
Comparative Analysis of Chloroplast Genomes of Four Medicinal Capparaceae Species: Genome Structures, Phylogenetic Relationships and Adaptive Evolution

Abstract
:1. Introduction
2. Results
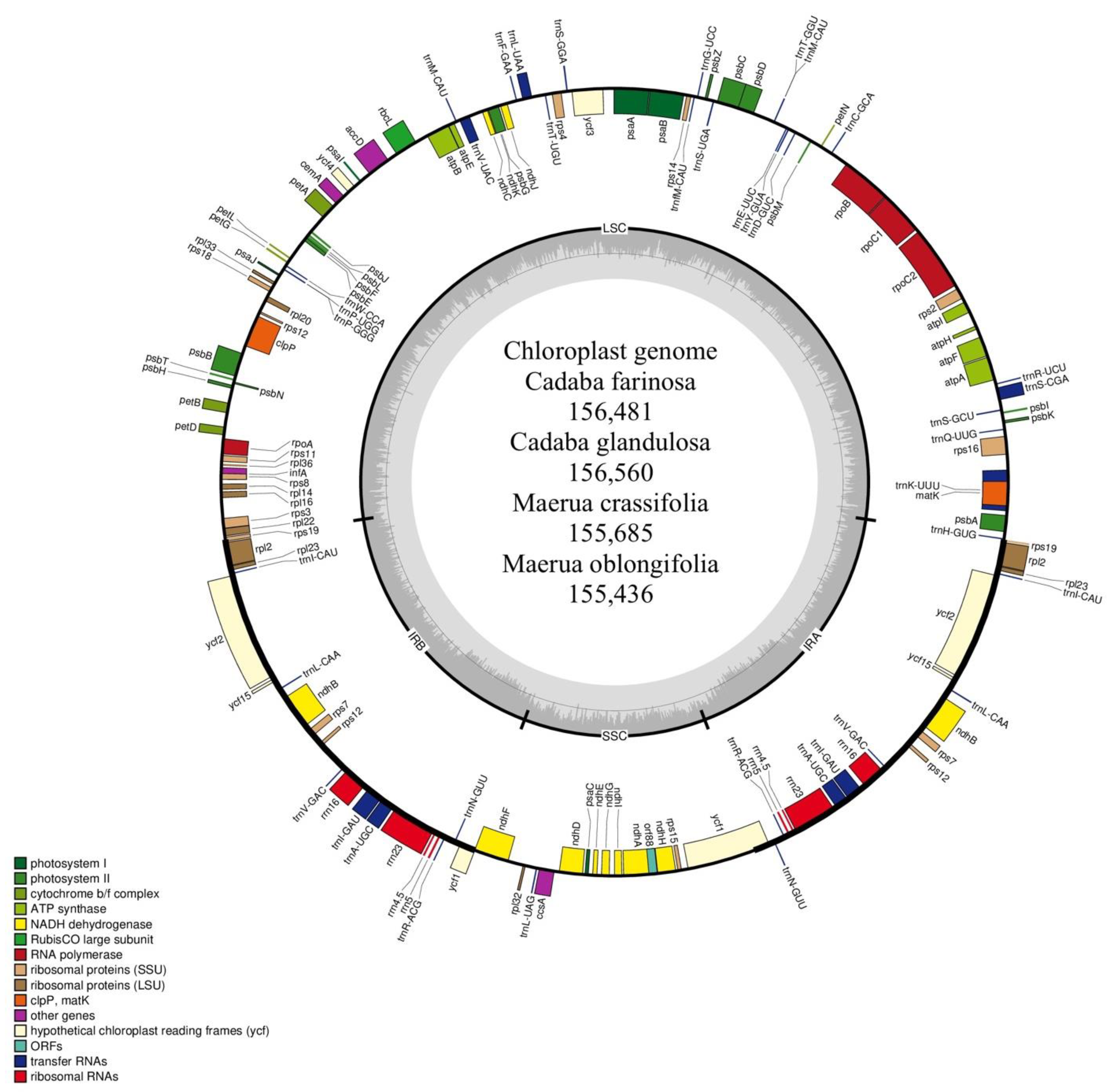
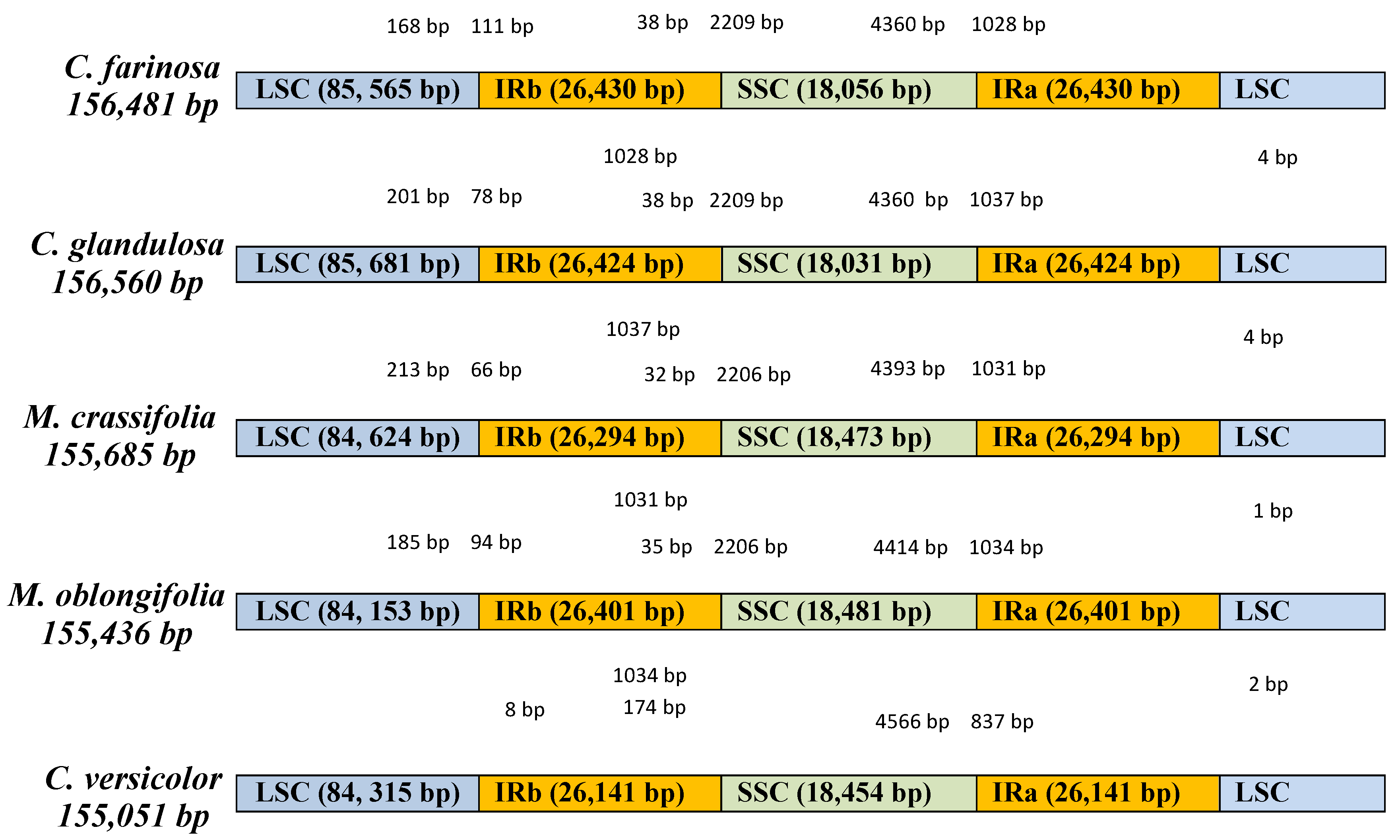
2.1. Characteristics of Four Chloroplast Genomes
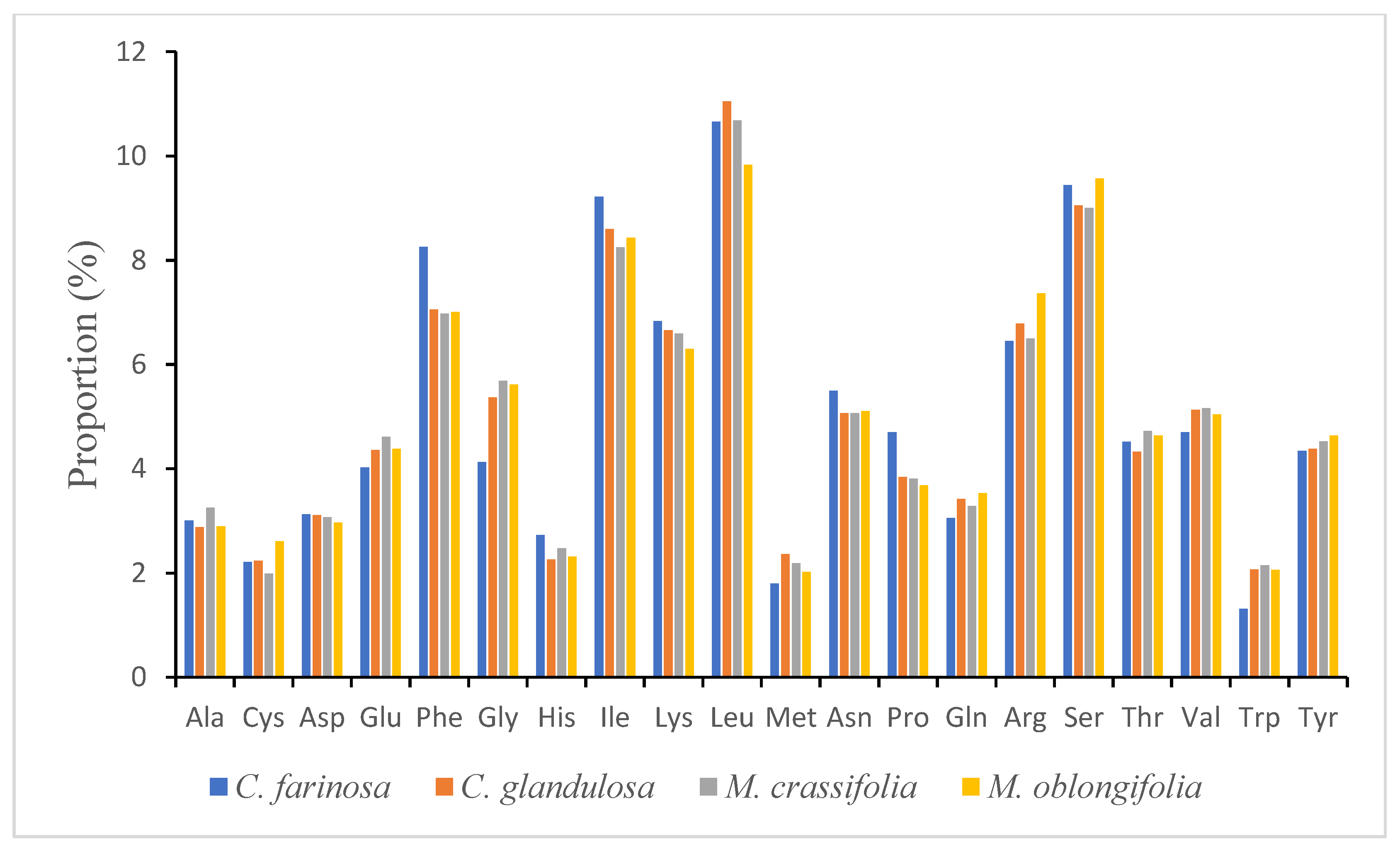
2.2. Codon Usage
2.3. RNA Editing Sites
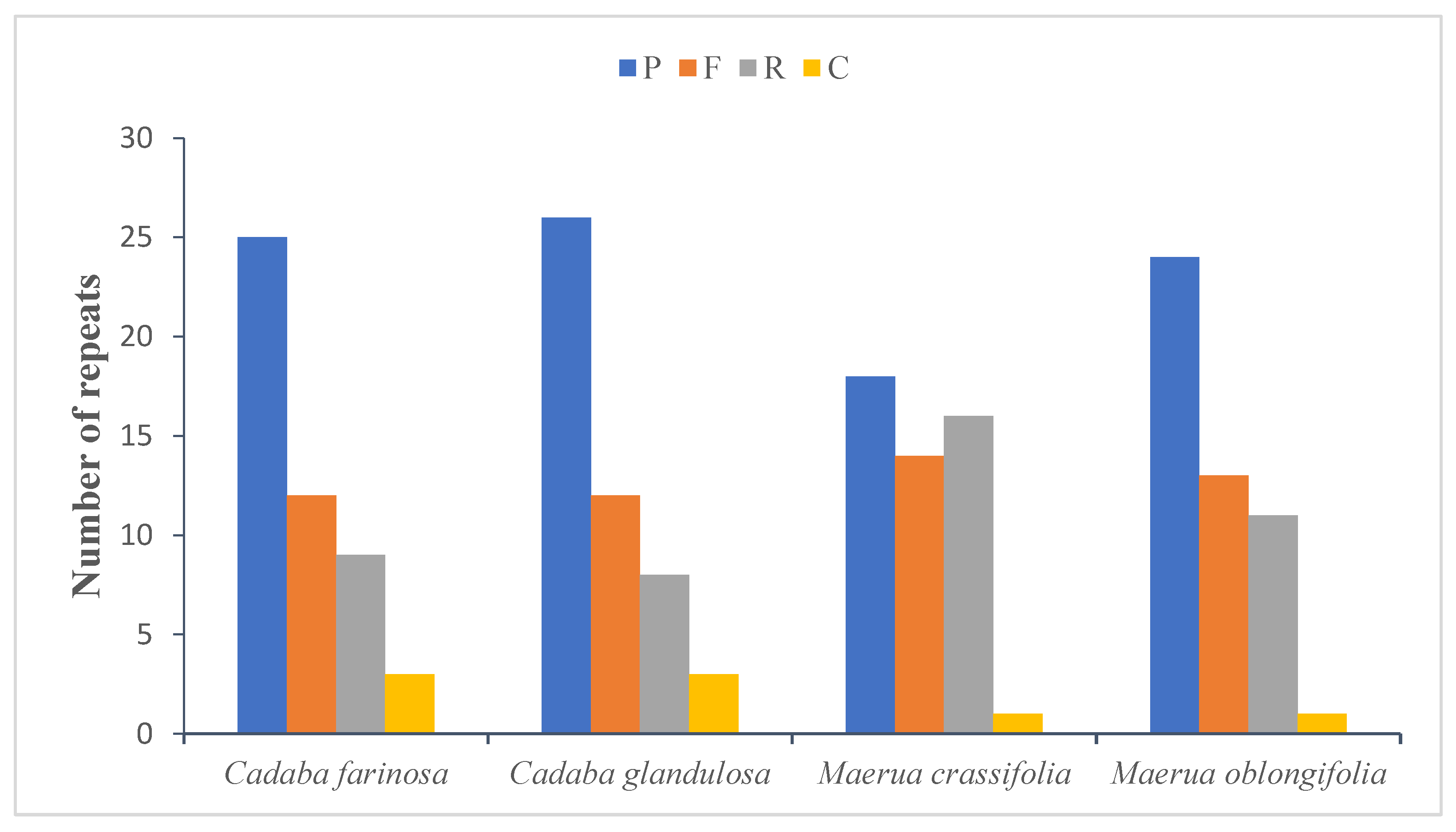
2.4. Repeat Analysis
2.4.1. Long Repeats
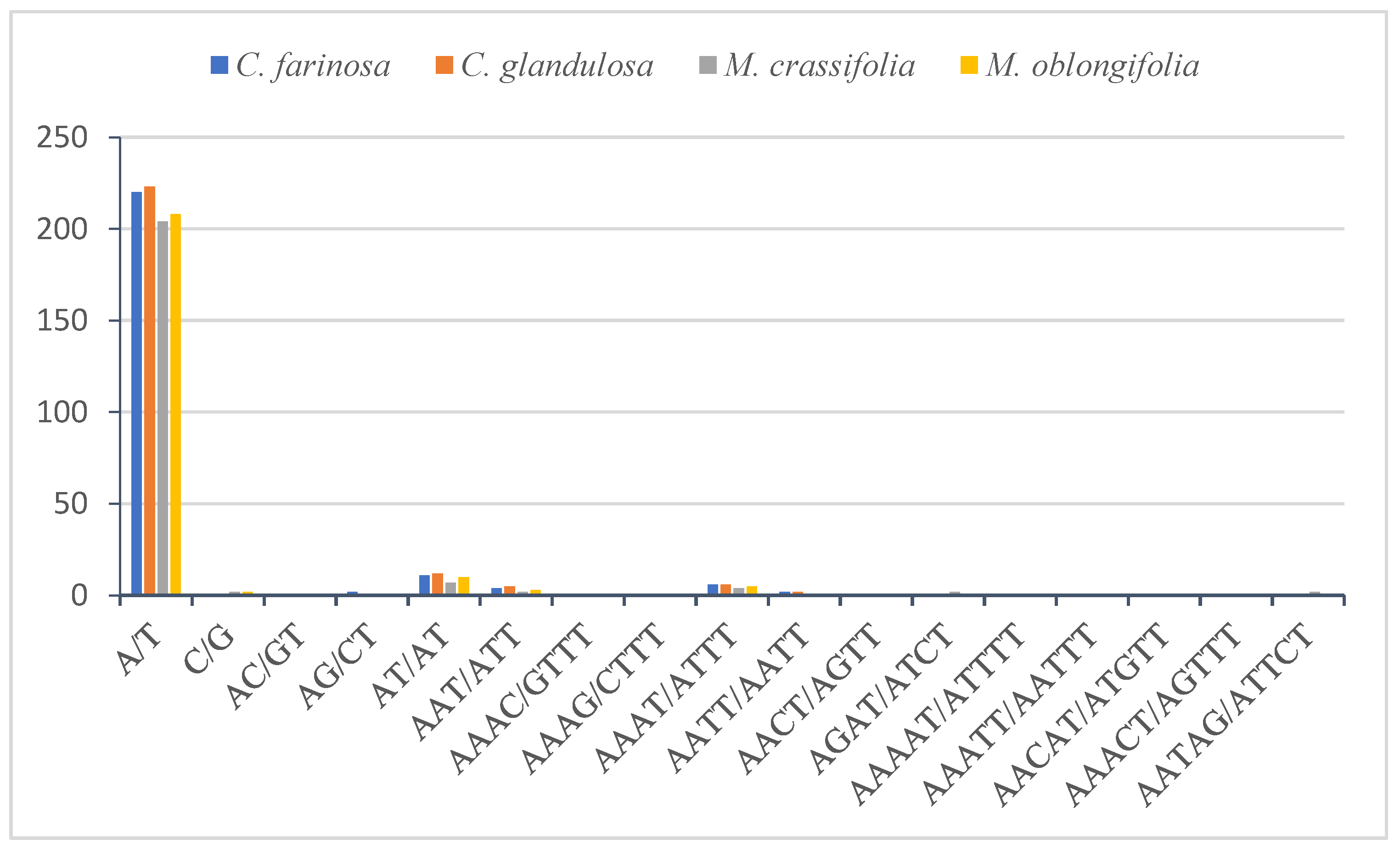
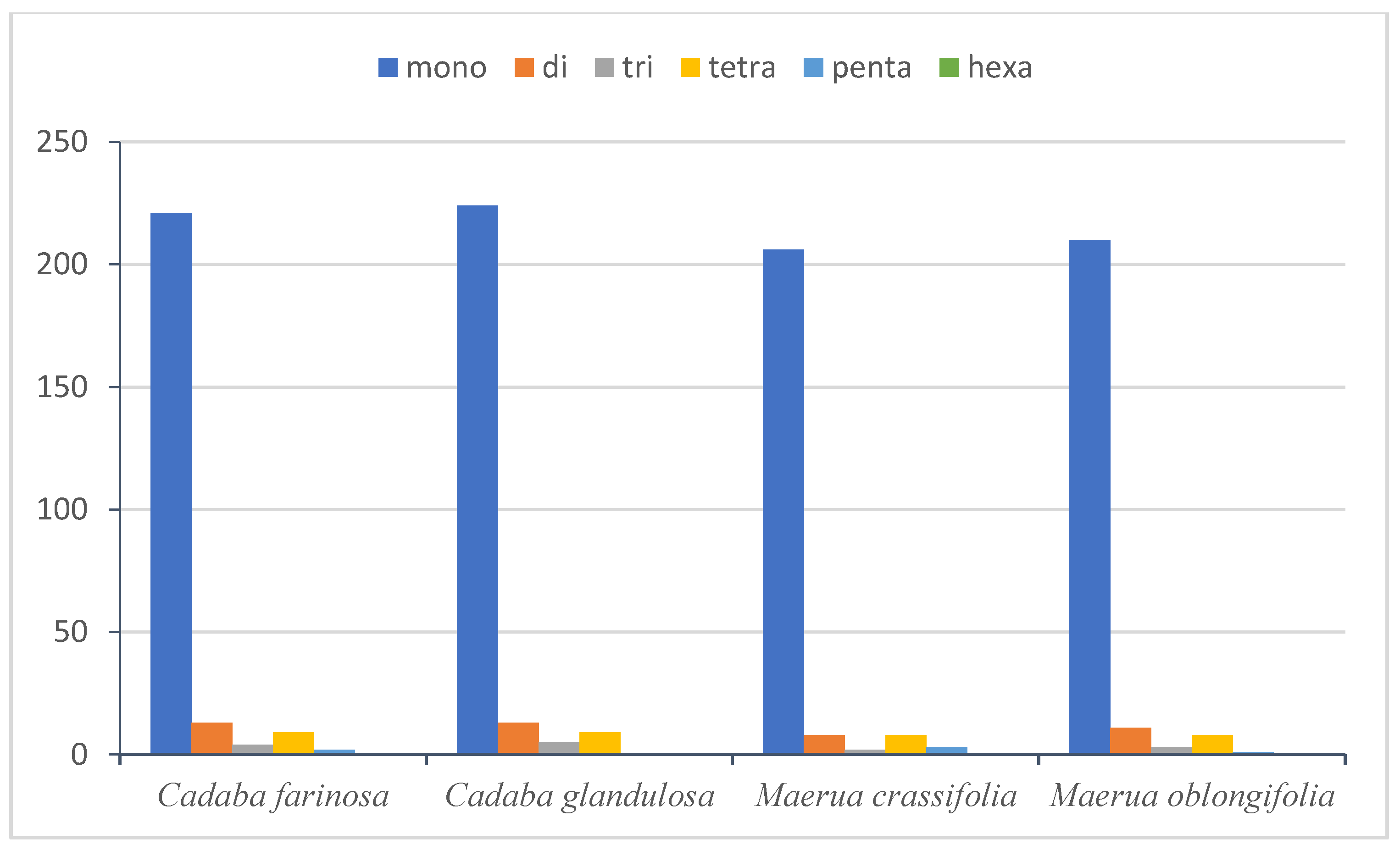
2.4.2. Simple Sequence Repeats (SSRs)
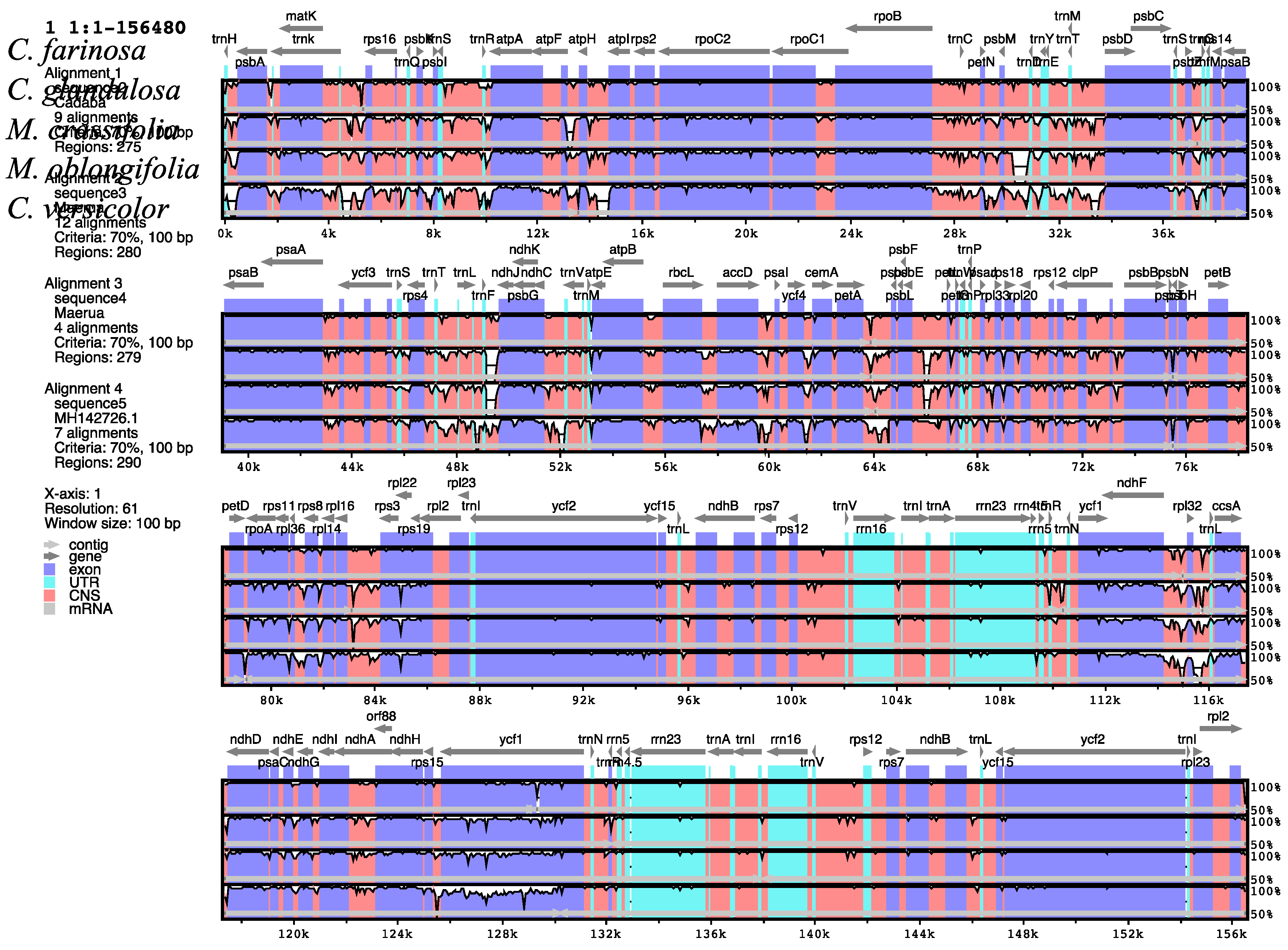
2.5. Comparative Analysis of the Capparaceae Species Cp Genome
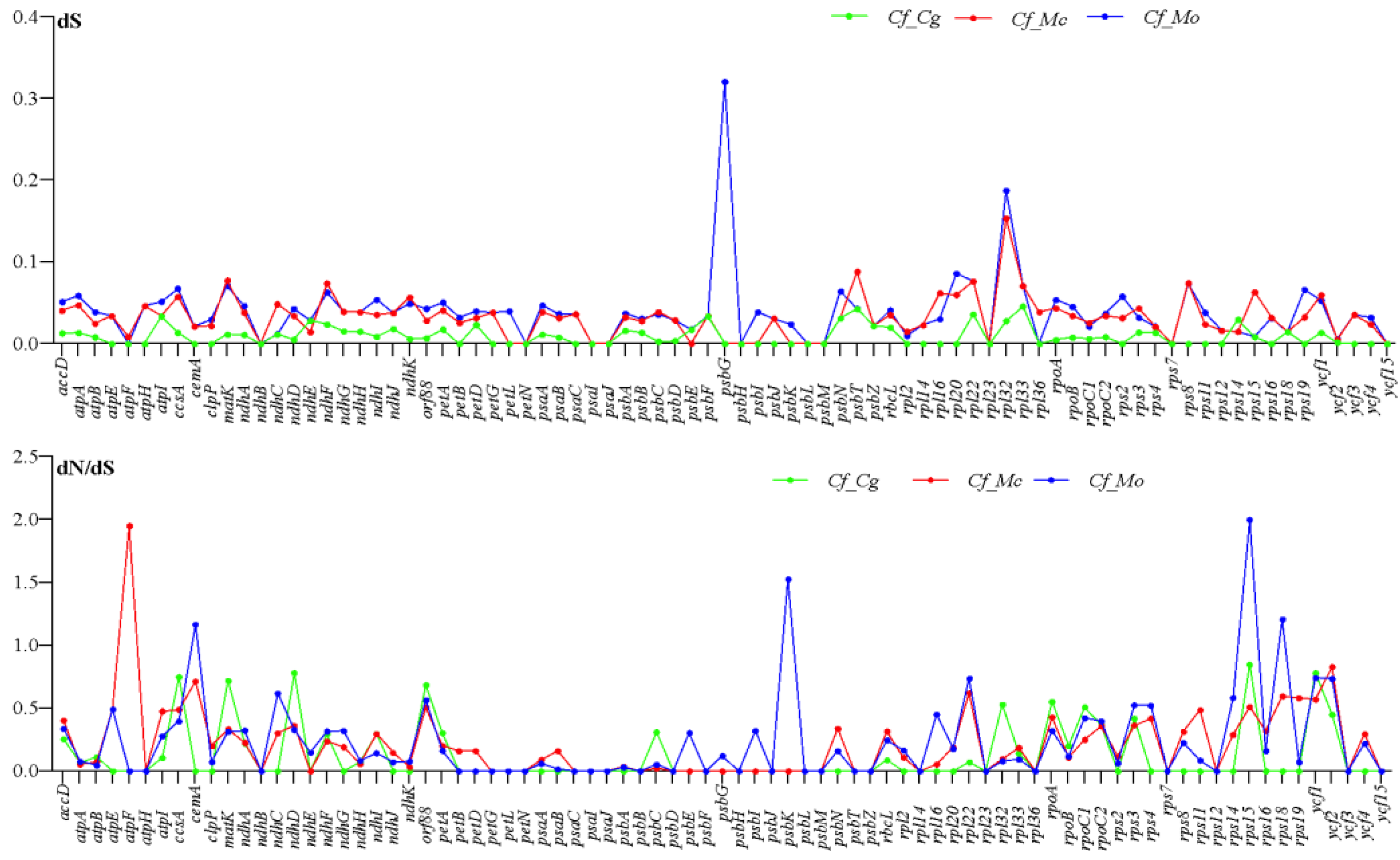
2.6. Divergence of Protein-Coding Gene Sequence
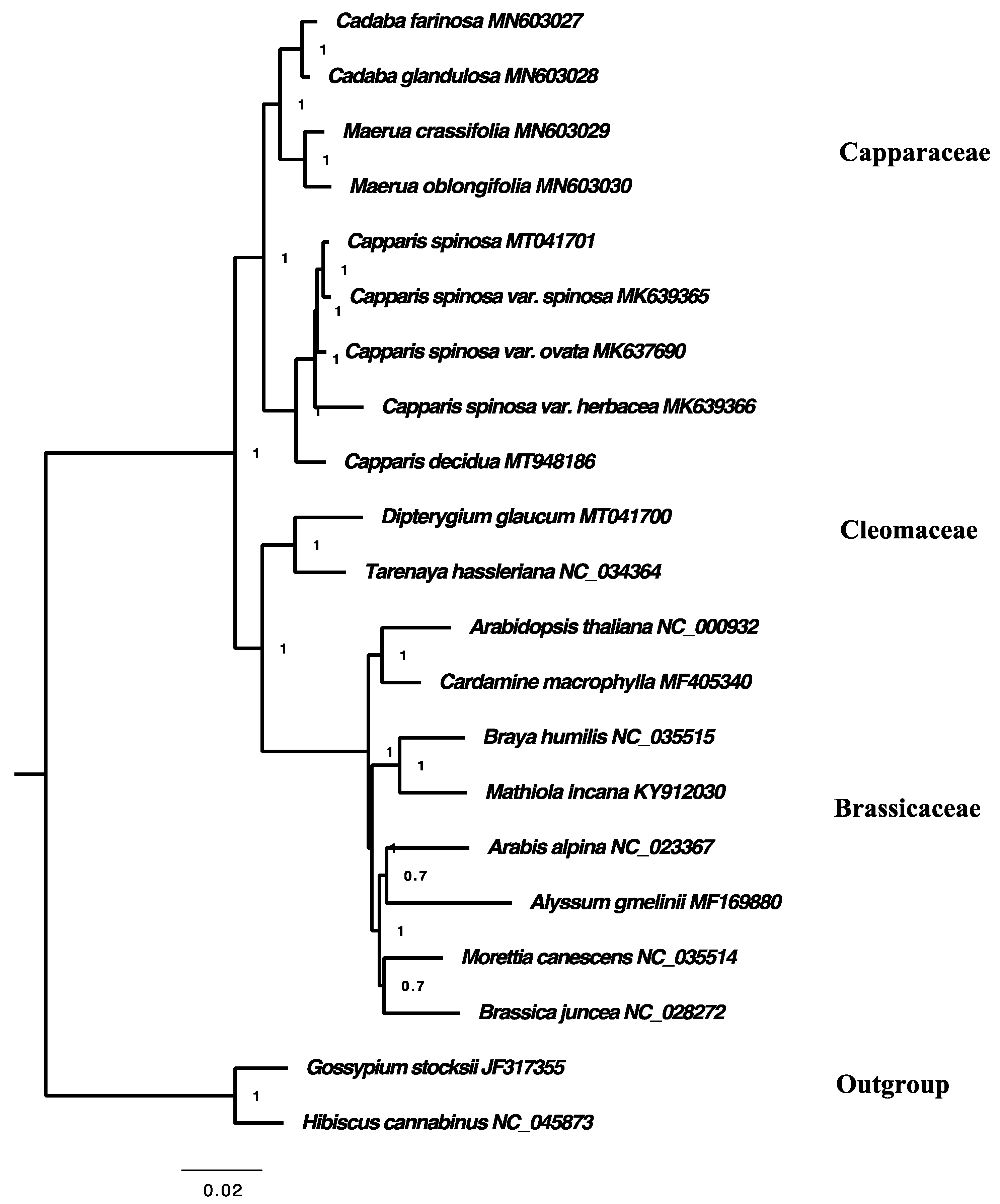
2.7. Phylogenetic Analysis
3. Materials and Methods
3.1. Plant Material and DNA Extraction
3.2. Library Construction, Sequencing and Assembly
3.3. Gene Annotation
3.4. Sequence Analysis
3.5. Genome Comparison
3.6. Characterization of Substitution Rate
3.7. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cronquist, A.; Takhtadzhiáǹ, A.L. An Integrated System of Classification of Flowering Plants; Columbia University Press: New York, NY, USA, 1981; p. 337. [Google Scholar]
- Mabberley, D.J. The Plant-Book: A Portable Dictionary of the Higher Plants; Cambridge University Press: Cambridge, UK, 1997. [Google Scholar]
- Heywood, V.H.; Brummitt, R.K.; Culham, A.; Seberg, O. Flowering Plant Families of the World. Royal Botanic Gardens, Kew. Int. J. Pharm. 2007, 11, 874–887. [Google Scholar]
- Gibbs, R.D. Chemotaxonomy of Flowering Plants; McGill-Queens University Press: London, UK, 1974; p. 761. [Google Scholar]
- Rahman, M.; Mossa, J.S.; Al-Said, M.S.; Al-Yahya, M.A. Medicinal plant diversity in the flora of Saudi Arabia 1: A report on seven plant families. Fitoterapia 2004, 75, 149–161. [Google Scholar] [CrossRef]
- Telrandhe, U.B.; Uplanchiwar, V. Phyto-Pharmacological Perspective of Cadaba farinosa forsk. Am. J. Phytomed. Clin. Ther. 2013, 1, 11–22. [Google Scholar]
- Agize, M.; Demissew, S.; Asfaw, Z. Ethnobotany of Medicinal Plants in Loma and Gena Bosa Districts (Woredas) of Dawro Zone, Southern Ethiopia. Topclass J. Herb. Med. 2013, 2, 194–212. [Google Scholar]
- Laxmichand, B.H.; Modi, N.R. A Comprehensive Review on Maerua Oblongifolia (Forsk.) A. Rich. Int. J. Res. Advent Technol. 2019, 4, 2321–9637. [Google Scholar]
