
Cleaning the Medicago Microarray Database to Improve Gene Function Analysis


Abstract
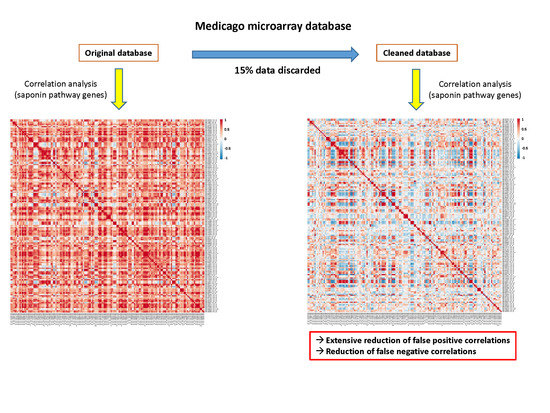
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Data
2.2. Sum of the Expression Values
2.3. Pearson Correlation Coefficient
2.4. Cleaning the Database
- −
- Replicates with large variation in expression values as detected by the Pearson correlation coefficients (calculated between replicate pairs). We removed replicates whose sample pairs coefficients were below the 0.90 threshold (Table S7). The number of replicates removed for each sample is as follows: 3 for RT_Myc_3wks_infection, 3 for GiantCell, 3 for GallTissue_GiantCell, 2 for RT_LCM_arbuscular, 2 for RT_LCM_cortical, 2 for RT_LCM_adjacent, 1 for Nod_Naut1_SalsC, 1 for RT_CRR_72hpi, 1 for RT_CRR_96hpi, 1 for Root_A17_control. (19 columns in total).
- −
- Samples with extremely low (<1 × 106) sums of the expression values. All replicates were removed (Table S4B,C). (30 columns).
- −
- Duplicated data. Replicates were removed at alternated lines (Tables S5 and S7). (24 columns).
- −
- Experiments with one measurement for each sample, so-called “single” replicate (Table S6). (30 columns).
2.5. AgriGO
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Microarray Data
5.2. R
5.3. AgriGO
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Richard, A.; Louise, P.H. “Omic” technologies: Genomics, transcriptomics, proteomics and metabolomics. Obstet. Gynaecol. 2011, 13, 189–195. [Google Scholar]
- Huang, X.Y.; Salt, D.E.E. Plant Ionomics: From Elemental Profiling to Environmental Adaptation. Mol. Plant 2016, 9, 787–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quackenbush, J. Microarray data normalization and transformation. Nat. Genet. 2002, 32, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Park, T.; Yi, S.G.; Kang, S.H.; Lee, S.Y.; Lee, Y.S.; Simon, R. Evaluation of normalization methods for microarray data. BMC Bioinform. 2003, 4. [Google Scholar] [CrossRef] [Green Version]
- Slonim, D.K.; Yanai, I. Getting started in gene expression microarray analysis. PLoS Comput. Biol. 2009, 5. [Google Scholar] [CrossRef]
- Bumgarner, R. Overview of DNA microarrays: Types, applications, and their future. Curr. Protoc. Mol. Biol. 2013, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ledford, H. The death of microarrays? Nature 2008, 455, 847. [Google Scholar] [CrossRef]
- Edgar, R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, 991–995. [Google Scholar] [CrossRef] [Green Version]
- Huala, E.; Dickerman, A.W.; Garcia-Hernandez, M.; Weems, D.; Reiser, L.; LaFond, F.; Hanley, D.; Kiphart, D.; Zhuang, M.; Huang, W.; et al. The Arabidopsis Information Resource (TAIR): A comprehensive database and web-based information retrieval, analysis, and visualization system for a model plant. Nucleic Acids Res. 2001, 29, 102–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedito, V.A.; Torres-Jerez, I.; Murray, J.D.; Andriankaja, A.; Allen, S.; Kakar, K.; Wandrey, M.; Verdier, J.; Zuber, H.; Ott, T.; et al. A gene expression atlas of the model legume Medicago truncatula. Plant J. 2008, 55, 504–513. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Benedito, V.A.; Wang, M.; Murray, J.D.; Zhao, P.X.; Tang, Y.; Udvardi, M.K. The Medicago truncatula gene expression atlas web server. BMC Bioinform. 2009, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gholami, A.; De Geyter, N.; Pollier, J.; Goormachtig, S.; Goossens, A. Natural product biosynthesis in Medicago species. Nat. Prod. Rep. 2014, 31, 356–380. [Google Scholar] [CrossRef]
