih EM. Reference-Guided De Novo Genome Assembly to Dissect a QTL Region for Submergence Tolerance Derived from Ciherang-Sub1. Plants. 2021; 10(12):2740. https://doi.org/10.3390/plants10122740
Chicago/Turabian StyleLiang, Yuya, Shichen Wang, Chersty L. Harper, Nithya K. Subramanian, Rodante E. Tabien, Charles D. Johnson, Julia Bailey-Serres, and Endang M. Septiningsih. 2021. "Reference-Guided De Novo Genome Assembly to Dissect a QTL Region for Submergence Tolerance Derived from Ciherang-Sub1" Plants 10, no. 12: 2740. https://doi.org/10.3390/plants10122740
APA StyleLiang, Y., Wang, S., Harper, C. L., Subramanian, N. K., Tabien, R. E., Johnson, C. D., Bailey-Serres, J., & Septiningsih, E. M. (2021). Reference-Guided De Novo Genome Assembly to Dissect a QTL Region for Submergence Tolerance Derived from Ciherang-Sub1. Plants, 10(12), 2740. https://doi.org/10.3390/plants10122740