- Burkill, H.M.; Dalziel, J.M.; Hutchinson, J. Entry for Maerua crassifolia Forssk. [family CAPPARACEAE]. In Useful Plants of West Tropical Africa, 2nd ed.; Royal Botanic Gardens: Kew, UK, 1985. [Google Scholar]
- Akuodor, G.C.; Ibrahim, J.A.; Akpan, J.L.; Okorie, A.U.; Ezeokpo, B.C. Phytochemical and Anti-diarrhoeal Properties of Methanolic Leaf Extract of Maerua crassifolia Forssk. Eur. J. Med. Plants 2014, 4, 1223–1231. [Google Scholar] [CrossRef]
- Ckilaka, K.; Akuodor, G.; Akpan, J.; Ogiji, E.; Eze, C.; Ezeokpo, B.C. Antibacterial and antioxidant activities of methanolic leaf extract of Maerua crassifolia. J. Appl. Pharm. Sci. 2015, 5, 147–150. [Google Scholar] [CrossRef] [Green Version]
- Bentham, G.; Hooker, J.D. Genera Plantarum ad Eemplaria Imprimis in Herbariis Kewensibus Servata Definite; Reeve and Co.: London, UK, 1883. [Google Scholar]
- Engler, A.; Prantl, K. Die Naturlichen Pflanzenfamilien; Wilhelm Engelmann: Leipzig, Germany, 1915. [Google Scholar]
- Hutchinson, J. The Genera of Flowering Plants; Clarendon Press: Oxford, UK, 1967. [Google Scholar]
- Takhtajan, A. Origins of Angiospermous Plants; American Institute of Biological Science: Washington, DC, USA, 1954. [Google Scholar]
- Nee, M. Diversity and Classification of Flowering Plants A. Takhtajan. Brittonia 1998, 50, 191–192. [Google Scholar] [CrossRef]
- APG I (Angiosperm Phylogeny Group). An ordinal classification for the families of flowering plants. Ann. Mo. Bot. Gard. 1998, 85, 531–553. [Google Scholar] [CrossRef] [Green Version]
- APG II (Angiosperm Phylogeny Group). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants. Bot. J. Linn. Soc. 2003, 141, 399–436. [Google Scholar] [CrossRef] [Green Version]
- APG III (Angiosperm Phylogeny Group). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants. Bot. J. Linn. Soc. 2009, 161, 105–121. [Google Scholar]
- APG IV (Angiosperm Phylogeny Group). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar]
- Iltis, H.H. Studies in the Capparidaceae. III. Evolution and Phylogeny of the Western North American Cleomoideae. Ann. Mo. Bot. Gard. 1957, 44, 77. [Google Scholar] [CrossRef]
- Al-Shehbaz, I.A. The biosystematics of the genus Thelypodium (Cruciferae). Contrib. Gray Herb. Harv. Univ. 1973, 204, 3–148. [Google Scholar]
- Al-Shehbaz, I.A. The tribes of Cruciferae (Brassicaceae) in the southeastern United States. J. Arn. Arbor. 1984, 65, 343–373. [Google Scholar]
