
Genome-Wide Identification and Genetic Variations of the Starch Synthase Gene Family in Rice

 and
and 
Abstract
:1. Introduction
2. Results
2.1. Identification of SS Genes in Rice Genome
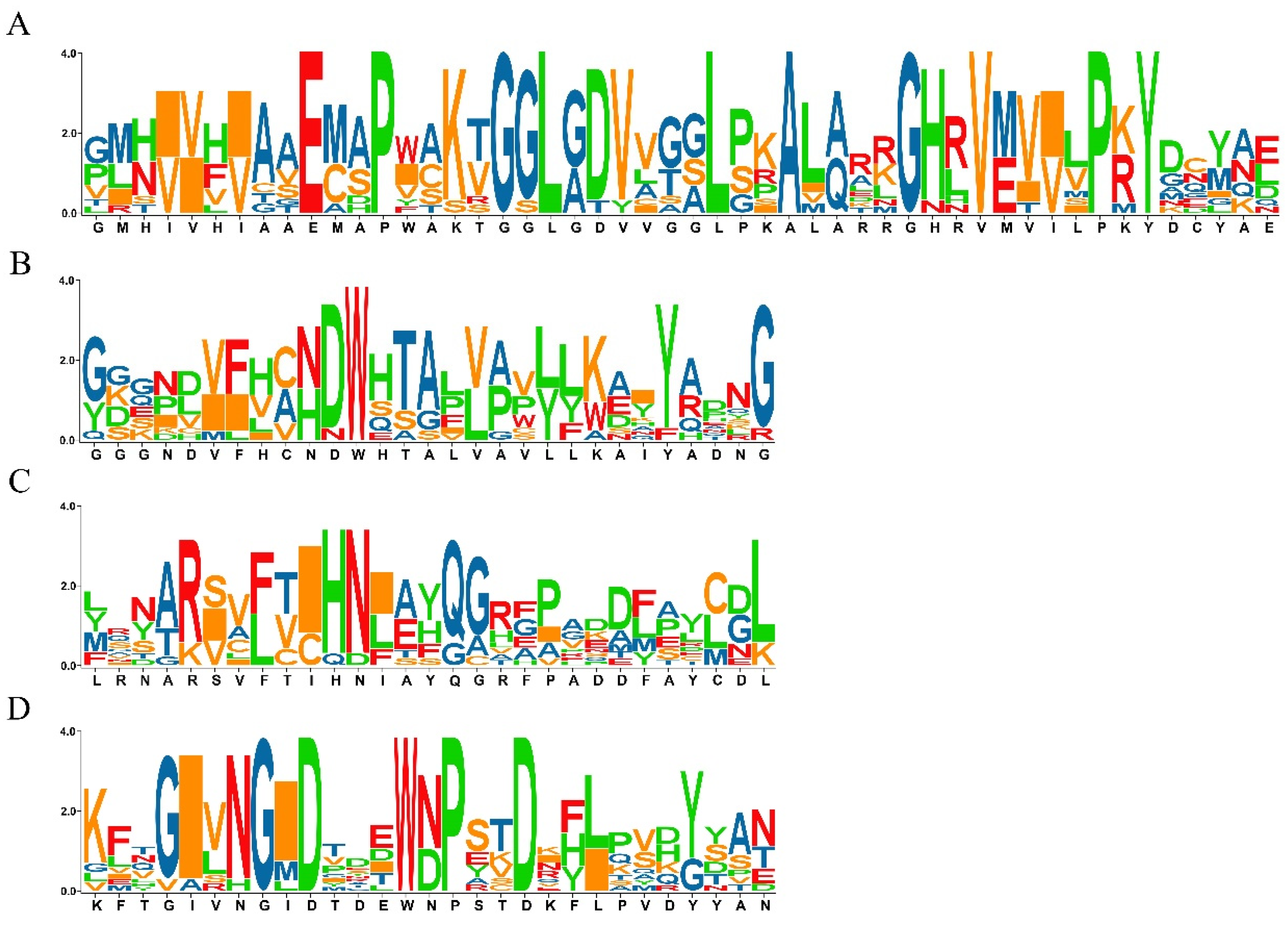
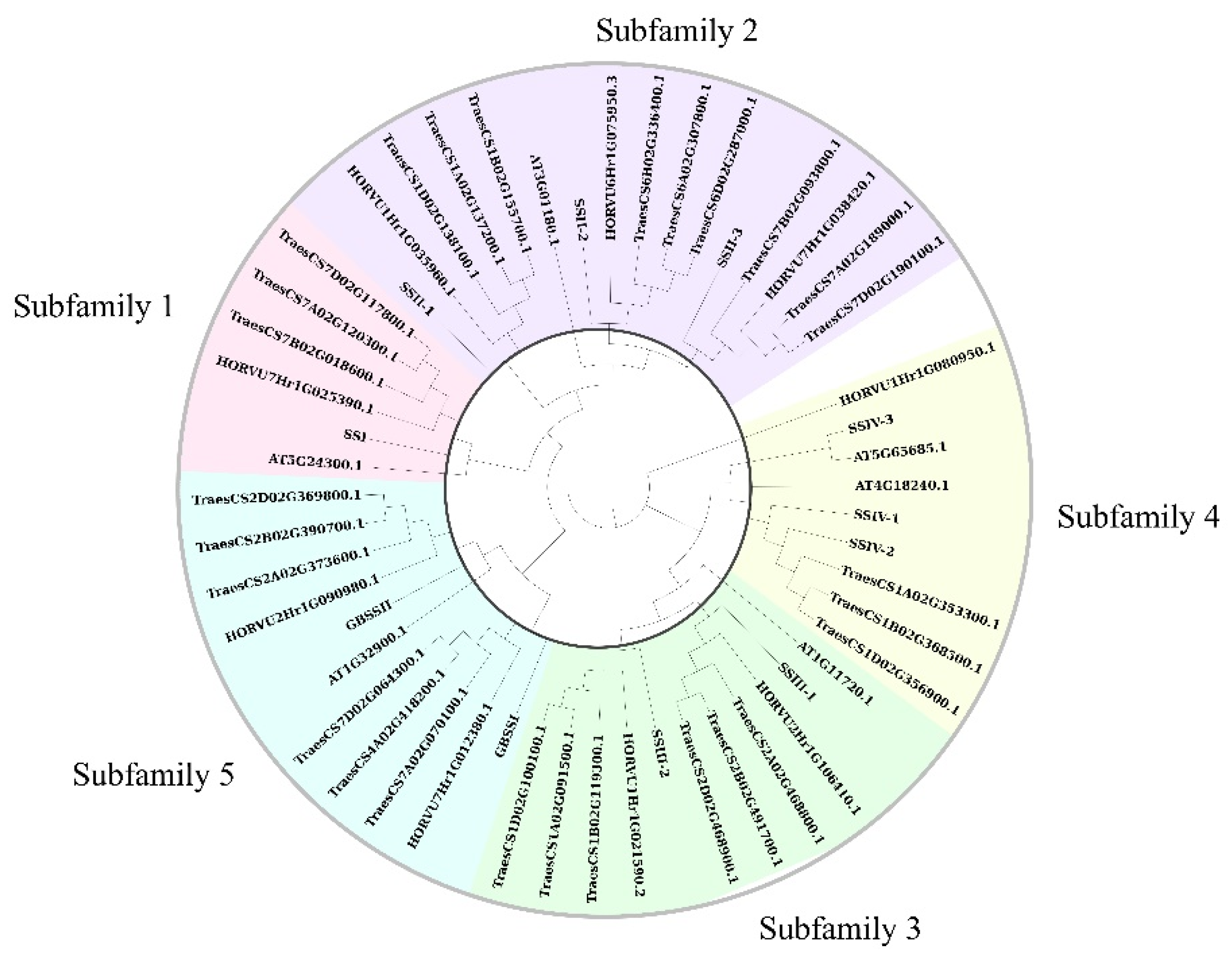
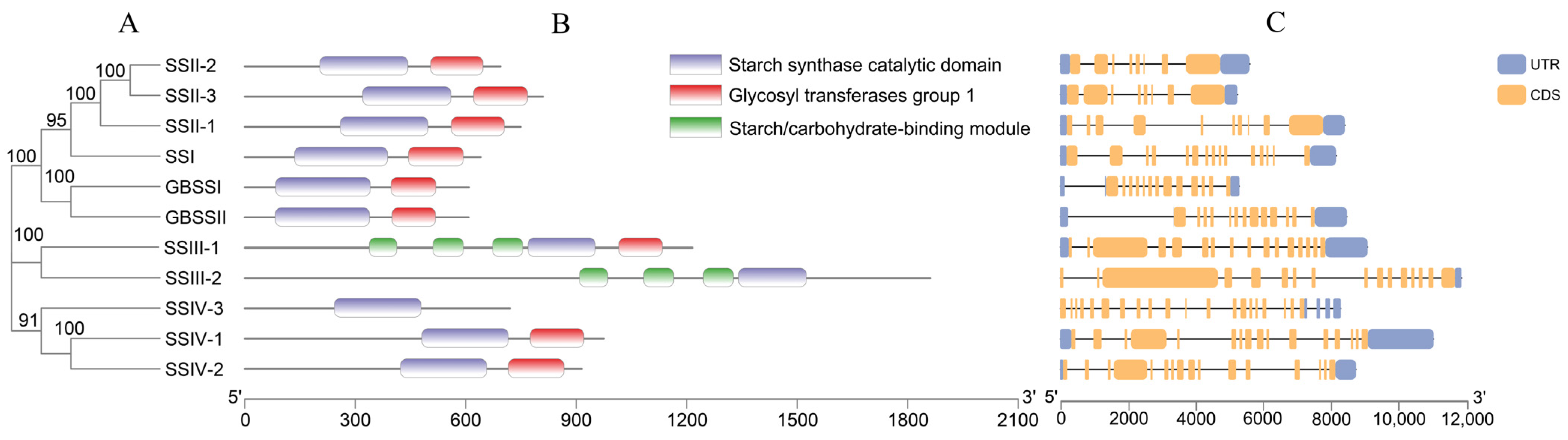
2.2. Analysis of Phylogenetic Relationship and Gene Structure
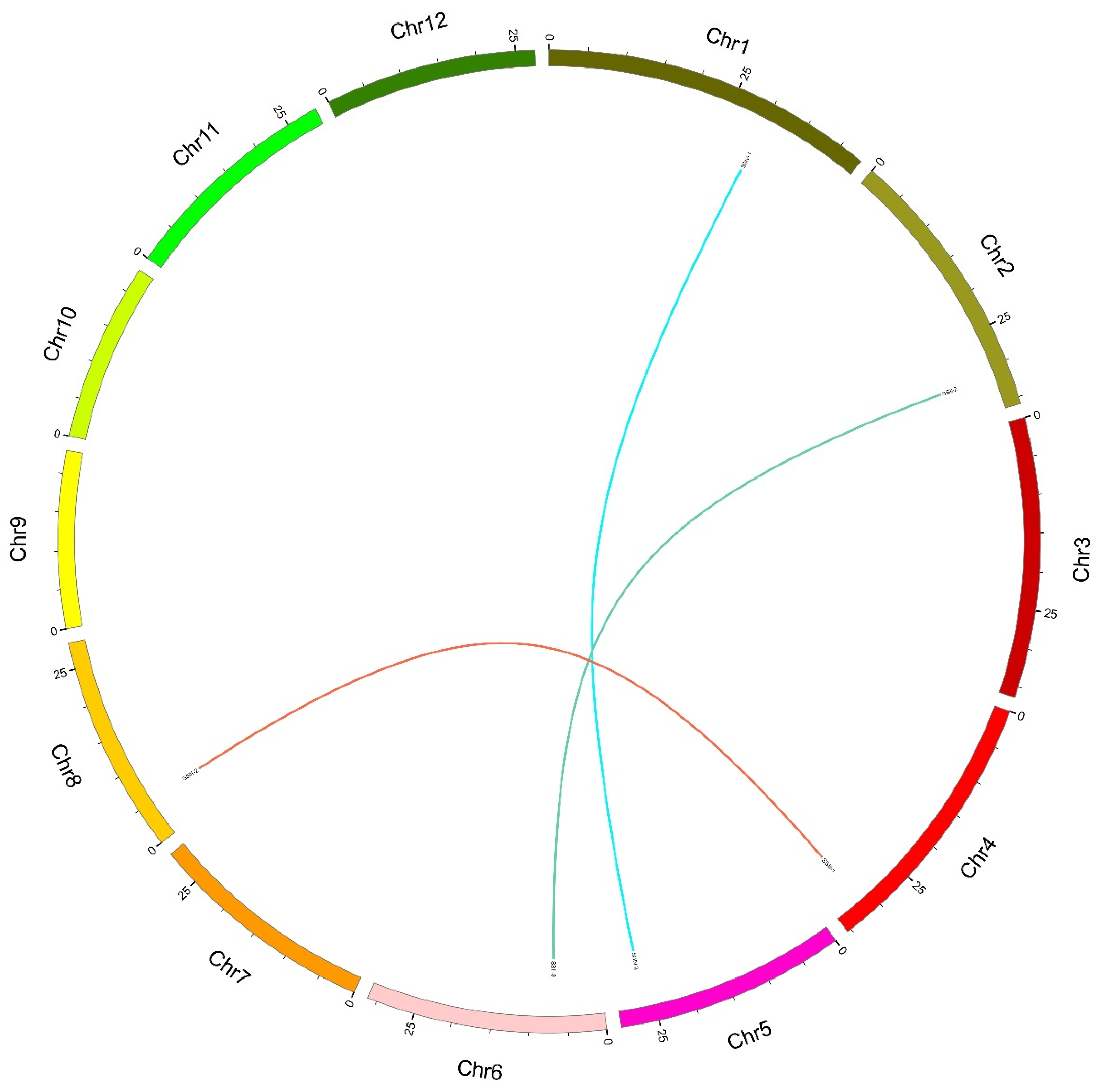
2.3. Synteny Analysis of SS Genes between Rice and Other Species
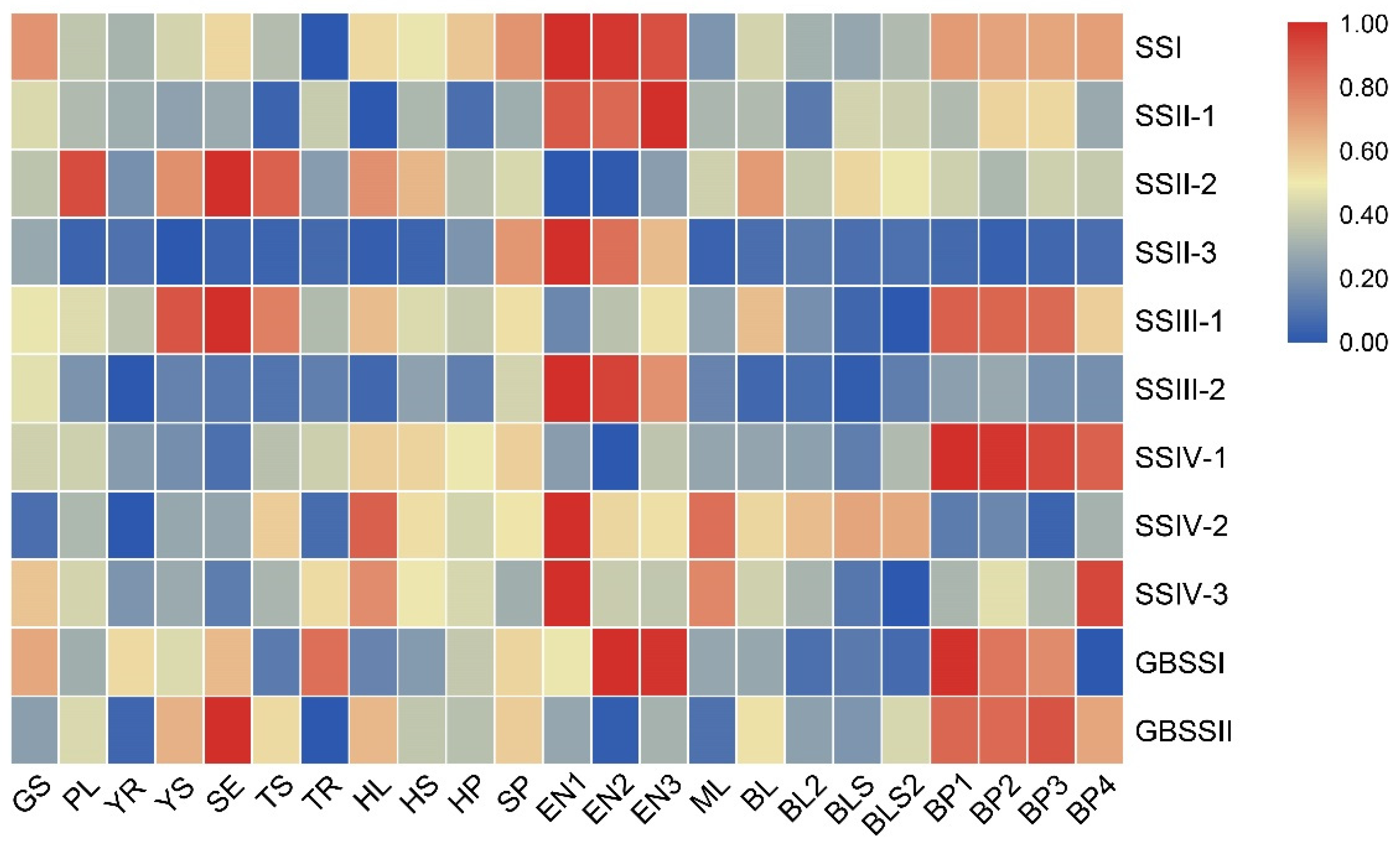
2.4. Comprehensive Analysis of the Expression Profiles of SS Genes
2.5. Prediction of Regulation Network by miRNA-Targeted SS Genes
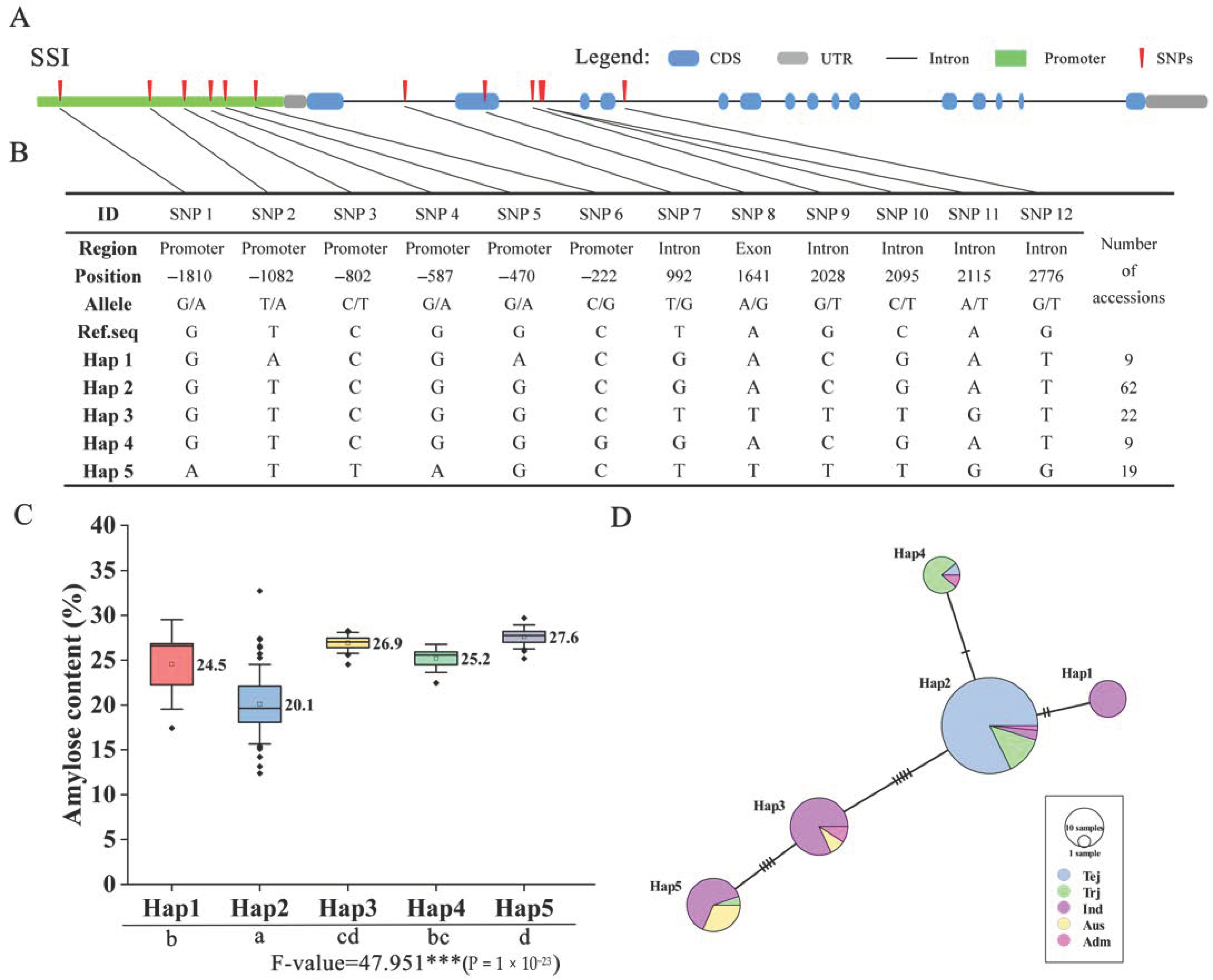
2.6. Haplotype Analysis for OsSS Genes
3. Discussion
4. Materials and Methods
4.1. Identification of SS Genes in the Rice Genome
4.2. Phylogenetic and Structure Analysis of OsSS Genes
4.3. Gene Duplication and Synteny Analysis
4.4. Expression Analysis of SS Genes
4.5. Prediction of Regulation Network for miRNA-Target SS Genes
4.6. Haplotype Analysis for OsSSs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| NJ | neighbor-joining |
| Ks | synonymous |
| Ka | nonsynonymous |
| SS | Starch synthase |
| GBSS | granule-bound starch synthase |
| AC | amylose content |
| Hap | haplotype |
| Tej | Temperate Japonica |
| Trj | Tropical Japonica |
| Ind | Indica |
| Adm | Admixture |
| GA | gibberellins |
References
- Xu, J.; Henry, A.; Sreenivasulu, N. Rice yield formation under high day and night temperatures—A prerequisite to ensure future food security. Plant Cell Environ. 2020, 43, 1595–1608. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, M.; Ouwerkerk, P.B. Molecular and environmental factors determining grain quality in rice. Food Energy Secur. 2012, 1, 111–132. [Google Scholar] [CrossRef]
- Patindol, J.; Wang, Y.J. Fine structures of starches from long-grain rice cultivars with different functionality. Cereal Chem. 2002, 79, 465–469. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Ham, T.-H.; Im, D.-E.; Lar, S.M.; Jang, S.-G.; Lee, J.; Mo, Y.; Jeung, J.-U.; Kim, S.T.; Kwon, S.-W. A New SNP in Rice Gene Encoding Pyruvate Phosphate Dikinase (PPDK) Associated with Floury Endosperm. Genes 2020, 11, 465. [Google Scholar] [CrossRef]