- Kang, Y.; Li, M.; Sinharoy, S.; Verdier, J. A snapshot of functional genetic studies in Medicago truncatula. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, D.G.; Bianchi, S.; Blondon, F.; Dattée, Y.; Duc, G.; Essad, S.; Flament, P.; Gallusci, P.; Génier, G.; Guy, P.; et al. Medicago truncatula, a model plant for studying the molecular genetics of the Rhizobium-legume symbiosis. Plant Mol. Biol. Rep. 1990, 8, 40–49. [Google Scholar] [CrossRef]
- Graham, P.H.; Vance, C.P. Update on Legume Utilization Legumes: Importance and Constraints to Greater Use. Plant Physiol. 2003, 131, 872–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, N.D.; Debellé, F.; Oldroyd, G.E.D.; Geurts, R.; Cannon, S.B.; Udvardi, M.K.; Que, F. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 2011, 480, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Dai, X.; Liu, T.; Zhao, P.X. LegumeIP: An integrative database for comparative genomics and transcriptomics of model legumes. Nucleic Acids Res. 2012, 40, 1221–1229. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Verdier, J.; Benedito, V.A.; Tang, Y.; Murray, J.D.; Ge, Y.; Becker, J.D.; Carvalho, H.; Rogers, C.; Udvardi, M.; et al. LegumeGRN: A Gene Regulatory Network Prediction Server for Functional and Comparative Studies. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Dalma-Weiszhausz, D.D.; Warrington, J.; Tanimoto, E.Y.; Miyada, C.G. The Affymetrix GeneChip® Platform: An Overview. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2006; Volume 410, pp. 3–28. ISBN 0121828158. [Google Scholar]
- Franzese, M.; Iuliano, A. Correlation analysis. In Encyclopedia of Bioinformatics and Computational Biology: ABC of Bioinformatics; Elsevier: Amsterdam, The Netherlands, 2018; Volume 1–3, pp. 706–721. ISBN 9780128114322. [Google Scholar]
- Akoglu, H. User’s guide to correlation coefficients. Turk. J. Emerg. Med. 2018, 18, 91–93. [Google Scholar] [CrossRef]
- Murgia, I.; Tarantino, D.; Soave, C.; Morandini, P. Arabidopsis CYP82C4 expression is dependent on Fe availability and circadian rhythm, and correlates with genes involved in the early Fe deficiency response. J. Plant Physiol. 2011, 168, 894–902. [Google Scholar] [CrossRef]
- Månsson, R.; Tsapogas, P.; Åkerlund, M.; Lagergren, A.; Gisler, R.; Sigvardsson, M. Pearson Correlation Analysis of Microarray Data Allows for the Identification of Genetic Targets for Early B-cell Factor. J. Biol. Chem. 2004, 279, 17905–17913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zermiani, M.; Begheldo, M.; Nonis, A.; Palme, K.; Mizzi, L.; Morandini, P.; Nonis, A.; Ruperti, B. Identification of the arabidopsis RAM/MOR signalling network: Adding new regulatory players in plant stem cell maintenance and cell polarization. Ann. Bot. 2015, 116, 69–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beekweelder, J.; van Leeuwen, W.; van Dam, N.M.; Bertossi, M.; Grandi, V.; Mizzi, L.; Soloviev, M.; Szabados, L.; Molthoff, J.W.; Schipper, B.; et al. The impact of the absence of aliphatic glucosinolates on insect herbivory in Arabidopsis. PLoS ONE 2008, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naoumkina, M.A.; Modolo, L.V.; Huhman, D.V.; Urbanczyk-Wochniak, E.; Tang, Y.; Sumner, L.W.; Dixon, R.A. Genomic and coexpression analyses predict multiple genes involved in triterpene saponin biosynthesis in Medicago truncatula. Plant Cell 2010, 22, 850–866. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yon Rhee, S.; Wood, V.; Dolinski, K.; Draghici, S. Use and misuse of the gene ontology annotations. Nat. Rev. Genet. 2008, 9, 509–515. [Google Scholar] [CrossRef]