- Dalhgren, R. A System of classification of the angiosperms to be used to demonstrate the distribution of characters. Bot. Not. 1975, 128, 119–147. [Google Scholar]
- Takhtajan, A.L. Outline of the classification of flowering plants (magnoliophyta). Bot. Rev. 1980, 46, 225–359. [Google Scholar] [CrossRef]
- Hauser, L.A.; Crovello, T.J. Numerical Analysis of Generic Relationships in Thelypodieae (Brassicaceae). Syst. Bot. 1982, 7, 249. [Google Scholar] [CrossRef]
- Rodman, J.; Price, R.A.; Karol, K.; Conti, E.; Systma, K.J.; Palmer, J.D. Nucleotide Sequences of the rbcL Gene Indicate Monophyly of Mustard Oil Plants. Ann. Mo. Bot. Gard. 1993, 80, 686. [Google Scholar] [CrossRef]
- Rodman, J.E.; Karol, K.G.; Price, R.A.; Sytsma, K.J. Molecules, Morphology, and Dahlgren’s Expanded Order Capparales. Syst. Bot. 1996, 21, 289. [Google Scholar] [CrossRef]
- Rodman, J.E.; Soltis, P.S.; Soltis, D.E.; Sytsma, K.J.; Karol, K.G. Parallel evolution of glucosinolate biosynthesis inferred from congruent nuclear and plastid gene phylogenies. Am. J. Bot. 1998, 85, 997–1006. [Google Scholar] [CrossRef] [Green Version]
- Schmid, R.; Rollins, R.C. The Cruciferae of Continental North America: Systematics of the Mustard Family from the Arctic to Panama. TAXON 1994, 43, 153. [Google Scholar] [CrossRef]
- Judd, W.S.; Sanders, R.W.; Donoghue, M.J. Angiosperm family pairs: Preliminary phylogenetic analyses. Harv. Pap. Bot. 1994, 5, 1–51. [Google Scholar]
- Judd, W.S.; Campbell, C.S.; Kellogg, E.A.; Stevens, P.F.; Donoghue, M.J. Plant Systematics: A Phylogenetic Approach, 3rd ed.; Sinauer Associates: Sunderland, MA, USA, 2007. [Google Scholar]
- Hall, J.; Sytsma, K.J.; Iltis, H.H. Phylogeny of Capparaceae and Brassicaceae based on chloroplast sequence data. Am. J. Bot. 2002, 89, 1826–1842. [Google Scholar] [CrossRef] [PubMed]
- Airy Shaw, H.K. Diagnoses of new families, new names, etc., for the seventh edition of Willis’s ‘Dictionary’. Kew Bull. 1965, 18, 249–273. [Google Scholar] [CrossRef]
- Pax, F.; Hoffmann, K. Capparidaceae. In Die Naturlichen Pflanzenfamilien, 2nd ed.; Engler, A., Prantl, K., Eds.; Wilhelm Engelmann: Leipzig, Germany, 1936; Volume 17, pp. 146–223. [Google Scholar]
- Hedge, I.C.; Kjaer, A.; Malver, O. Dipterygium—Cruciferaeor Capparaceae? R. Bot. Gard. Edinb. 1980, 38, 247–250. [Google Scholar]