- Wani, A.A.; Singh, P.; Shah, M.A.; Schweiggert-Weisz, U.; Gul, K.; Wani, I.A. Rice starch diversity: Effects on structural, morphological, thermal, and physicochemical properties—A review. Compr. Rev. Food Sci. Food Saf. 2012, 11, 417–436. [Google Scholar] [CrossRef]
- Wickramasinghe, H.A.M.; Noda, T. Physicochemical properties of starches from Sri Lankan rice varieties. Food Sci. Technol. Res. 2008, 14, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Xu, S.; Xu, L.; You, H.; Xiang, X. Effects of Wx and its interaction with SSIII-2 on rice eating and cooking qualities. Front. Plant Sci. 2018, 9, 456. [Google Scholar] [CrossRef]
- Pijning, T.; Vujičić-Žagar, A.; Kralj, S.; Dijkhuizen, L.; Dijkstra, B.W. Structure of the α-1, 6/α-1, 4-specific glucansucrase GTFA from Lactobacillus reuteri 121. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 1448–1454. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yu, L.; Xie, F.; Chen, L. Gelatinization of cornstarch with different amylose/amylopectin content. Carbohydr. Polym. 2006, 65, 357–363. [Google Scholar] [CrossRef]
- Tang, X.-J.; Peng, C.; Zhang, J.; Cai, Y.; You, X.-M.; Kong, F.; Yan, H.-G.; Wang, G.-X.; Wang, L.; Jin, J. ADP-glucose pyrophosphorylase large subunit 2 is essential for storage substance accumulation and subunit interactions in rice endosperm. Plant Sci. 2016, 249, 70–83. [Google Scholar] [CrossRef]
- Li, N.; Zhang, S.; Zhao, Y.; Li, B.; Zhang, J. Over-expression of AGPase genes enhances seed weight and starch content in transgenic maize. Planta 2011, 233, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Terao, T. A comprehensive expression analysis of the starch synthase gene family in rice (Oryza sativa L.). Planta 2004, 220, 9–16. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, L.; Liu, G.; Meng, X.; Jing, Y.; Shu, X.; Kong, X.; Sun, J.; Yu, H.; Smith, S.M. Critical roles of soluble starch synthase SSIIIa and granule-bound starch synthase Waxy in synthesizing resistant starch in rice. Proc. Natl. Acad. Sci. USA 2016, 113, 12844–12849. [Google Scholar] [CrossRef] [Green Version]
- Prathap, V.; Tyagi, A. Correlation between expression and activity of ADP glucose pyrophosphorylase and starch synthase and their role in starch accumulation during grain filling under drought stress in rice. Plant Physiol. Biochem. 2020, 157, 239–243. [Google Scholar]
