1. Introduction
Due to the unpredictability of environmental conditions and the inability of plants to move in order to avoid unfavorable conditions, a number of abiotic stress factors threaten plant productivity and sustainability [
1,
2] Salinity is one of the main environmental stressors limiting crop production globally [
3], it causes oxidative damages, ion toxicity, and nutrition imbalance [
2,
4,
5,
6]. According to the United Nations (UN) Environment Program (UNEP), worldwide, approximately 50% of agricultural lands are now characterized as saline soils [
6]. Additionally, this area increases every day due to inadequate irrigation practices and it aggravates the salinity problem [
7].
Survival under this stress requires the integration of adaptive metabolic, physiological, and molecular responses. In response to salinity, plants employ different strategies and mechanisms to accumulate organic solutes in the tissue, and provide tissue tolerance to high salt concentrations [
7,
8,
9,
10] These organic solutes can protect plants against short term and high intensity salt stress [
11]. Moreover, it is known that soluble protein content is an important indicator of physiological status of plants [
12]. Thus, understanding the different adaption mechanisms to environmental stresses may lead to novel strategies for plant improvement.
According to the ability to grow on high salt medium, plants have been classified as glycophytes or halophytes, where glycophytes cannot grow well under salt stress condition whereas halophytes grow well under high salinity. Most crop species are glycophytes and cannot tolerate salt stress [
13,
14], so to cope with this stress, plants have evolved complex mechanisms and elaborated signaling network that perceives signals from their surroundings and appropriately responds to environmental changes by modulating the expression of responsive genes [
15]. These genes encode two major groups of proteins: functional and regulatory proteins. The main regulators of abiotic stress mediated gene expression are transcription factors (TFs), which have the capacity to recognize and bind specifically to cis-elements in the promoters of stress-responsive genes, and regulate their transcription [
15,
16,
17], TFs function as terminal transducers and directly modulate gene expression of an array of downstream genes [
18,
19,
20,
21] Due to this property, the manipulation of TFs is a very useful strategy for imparting multiple stress tolerance in plants [
22,
23,
24]. The APETALA2/ethylene responsive factor (AP2/ERF) superfamily is one of the largest groups of transcription factors in plants [
25]. This family is characterized by the presence of a common domain of about 60–70 amino acids residues known as the AP2 domain. A simple classification based on the copy number of AP2 domains yielded four families: AP2, ERF, RAV and soloist [
26,
27]. The AP2 family owns duplicated AP2/ERF domains, whereas the ERF family exhibits a single AP2/ERF domain, the RAV family has one B3 domain and one AP2/ERF domain and the Soloist family contains a small group of TFs with a highly divergent single AP2 domain (AP2-like domain) and gene structure.
The ERF family is further subdivided into the ERF and dehydration responsive element binding proteins (DREB) subfamilies on the basis of the similarities in amino acid residues of the AP2 domain [
28]. The TFs from the ERF and DREB subfamilies, are closely associated with responses to environmental stress, such as pathogen and disease stimuli [
28,
29], salinity [
30,
31], drought [
31], and freezing [
32,
33]. With more extensive plant genome sequences, AP2/ERF gene families have been identified in various plants, such as Arabidopsis [
34], soybean [
35], rice [
36], potato [
25],
Medicago truncatula [
37], barley [
38], and
Ammopiptanthus nanus [
39], among others.
Moreover, legumes are the plants in family Fabaceae or Leguminosae, which are playing a crucial role in crop rotation due to their symbiotic nitrogen-fixing bacteria in structures [
40,
41]. Fabaceae are primarily grown for human consumption, for livestock forage and silage, and as soil-enhancing green manure [
42,
43]. Forage crops are the backbone of sustainable agriculture; they are often grown in less favorable areas and thus require sophisticated protective mechanisms to withstand severe environmental conditions [
43]. The transcription factors play a crucial role in enhancing some legume species to adapt during abiotic stresses. Thus, we have been interested in characterizing certain transcription factor genes from
M. truncatula, which is an omni-Mediterranean forage legume species and a model plant for legume biological studies in view of its small diploid genome, self-fertile nature, relatively short life cycle and high genetic transformation efficiency [
42,
44]. Due to these characteristics and to the fact that it is a close relative of alfalfa and clovers,
M. truncatula has become the focus of intensive research around the globe, aimed at identifying and characterizing major stress responsive genes using modern tools, such as genomics as well as genetic transformation [
44,
45,
46].
This study aims to (i) analyze the morpho-physiological and biochemical responses of four M. truncatula lines under salt stress, and (ii) to explore the expression of an ERF gene in M. truncatula under salt stress.
4. Material and Methods
4.1. Plant Material and Culture Conditions
Four lines of M. truncatula were used. They include the two Tunisian lines TN1.11 and TN6.18, the Moroccan line A10, lines belonging to a collection of genotypes used in the M. truncatula HapMap project, and Jemalong A17 (JA17) from the Australian collection, which was used to obtain the reference genome for M. truncatula. Three biological replicates were used in all analysis.