- Doweld, A.; Reveal, J.L. New suprageneric names for vascular plants. Phytologia 2008, 90, 416–417. [Google Scholar]
- Grevich, J.J.; Daniell, H. Chloroplast Genetic Engineering: Recent Advances and Future Perspectives. Crit. Rev. Plant Sci. 2005, 24, 83–107. [Google Scholar] [CrossRef]
- Neuhaus, H.; Emes, M. Nonphotosynthetic metabolism in plastids. Ann. Rev. Plant Biol. 2000, 51, 111–140. [Google Scholar] [CrossRef] [PubMed]
- WickeGerald, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Raubeson, L.; Jansen, R.; Henry, R.J. (Eds.) Chloroplast genomes of plants. In Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants; CABI: London, UK, 2005; pp. 45–68. [Google Scholar]
- Greiner, S.; Sobanski, J.; Bock, R. Why are most organelle genomes transmitted maternally? BioEssays 2015, 37, 80–94. [Google Scholar] [CrossRef] [PubMed]
- GuisingerTimothy, M.M.; Chumley, T.W.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Implications of the Plastid Genome Sequence of Typha (Typhaceae, Poales) for Understanding Genome Evolution in Poaceae. J. Mol. Evol. 2010, 70, 149–166. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.-B.; Tang, M.; Li, H.-T.; Zhang, Z.-R.; Li, D.-Z. Complete chloroplast genome of the genus Cymbidium: Lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 2013, 13, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzahrani, D.; Albokhari, E.; Yaradua, S.; Abba, A. The complete plastome sequence for the medicinal species Capparis spinosa L. (Capparaceae). Gene Rep. 2021, 23, 101059. [Google Scholar] [CrossRef]
- Maurya, S.; Darshetkar, A.M.; Datar, M.N.; Tamhankar, S.; Li, P.; Choudhary, R.K. Plastome data provide insights into intra and interspecific diversity and ndh gene loss in Capparis (Capparaceae). Phytotaxa 2020, 432, 206–220. [Google Scholar] [CrossRef]
- Chen, H.; Shao, J.; Zhang, H.; Jiang, M.; Huang, L.; Zhang, Z.; Yang, D.; He, M.; Ronaghi, M.; Luo, X.; et al. Sequencing and Analysis of Strobilanthes cusia (Nees) Kuntze Chloroplast Genome Revealed the Rare Simultaneous Contraction and Expansion of the Inverted Repeat Region in Angiosperm. Front. Plant Sci. 2018, 9, 324. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Lin, H.C.; Lin, I.P.; Chow, T.Y.; Chen, H.H.; Chen, W.H.; Cheng, C.H.; Lin, C.U.; Liu, S.M.; Chang, C.C.; et al. The chloroplast genome of Phalaenopsis aphrodite (Orchidaceae): Comparative analysis of evolutionary rate with that of grasses and its phylogenetic implications. Mol. Biol. Evol. 2006, 23, 279–291. [Google Scholar] [CrossRef] [Green Version]