- Su, Y.; Rao, Y.; Hu, S.; Yang, Y.; Gao, Z.; Zhang, G.; Liu, J.; Hu, J.; Yan, M.; Dong, G. Map-based cloning proves qGC-6, a major QTL for gel consistency of japonica/indica cross, responds by Waxy in rice (Oryza sativa L.). Theor. Appl. Genet. 2011, 123, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Vrinten, P.L.; Nakamura, T. Wheat granule-bound starch synthase I and II are encoded by separate genes that are expressed in different tissues. Plant Physiol. 2000, 122, 255–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, N.; Yoshida, M.; Asakura, N.; Ohdan, T.; Miyao, A.; Hirochika, H.; Nakamura, Y. Function and characterization of starch synthase I using mutants in rice. Plant Physiol. 2006, 140, 1070–1084. [Google Scholar] [CrossRef] [Green Version]
- Ohdan, T.; Francisco, P.B., Jr.; Sawada, T.; Hirose, T.; Terao, T.; Satoh, H.; Nakamura, Y. Expression profiling of genes involved in starch synthesis in sink and source organs of rice. J. Exp. Bot. 2005, 56, 3229–3244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crofts, N.; Sugimoto, K.; Oitome, N.F.; Nakamura, Y.; Fujita, N. Differences in specificity and compensatory functions among three major starch synthases determine the structure of amylopectin in rice endosperm. Plant Mol. Biol. 2017, 94, 399–417. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, J.R.; Springer, F.; Buléon, A.; Müller-Röber, B.; Willmitzer, L.; Kossmann, J. The influence of alterations in ADP-glucose pyrophosphorylase activities on starch structure and composition in potato tubers. Planta 1999, 209, 230–238. [Google Scholar] [CrossRef]
- Pan, G.; Li, Z.; Yin, M.; Huang, S.; Tao, J.; Chen, A.; Li, J.; Tang, H.; Chang, L.; Deng, Y. Genome-wide identification, expression, and sequence analysis of CONSTANS-like gene family in cannabis reveals a potential role in plant flowering time regulation. BMC Plant. Biol. 2021, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Cheng, Z.; Zhang, X.; Guo, X.; Su, N.; Jiang, L.; Mao, L.; Wan, J. Double repression of soluble starch synthase genes SSIIa and SSIIIa in rice (Oryza sativa L.) uncovers interactive effects on the physicochemical properties of starch. Genome 2011, 54, 448–459. [Google Scholar] [CrossRef]