- Gaude, N.; Bortfeld, S.; Duensing, N.; Lohse, M.; Krajinski, F. Arbuscule-containing and non-colonized cortical cells of mycorrhizal roots undergo extensive and specific reprogramming during arbuscular mycorrhizal development. Plant J. 2012, 69, 510–528. [Google Scholar] [CrossRef]
- Hogekamp, C.; Küster, H. A roadmap of cell-type specific gene expression during sequential stages of the arbuscular mycorrhiza symbiosis. BMC Genom. 2013, 14. [Google Scholar] [CrossRef] [Green Version]
- Limpens, E.; Moling, S.; Hooiveld, G.; Pereira, P.A.; Bisseling, T.; Becker, J.D.; Küster, H. Cell- and Tissue-Specific Transcriptome Analyses of Medicago truncatula Root Nodules. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Ortu, G.; Balestrini, R.; Pereira, P.A.; Becker, J.D.; Küster, H.; Bonfante, P. Plant genes related to gibberellin biosynthesis and signaling are differentially regulated during the early Stages of AM fungal interactions. Mol. Plant 2012, 5, 951–954. [Google Scholar] [CrossRef] [Green Version]
- Seabra, A.R.; Pereira, P.A.; Becker, J.D.; Carvalho, H.G. Inhibition of glutamine synthetase by phosphinothricin leads to transcriptome reprograming in root nodules of Medicago truncatula. Mol. Plant Microbe Interact. 2012, 25, 976–992. [Google Scholar] [CrossRef] [Green Version]
- Vranová, E.; Coman, D.; Gruissem, W. Structure and Dynamics of the Isoprenoid Pathway Network. Mol. Plant 2012, 5, 318–333. [Google Scholar] [CrossRef] [Green Version]
- Dubey, V.S.; Bhalla, R.; Luhtra, R. An overview of the non-mevalonate pathway for terpenoid biosynthesis in plants. J. Biosci. 2003, 28, 637–646. [Google Scholar] [CrossRef]
- Savage, G.P. Saponins. In Encyclopedia of Food Science and Nutrition, 2nd ed.; Academic Press: Cambridge, MA, USA, 2003; pp. 5095–5098. ISBN 9780122270550. [Google Scholar] [CrossRef]
- Tava, A.; Scotti, C.; Avato, P. Biosynthesis of saponins in the genus Medicago. Phytochem. Rev. 2011, 10, 459–469. [Google Scholar] [CrossRef]
- Liu, C.; Ha, C.M.; Dixon, R.A. Functional genomics in the study of metabolic pathways in Medicago truncatula: An overview. Methods Mol. Biol. 2018, 1822, 315–337. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Y.; Sui, N. Transcriptional regulation of bHLH during plant response to stress. Biochem. Biophys. Res. Commun. 2018, 503, 397–401. [Google Scholar] [CrossRef]
- Toledo-Ortiz, G.; Huq, E.; Quail, P.H. The Arabidopsis Basic/Helix-Loop-Helix Transcription Factor Family. Plant Cell 2003, 15, 1749–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, F.; Yang, D.C.; Meng, Y.Q.; Jin, J.; Gao, G. PlantRegMap: Charting functional regulatory maps in plants. Nucleic Acids Res. 2020, 48, D1104–D1113. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, 64–70. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. AgriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef]
- Emmert-Buck, M.R.; Bonner, R.F.; Smith, P.D.; Chuaqui, R.F.; Zhuang, Z.; Goldstein, S.; Weiss, R.A.; Liotta, L.A. Laser Capture Microdissection. Science 1996, 274, 998–1001. [Google Scholar] [CrossRef]
- Wina, E.; Muetzel, S.; Becker, K. The impact of saponins or saponin-containing plant materials on ruminant production—A review. J. Agric. Food Chem. 2005, 53, 8093–8105. [Google Scholar] [CrossRef]
- Carelli, M.; Biazzi, E.; Panara, F.; Tava, A.; Scaramelli, L.; Porceddu, A.; Graham, N.; Odoardi, M.; Piano, E.; Arcioni, S.; et al. Medicago truncatula CYP716A12 is a multifunctional oxidase involved in the biosynthesis of hemolytic saponins. Plant Cell 2011, 23, 3070–3081. [Google Scholar] [CrossRef] [Green Version]