- Raman, G.; Park, S. The complete chloroplast genome sequence of Ampelopsis: Gene organization, comparative analysis, and phylogenetic relationships to other angiosperms. Front. Plant Sci. 2016, 341, 7. [Google Scholar] [CrossRef] [Green Version]
- Park, I.; Kim, W.J.; Yeo, S.-M.; Choi, G.; Kang, Y.-M.; Piao, R.; Moon, B.C. The Complete Chloroplast Genome Sequences of Fritillaria ussuriensis Maxim. And Fritillaria cirrhosa D. Don, and Comparative Analysis with Other Fritillaria Species. Molecules 2017, 22, 982. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Lin, F.; Huang, P.; Guo, W.; Zheng, Y. Complete Chloroplast Genome Sequence of Decaisnea insignis: Genome Organization, Genomic Resources and Comparative Analysis. Sci. Rep. 2017, 7, 10073. [Google Scholar] [CrossRef]
- Liu, X.; Li, Y.; Yang, H.; Zhou, B. Chloroplast Genome of the Folk Medicine and Vegetable Plant Talinum paniculatum (Jacq.) Gaertn.: Gene Organization, Comparative and Phylogenetic Analysis. Molecules 2018, 23, 857. [Google Scholar] [CrossRef] [Green Version]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef] [PubMed]
- Bundschuh, R.; Altmüller, J.; Becker, C.; Nürnberg, P.; Gott, J.M. Complete characterization of the edited transcriptome of the mitochondrion of Physarum polycephalum using deep sequencing of RNA. Nucleic Acids Res. 2011, 39, 6044–6055. [Google Scholar] [CrossRef] [Green Version]
- Zeng, W.-H.; Liao, S.-C.; Chang, C.-C. Identification of RNA Editing Sites in Chloroplast Transcripts of Phalaenopsis aphrodite and Comparative Analysis with Those of Other Seed Plants. Plant Cell Physiol. 2007, 48, 362–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Hou, B.-W.; Niu, Z.-T.; Liu, W.; Xue, Q.-Y.; Ding, X.-Y. Comparative Chloroplast Genomes of Photosynthetic Orchids: Insights into Evolution of the Orchidaceae and Development of Molecular Markers for Phylogenetic Applications. PLoS ONE 2014, 9, e99016. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yu, H.; Wang, J.; Lei, W.; Gao, J.; Qiu, X.; Wang, J. The Complete Chloroplast Genome Sequences of the Medicinal Plant Forsythia suspensa (Oleaceae). Int. J. Mol. Sci. 2017, 18, 2288. [Google Scholar] [CrossRef] [Green Version]
- Kumbhar, F.; Nie, X.; Xing, G.; Zhao, X.; Lin, Y.; Wang, S.; Weining, S. Identification and characterisation of RNA editing sites in chloroplast transcripts of einkorn wheat (Triticum monococcum). Ann. Appl. Biol. 2018, 172, 197–207. [Google Scholar] [CrossRef]