- Ryoo, N.; Yu, C.; Park, C.-S.; Baik, M.-Y.; Park, I.M.; Cho, M.-H.; Bhoo, S.H.; An, G.; Hahn, T.-R.; Jeon, J.-S. Knockout of a starch synthase gene OsSSIIIa/Flo5 causes white-core floury endosperm in rice (Oryza sativa L.). Plant Cell Rep. 2007, 26, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Qian, Q.; Liu, Q.; Yan, M.; Liu, X.; Yan, C.; Liu, G.; Gao, Z.; Tang, S.; Zeng, D. Allelic diversities in rice starch biosynthesis lead to a diverse array of rice eating and cooking qualities. Proc. Natl. Acad. Sci. USA 2009, 106, 21760–21765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.-M.; Abdula, S.E.; Lee, H.-J.; Cho, Y.-C.; Han, L.-Z.; Koh, H.-J.; Cho, Y.-G. Molecular aspect of good eating quality formation in japonica rice. PLoS ONE 2011, 6, e18385. [Google Scholar] [CrossRef] [Green Version]
- Pandey, M.K.; Rani, N.S.; Madhav, M.S.; Sundaram, R.; Varaprasad, G.; Sivaranjani, A.; Bohra, A.; Kumar, G.R.; Kumar, A. Different isoforms of starch-synthesizing enzymes controlling amylose and amylopectin content in rice (Oryza sativa L.). Biotechnol. Adv. 2012, 30, 1697–1706. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Yu, W.; Zhang, C.; Zhu, Y.; Xu, J.; Li, E.; Gilbert, R.G.; Liu, Q. New insights into amylose and amylopectin biosynthesis in rice endosperm. Carbohydr. Polym. 2020, 230, 115656. [Google Scholar] [CrossRef]
- Gonzalez, Z.; Pérez, E. Effect of acetylation on some properties of rice starch. Starch 2002, 54, 148–154. [Google Scholar] [CrossRef]
- Gao, Z.; Zeng, D.; Cui, X.; Zhou, Y.; Yan, M.; Huang, D.; Li, J.; Qian, Q. Map-based cloning of the ALK gene, which controls the gelatinization temperature of rice. Sci. China Ser. C Life Sci. 2003, 46, 661–668. [Google Scholar] [CrossRef]
- He, P.; Li, S.; Qian, Q.; Ma, Y.; Li, J.; Wang, W.; Chen, Y.; Zhu, L. Genetic analysis of rice grain quality. Theor. Appl. Genet. 1999, 98, 502–508. [Google Scholar] [CrossRef]
- Olmstead, J.W.; Sebolt, A.M.; Cabrera, A.; Sooriyapathirana, S.S.; Hammar, S.; Iriarte, G.; Wang, D.; Chen, C.Y.; van der Knaap, E.; Iezzoni, A.F. Construction of an intra-specific sweet cherry (Prunus avium L.) genetic linkage map and synteny analysis with the Prunus reference map. Tree Genet. Genomes 2008, 4, 897–910. [Google Scholar] [CrossRef]