- Farag, M.A.; Deavours, B.E.; de Fáltima, Â.; Naoumkina, M.; Dixon, R.A.; Sumner, L.W. Integrated metabolite and transcript profiling identify a biosynthetic mechanism for hispidol in Medicago truncatula cell cultures. Plant Physiol. 2009, 151, 1096–1113. [Google Scholar] [CrossRef] [Green Version]
- Naoumkina, M.A.; He, X.; Dixon, R.A. Elicitor-induced transcription factors for metabolic reprogramming of secondary metabolism in Medicago truncatula. BMC Plant Biol. 2008, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Jacob, L.; Gagnon-Bartsch, J.A.; Speed, T.P. Correcting gene expression data when neither the unwanted variation nor the factor of interest are observed. Biostatistics 2016, 17, 16–28. [Google Scholar] [CrossRef] [Green Version]
- Freytag, S.; Gagnon-Bartsch, J.; Speed, T.P. Systematic noise degrades gene co-expression signals but can be corrected. BMC Bioinform. 2015, 16, 309. [Google Scholar] [CrossRef]
- Varma, S. Blind estimation and correction of microarray batch effect. PLoS ONE 2020, 15, e0231446. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Wickham, H. ggplot2: Create Elegant Data Visualisations Using the Grammar of Graphics, R. Package Version 3.3.2; 2009. Available online: https://cran.r-project.org/web/packages/ggplot2/index.html (accessed on 15 April 2021).
- Kolde, R. pheatmap: Pretty Heatmaps, R Package Version 1.0.12; 2019. Available online: https://cran.r-project.org/web/packages/pheatmap/index.html (accessed on 15 April 2021).
- Dowle, M. Package “Data.Table”, R Package Version 1.14.0; 2021. Available online: https://cran.r-project.org/web/packages/data.table/index.html (accessed on 15 April 2021).
- Harrell, F.E., Jr. Package “Hmisc”, R Package Version 4.5-0; 2021. Available online: https://cran.r-project.org/web/packages/Hmisc/index.html (accessed on 15 April 2021).
- Neuwirth, E. Package “RColorBrewer”, R Package Version 1.1-2; 2014. Available online: https://cran.r-project.org/web/packages/RColorBrewer/index.html (accessed on 15 April 2021).
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heath, K.D.; Burke, P.V.; Stinchcombe, J.R. Coevolutionary genetic variation in the legume-rhizobium transcriptome. Mol. Ecol. 2012, 21, 4735–4747. [Google Scholar] [CrossRef] [PubMed]
- Uppalapati, S.R.; Marek, S.M.; Lee, H.K.; Nakashima, J.; Tang, Y.; Sledge, M.K.; Dixon, R.A.; Mysore, K.S. Global gene expression profiling during Medicago truncatula-Phymatotrichopsis omnivora interaction reveals a role for jasmonic acid, ethylene, and the flavonoid pathway in disease development. Mol. Plant Microbe Interact. 2009, 22, 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marzorati, F.; Wang, C.; Pavesi, G.; Mizzi, L.; Morandini, P. Cleaning the Medicago Microarray Database to Improve Gene Function Analysis. Plants 2021, 10, 1240. https://doi.org/10.3390/plants10061240
Marzorati F, Wang C, Pavesi G, Mizzi L, Morandini P. Cleaning the Medicago Microarray Database to Improve Gene Function Analysis. Plants. 2021; 10(6):1240. https://doi.org/10.3390/plants10061240
Chicago/Turabian StyleMarzorati, Francesca, Chu Wang, Giulio Pavesi, Luca Mizzi, and Piero Morandini. 2021. "Cleaning the Medicago Microarray Database to Improve Gene Function Analysis" Plants 10, no. 6: 1240. https://doi.org/10.3390/plants10061240
APA StyleMarzorati, F., Wang, C., Pavesi, G., Mizzi, L., & Morandini, P. (2021). Cleaning the Medicago Microarray Database to Improve Gene Function Analysis. Plants, 10(6), 1240. https://doi.org/10.3390/plants10061240