- Park, M.; Park, H.; Lee, H.; Lee, B.-H.; Lee, J. The Complete Plastome Sequence of an Antarctic Bryophyte Sanionia uncinata (Hedw.) Loeske. Int. J. Mol. Sci. 2018, 19, 709. [Google Scholar] [CrossRef] [Green Version]
- Magdalena, G.N.; Ewa, F.; Wojciech, P. Cucumber, melon, pumpkin, and squash: Are rules of editing in flowering plants chloroplast genes so well-known indeed? Gene 2009, 434, 1–8. [Google Scholar]
- Huang, Y.-Y.; Matzke, A.J.M.; Matzke, M. Complete Sequence and Comparative Analysis of the Chloroplast Genome of Coconut Palm (Cocos nucifera). PLoS ONE 2013, 8, e74736. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xu, W.; Zou, W.; Jiang, D.; Liu, X. Complete chloroplast genome sequences of two endangered Phoebe (Lauraceae) species. Bot. Stud. 2017, 58, 1–10. [Google Scholar] [CrossRef]
- Greiner, S.; Wang, X.; Rauwolf, U.; Silber, M.V.; Mayer, K.; Meurer, J.; Haberer, G.; Herrmann, R.G. The complete nucleotide sequences of the five genetically distinct plastid genomes of Oenothera, subsection Oenothera: I. Sequence evaluation and plastome evolution. Nucleic Acids Res. 2008, 36, 2366–2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Wang, S.; Ding, Y.; Xu, J.; Li, M.F.; Zhu, S.; Chen, N. Chloroplast Genomic Resource of Paris for Species Discrimination. Sci. Rep. 2017, 7, 3427. [Google Scholar] [CrossRef] [Green Version]
- Kaila, T.; Chaduvla, P.K.; Rawal, H.C.; Saxena, S.; Tyagi, A.; Mithra, S.V.A.; Solanke, A.U.; Kalia, P.; Sharma, T.R.; Singh, N.K.; et al. Chloroplast Genome sequence of Cluster bean (Cyamopsis tetragonoloba L.): Genome structure and comparative analysis. Genes 2017, 8, 212. [Google Scholar] [CrossRef] [Green Version]
- Bryan, G.J.; McNicoll, J.; Ramsay, G.; Meyer, R.C.; De Jong, W.S. Polymorphic simple sequence repeat markers in chloroplast genomes of Solanaceous plants. Theor. Appl. Genet. 1999, 99, 859–867. [Google Scholar] [CrossRef]
- Provan, J. Novel chloroplast microsatellites reveal cytoplasmic variation in Arabidopsis thaliana. Mol. Ecol. 2000, 9, 2183–2185. [Google Scholar] [CrossRef] [PubMed]
- Ebert, D.; Peakall, R. Chloroplast simple sequence repeats (cpSSRs): Technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Mol. Ecol. Resour. 2009, 9, 673–690. [Google Scholar] [CrossRef]