- Hammoudi, V.; Vlachakis, G.; Schranz, M.E.; van den Burg, H.A. Whole-genome duplications followed by tandem duplications drive diversification of the protein modifier SUMO in Angiosperms. New Phytol. 2016, 211, 172–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakatsu, T.; Yamamoto, M.P.; Touno, S.M.; Yasuda, H.; Takaiwa, F. Compensation and interaction between RISBZ1 and RPBF during grain filling in rice. Plant J. 2009, 59, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhu, C.; Pang, J.; Zhang, X.; Yang, C.; Xia, G.; Tian, Y.; He, C. OsLOL1, a C2C2-type zinc finger protein, interacts with OsbZIP58 to promote seed germination through the modulation of gibberellin biosynthesis in Oryza sativa. Plant J. 2014, 80, 1118–1130. [Google Scholar] [CrossRef]
- Archak, S.; Nagaraju, J. Computational prediction of rice (Oryza sativa) miRNA targets. Genom. Proteom. Bioinform. 2007, 5, 196–206. [Google Scholar] [CrossRef] [Green Version]
- Garris, A.J.; McCouch, S.R.; Kresovich, S. Population structure and its effect on haplotype diversity and linkage disequilibrium surrounding the xa5 locus of rice (Oryza sativa L.). Genetics 2003, 165, 759–769. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.; Tosatto, S.C.; Paladin, L.; Raj, S.; Richardson, L.J. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in bipolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar] [PubMed]
- Yu, C.S.; Chen, Y.C.; Lu, C.H.; Hwang, J.K. Prediction of protein subcellular localization. Proteins Struct. Funct. Bioinf. 2006, 64, 643–651. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-h.; Jin, H.; Marler, B.; Guo, H. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, L.; Yang, G.; Yan, J.; Pan, Y.; Nie, X. Genome-wide identification, expression profiles and regulatory network of MAPK cascade gene family in barley. BMC Genom. 2019, 20, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, L.-L.; Xing, F.; Kudrna, D.A.; Yao, W.; Copetti, D.; Mu, T.; Li, W.; Song, J.-M.; Xie, W. Extensive sequence divergence between the reference genomes of two elite indica rice varieties Zhenshan97 and Minghui63. Proc. Natl. Acad. Sci. USA 2016, 113, E5163–E5171. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.-L.; Xue, H.-W. Global analysis of expression profiles of rice receptor-like kinase genes. Mol. Plant 2012, 5, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Xing, M.-Q.; Zhang, Y.-J.; Zhou, S.-R.; Hu, W.-Y.; Wu, X.-T.; Ye, Y.-J.; Wu, X.-X.; Xiao, Y.-P.; Li, X.; Xue, H.-W. Global analysis reveals the crucial roles of DNA methylation during rice seed development. Plant Physiol. 2015, 168, 1417–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, E.; Wang, J.; Zhu, X.; Hao, W.; Wang, L.; Li, Q.; Zhang, L.; He, W.; Lu, B.; Lin, H. Control of rice grain-filling and yield by a gene with a potential signature of domestication. Nat. Genet. 2008, 40, 1370–1374. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Zhao, W.-G.; Chung, J.-W.; Kwon, S.-W.; Lee, J.-H.; Ma, K.-H.; Park, Y.-J. Association analysis of physicochemical traits on eating quality in rice (Oryza sativa L.). Euphytica 2013, 191, 9–21. [Google Scholar] [CrossRef]
- Kim, T.-S.; He, Q.; Kim, K.-W.; Yoon, M.-Y.; Ra, W.-H.; Li, F.P.; Tong, W.; Yu, J.; Oo, W.H.; Choi, B. Genome-wide resequencing of KRICE_CORE reveals their potential for future breeding, as well as functional and evolutionary studies in the post-genomic era. BMC Genom. 2016, 17, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Jin, J.; Guo, A.-Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leigh, J.W.; Bryant, D. popart: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | RAP-ID | MSU-ID | Chr. | Start | End | Gene Length | Cds Length | PI | MW |
|---|---|---|---|---|---|---|---|---|---|
| SSI | Os06g0160700 | LOC_Os06g06560 | Chr6 | 3,079,059 | 3,086,808 | 7750 | 1926 | 5.71 | 59,234.43 |
| SSII-1 | Os10g0437600 | LOC_Os10g30156 | Chr10 | 15,673,128 | 15,681,124 | 7997 | 2250 | 5.39 | 78,907.22 |
| SSII-2 | Os02g0744700 | LOC_Os02g51070 | Chr2 | 31,232,888 | 31,238,210 | 5323 | 2085 | 6.04 | 75,623.36 |
| SSII-3 | Os06g0229800 | LOC_Os06g12450 | Chr6 | 6,748,358 | 6,753,338 | 4981 | 2433 | 5.28 | 86,809.55 |
| SSIII-1 | Os04g0624600 | LOC_Os04g53310 | Chr4 | 31,750,955 | 31,759,581 | 8627 | 3651 | 5.42 | 137,944.3 |
| SSIII-2 | Os08g0191433 | LOC_Os08g09230 | Chr8 | 5,352,105 | 5,363,367 | 11263 | 5586 | 4.96 | 205,368.8 |
| SSIV-1 | Os01g0720600 | LOC_Os01g52250 | Chr1 | 30,030,997 | 30,041,476 | 10480 | 2928 | 5.93 | 100,337.5 |