- Philippe, H.; Delsuc, F.; Brinkmann, H.; Lartillot, N. Phylogenomics, Annual Review of Ecology. Evol. Syst. 2005, 36, 541–562. [Google Scholar] [CrossRef]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 1–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [Green Version]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [Green Version]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.S.; Dubchak, I. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 2000, 16, 1046–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, J. Cases in which parsimony or compatibility methods will be positively misleading. Syst. Zool. 1978, 27, 401–410. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Systematic 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posada, D. jModelTest: Phylogenetic Model Averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | C. farinosa | C. glandulosa | M. crassifolia | M. oblongifolia |
|---|---|---|---|---|
| Genome size (bp) | 156,481 | 156,560 | 155,685 | 155,436 |
| IR (bp) | 26,430 | 26,424 | 26,294 | 26,401 |
| LSC (bp) | 85,565 | 85,681 | 84,624 | 84,153 |
| SSC (bp) | 18,056 | 18,031 | 18,473 | 18,481 |
| Total number of genes | 138 | 137 | 138 | 138 |
| rRNA | 4 | 4 | 4 | 4 |
| tRNA | 31 | 31 | 31 | 31 |
| Protein-coding genes | 81 | 80 | 81 | 81 |
| A% | 31 | 31 | 31 | 31 |
| C% | 18 | 18 | 18 | 18 |
| T% | 32 | 32 | 32 | 32 |
| G% | 17 | 17 | 17 | 17 |
| Category | Gene Groups | Gene Names |
|---|---|---|
| RNA genes | Ribosomal RNA genes (rRNA) | rrn5, rrn4.5, rrn16, rrn23 |
| Transfer RNA genes (tRNA) | trnH-GUG, trnK-UUU+, trnQ-UUG, trnS-GCU, trnS-CGA+, trnR-UCU, trnC-GCA, trnD-GUC, trnY-GUA, trnE-UUC, trnT-GGU, trnS-UGA, trnG-GCC, trnfM-CAU, trnS-GGA, trnT-UGU, trnL-UAA+, trnF-GAA, trnV-UAC+, trnM-CAU, trnW-CCA, trnP-UGG, trnP-GGG, trnI-CAUa, trnL-CAAa, trnV-GACa, trnI-GAU+,a, trnA-UGC+,a, trnR-ACGa, trnN-GUUa, trnL-UAG | |
| Ribosomal proteins | Small ribosomal subunit | rps2, rps3, rps4, rps7a, rps8, rps11, rps12a, rps14, rps15, rps16+, rps18, rps19 |
| Transcription | Large ribosomal subunit | rpl2+,a, rpl14, rpl16, rpl20, rpl22, rpl23a, rpl32, rpl33, rpl36 |
| DNA dependent RNA polymerase | rpoA, rpoB, rpoC1+, rpoC2 | |
| Protein-coding genes | Photosystem I | psaA, psaB, psaC, psaI, psaJ, ycf3++ |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbG, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ, ndhK * | |
| Subunit of cytochrome | petA, petB, petD, petG, petL, petN | |