| SSIV-2 | Os05g0533600 | LOC_Os05g45720 | Chr5 | 26,485,807 | 26,494,112 | 8306 | 2748 | 6.03 | 104,178.5 |
| SSIV-3 | Os02g0807100 | LOC_Os02g56320 | Chr2 | 34,475,930 | 34,483,804 | 7875 | 2163 | 6.18 | 81,317.8 |
| GBSSI | Os06g0133000 | LOC_Os06g04200 | Chr6 | 1,765,622 | 1,770,656 | 5035 | 1830 | 6.1 | 58,473.21 |
| GBSSII | Os07g0412100 | LOC_Os07g22930 | Chr7 | 12,916,277 | 12,924,325 | 8049 | 1827 | 6.26 | 67,354.78 |
| Gene | Comp. Result | Di-pep. Result | part-Comp. Result | chemotype. Result | Neighbor | Extracellular | Plasma Membrane | Cytoplasmic | Cytoskeletal | ER | Golgi | Lysosomal | Mitochondrial | Chloroplast | Peroxisomal | Vacuole | Nuclear | Predicted Location |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SSI | Chloroplast | Lysosomal | Chloroplast | Extracellular | Chloroplast | 0.414 | 0.399 | 0.96 | 0.017 | 0.155 | 0.029 | 0.834 | 0.503 | 1.233 | 0.323 | 0.048 | 0.085 | Chloroplast |
| SSII-1 | Cytoplasmic | Chloroplast | Chloroplast | Chloroplast | Chloroplast | 0.227 | 0.078 | 0.723 | 0.013 | 0.076 | 0.016 | 0.248 | 0.752 | 2.519 | 0.223 | 0.057 | 0.069 | Chloroplast |
| SSII-2 | Cytoplasmic | Chloroplast | Chloroplast | Chloroplast | Chloroplast | 0.053 | 0.015 | 0.539 | 0.01 | 0.071 | 0.011 | 0.018 | 0.441 | 3.628 | 0.087 | 0.081 | 0.046 | Chloroplast |
| SSII-3 | Cytoplasmic | Chloroplast | Chloroplast | Mitochondrial | Chloroplast | 0.135 | 0.199 | 0.87 | 0.032 | 0.119 | 0.056 | 0.037 | 0.468 | 2.5 | 0.186 | 0.099 | 0.3 | Chloroplast |
| SSIII-1 | Cytoplasmic | Nuclear | Cytoplasmic | Nuclear | Cytoplasmic | 0.079 | 0.225 | 2.15 | 0.043 | 0.074 | 0.046 | 0.009 | 0.228 | 0.452 | 0.076 | 0.018 | 1.6 | Cytoplasmic |
| SSIII-2 | Nuclear | Nuclear | Cytoplasmic | Nuclear | Cytoplasmic | 0.149 | 0.523 | 1.93 | 0.042 | 0.142 | 0.082 | 0.01 | 0.1 | 0.078 | 0.056 | 0.024 | 1.861 | Cytoplasmic |
| SSIV-1 | Cytoplasmic | Cytoplasmic | Cytoplasmic | Nuclear | Cytoplasmic | 0.144 | 0.086 | 3.034 | 0.026 | 0.053 | 0.116 | 0.011 | 0.326 | 0.274 | 0.097 | 0.012 | 0.82 | Cytoplasmic |
| SSIV-2 | Cytoplasmic | Cytoplasmic | Nuclear | Nuclear | Cytoplasmic | 0.11 | 0.113 | 2.109 | 0.066 | 0.04 | 0.141 | 0.013 | 0.517 | 0.278 | 0.116 | 0.009 | 1.487 | Cytoplasmic |
| SSIV-3 | Nuclear | Cytoplasmic | Cytoplasmic | Cytoplasmic | Cytoplasmic | 0.342 | 0.288 | 2.117 | 0.013 | 0.046 | 0.036 | 0.07 | 0.567 | 0.229 | 0.142 | 0.014 | 1.137 | Cytoplasmic |
| GBSSI | Chloroplast | Chloroplast | Chloroplast | Chloroplast | Chloroplast | 0.022 | 0.039 | 0.188 | 0.004 | 0.006 | 0.003 | 0.012 | 0.558 | 4.01 | 0.102 | 0.023 | 0.032 | Chloroplast |
| GBSSII | Cytoplasmic | Chloroplast | Chloroplast | Chloroplast | Chloroplast | 0.076 | 0.177 | 1.444 | 0.01 | 0.042 | 0.021 | 0.096 | 0.25 | 2.292 | 0.519 | 0.029 | 0.045 | Chloroplast |
| Homologous Genes in Rice Genome | Homologous Genes in Rice Genome | Ka | Ks | Ka/Ks | S | N | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Gene ID | Chr. | Start | End | Gene | Gene ID | Chr. | Start | End | |||||
| SSIV-1 | LOC_Os01g52250 | Chr1 | 30030997 | 30,041,476 | SSIV-2 | LOC_Os05g45720 | Chr5 | 26,485,807 | 26,494,112 | 0.19382 | 0.78169 | 0.24795 | 295.917 | 358.083 |
| SSII-2 | LOC_Os02g51070 | Chr2 | 31232888 | 31,238,210 | SSII-3 | LOC_Os06g12450 | Chr6 | 6,748,358 | 6,753,338 | 0.23731 | 0.71259 | 0.33303 | 236.083 | 317.917 |
| SSIII-1 | LOC_Os04g53310 | Chr4 | 31750955 | 31,759,581 | SSIII-2 | LOC_Os08g09230 | Chr8 | 5,352,105 | 5,363,367 | 0.23734 | 0.86354 | 0.27484 | 409.083 | 568.917 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Jang, S.-G.; Lar, S.M.; Lee, A.-R.; Cao, F.-Y.; Seo, J.; Kwon, S.-W. Genome-Wide Identification and Genetic Variations of the Starch Synthase Gene Family in Rice. Plants 2021, 10, 1154. https://doi.org/10.3390/plants10061154
Zhang H, Jang S-G, Lar SM, Lee A-R, Cao F-Y, Seo J, Kwon S-W. Genome-Wide Identification and Genetic Variations of the Starch Synthase Gene Family in Rice. Plants. 2021; 10(6):1154. https://doi.org/10.3390/plants10061154
Chicago/Turabian StyleZhang, Hongjia, Seong-Gyu Jang, San Mar Lar, Ah-Rim Lee, Fang-Yuan Cao, Jeonghwan Seo, and Soon-Wook Kwon. 2021. "Genome-Wide Identification and Genetic Variations of the Starch Synthase Gene Family in Rice" Plants 10, no. 6: 1154. https://doi.org/10.3390/plants10061154
APA StyleZhang, H., Jang, S.-G., Lar, S. M., Lee, A.-R., Cao, F.-Y., Seo, J., & Kwon, S.-W. (2021). Genome-Wide Identification and Genetic Variations of the Starch Synthase Gene Family in Rice. Plants, 10(6), 1154. https://doi.org/10.3390/plants10061154