| Subunit of synthase | atpA, atpB, atpE, atpF+, atpH, atpI | |
| Large subunit of Rubisco | rbcL | |
| NADH dehydrogenase | ndhA+, ndhB+,a, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ | |
| Other genes | ATP dependent protease subunit P | clpP++ |
| Chloroplast envelope membrane protein | cemA | |
| Maturase | matK | |
| Subunit of acetyl-CoA carboxylase | accD | |
| C-type cytochrome synthesis | ccsA | |
| Translation initiation factor | infA | |
| Hypothetical proteins | ycf2a, ycf4, ycf15a | |
| Component of the TIC complex | ycf1a |
| SSR Type | Repeat Unit | Species | |||
|---|---|---|---|---|---|
| C. farinosa | C. glandulosa | M. crassifolia | M. oblongifolia | ||
| Mono | A/T | 220 | 223 | 204 | 208 |
| C/G | 1 | 1 | 2 | 2 | |
| Di | AC/GT | 0 | 1 | 0 | 0 |
| AG/CT | 2 | 0 | 1 | 1 | |
| AT/AT | 11 | 12 | 7 | 10 | |
| Tri | AAT/ATT | 4 | 5 | 2 | 3 |
| Tetra | AAAC/GTTT | 0 | 0 | 1 | 0 |
| AAAG/CTTT | 0 | 0 | 0 | 1 | |
| AAAT/ATTT | 6 | 6 | 4 | 5 | |
| AATT/AATT | 2 | 2 | 1 | 0 | |
| AACT/AGTT | 0 | 0 | 0 | 1 | |
| AGAT/ATCT | 1 | 1 | 2 | 1 | |
| Penta | AAAAT/ATTTT | 1 | 0 | 0 | 0 |
| AAATT/AATTT | 0 | 0 | 1 | 0 | |
| AACAT/ATGTT | 0 | 0 | 0 | 1 | |
| AAACT/AGTTT | 1 | 0 | 0 | 0 | |
| AATAG/ATTCT | 0 | 0 | 2 | 0 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alzahrani, D.A.; Albokhari, E.J.; Yaradua, S.S.; Abba, A. Comparative Analysis of Chloroplast Genomes of Four Medicinal Capparaceae Species: Genome Structures, Phylogenetic Relationships and Adaptive Evolution. Plants 2021, 10, 1229. https://doi.org/10.3390/plants10061229
Alzahrani DA, Albokhari EJ, Yaradua SS, Abba A. Comparative Analysis of Chloroplast Genomes of Four Medicinal Capparaceae Species: Genome Structures, Phylogenetic Relationships and Adaptive Evolution. Plants. 2021; 10(6):1229. https://doi.org/10.3390/plants10061229
Chicago/Turabian StyleAlzahrani, Dhafer A., Enas J. Albokhari, Samaila S. Yaradua, and Abidina Abba. 2021. "Comparative Analysis of Chloroplast Genomes of Four Medicinal Capparaceae Species: Genome Structures, Phylogenetic Relationships and Adaptive Evolution" Plants 10, no. 6: 1229. https://doi.org/10.3390/plants10061229
APA StyleAlzahrani, D. A., Albokhari, E. J., Yaradua, S. S., & Abba, A. (2021). Comparative Analysis of Chloroplast Genomes of Four Medicinal Capparaceae Species: Genome Structures, Phylogenetic Relationships and Adaptive Evolution. Plants, 10(6), 1229. https://doi.org/10.3390/plants10061229