Seeds were scarified using sandpaper q60, then were kept for 72 h in the dark at 4 °C and thereafter transferred at 21 °C during 24 h for germination. The germinated seeds were sown into black pots with a two liters capacity filled with a mixture of sand and compost (3:1.
v/
v). Plants were grown in a growth chamber under controlled conditions at a temperature of 24 °C/18 °C (day/night), a relative humidity of 60–80%, and a photoperiod of 16/8 h. Plants were irrigated with Fahräeus nutritive solution [
89] once every 2 days, and after 21 days they were randomly divided into two groups one for the control treatment and the other for salt stress (100 mM NaCl).
4.2. Morphological and Photosynthetic Parameters
Fourteen parameters were measured for the studied lines including the length of stems (LS, cm), length of roots (LR, cm), number of leaves (NL), number of axes (NA), aerial fresh weight (AFW, g), aerial dry weight (ADW, g), root fresh weight (RFW, g), root dry weight (RDW, g), root dry weight and aerial dry weight ratio (RDW/ADW), relative water content (RWC, %), water content (WC), and the relative growth rate (RGR), chlorophyll a (Chla, mg.g−1 FW), and chlorophyll b (Chlb, mg.g−1 FW) content.
The relative water content (RWC) was calculated as follows: RWC (%) = 100 [(LFW−LDW)/(LTW−LDW)], where LFW is the leaf fresh weight, LDW is the leaf dry weight, and LTW is the leaf turgid weight. Briefly, the fresh weight of a young leaf is determined. The leaves were left floating on distilled water, in Petri dishes, for 24 h and the turgid weight is then recorded. After that, the leaf tissues were dried in an oven at 70 °C for 24 h and their dry weight was measured.
Relative growth rate (RGR) or the mean relative growth rate was determined as the rate of increase in total dry weight per unit of plant weight according to Hunt (1982) thus:
RGR = (In W2 − In W1/t2 − t1).
RGR in g.g−1 day−1,
where W is the total plant weight (g), t is the time (days), and the subscripts 1 and 2 are initial and final harvest of biomass yield.
4.3. MDA Determination
The extent of lipid peroxidation was estimated by determining the concentration of malondialdehyde [
90]. Briefly, 150 mg of fresh leaves were homogenized in TCA (0.1%) and centrifuged (13,000×
g, 20 min) at 4 °C. Thereafter, an aliquot of the supernatant was added to 0.5% (
w/
v) TBA in 20% (
w/
v) TCA and the mixture was incubated at 95 °C for 30 min. The reaction was stopped by transferring tubes to ice for 10 min followed by a centrifugation step at 10,000×
g for 10 min. The absorbance of supernatant was measured at 532 nm and the value for non-specific absorption at 600 nm was subtracted. The concentration of MDA was determined from the extinction coefficient of 155 mmol.L
−1cm
−1. Three independent extractions were made for each sample.
4.4. Measurement of Soluble Sugars Content
Soluble sugar content was quantified the method described by Yemm and Willis (1954) [
91]. Briefly, 25 mg of dry weight of leaves was homogenized with 5 mL of 80% ethanol for 30 min at 70 °C. The extract was separated by centrifugation at 6000×
g for 15 min. A second extraction was carried out under the same conditions as above. Then, the soluble sugars content of the aqueous extract was determined using the sulfuric acid anthrone colorimetric method. The soluble sugar content was revealed through absorbance measurement at 640 nm. The standard curve was drawn using glucose (1 to 14 µg/mL).
4.5. Measurement of Soluble Proteins
Fresh leaf material (150 mg) was homogenized with 2 mL buffer (1 M Tris-HCl pH8, 0.5 M EDTA, 100 mM PMSF. The homogenate was then centrifuged for 30 min at 12,000×
g at 4 °C. The protein concentration in the supernatant was measured by the protein assay using BSA as a standard [
92].
4.6. Gene Expression Assay by Quantitative Real-Time (RT-qPCR)
The coding sequence of MtERF1 was identified from the National Center for Biotechnology Information (NCBI) website (
http://www.ncbi.nlm.nih.gov/ (accessed on 15 August 2016)), its accession number is XM_003607935. Then this sequence was blasted against the
M. truncatula genome with expected values ≤ (1 × 10
−5). Finally, these proteins were submitted to the NCBI batch web CD-search tool (
http://www.ncbi. nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi, (accessed on 20 August 2016)). Pfam (
http://pfam.sanger.ac.uk/ (accessed on 3 March 2021)), and SMART (
http://smart.embl-hei delberg.de/ (accessed on 3 March 2021)) to confirm the presence and completeness of the AP2 domain and the primers were used to amplify the CDS regions of
MtERF1 and then it was sequenced to be sure if it is the right sequence.
To determine the expression pattern of
MtERF1 in
M. truncatula we analyzed its expression in three different organs: roots, stem, and leaves by real-time quantitative PCR (qRT-PCR) using
MtERF1 specific primers. Total RNA was extracted from the different tissues of the four studied lines according to the procedures described by [
93]. The concentration and purity of the total RNA were assessed with a NanoDrop-ND 1000 UV-Vis Spectrophotometer (NanoDrop Technologies, Auckland, New Zealand), using 1 μL of total RNA. RNA purity was estimated from the A260/A280 absorbance ratio. RNA samples were treated with recombinant RNase-free DNase I (Roche, Mannheim, Germany) for removing possible genomic DNA contamination. Then, 1 µg of each RNA extract was used to synthesize cDNA by using the iScript™. Reverse Transcription Supermix for RT-qPCR (Bio-Rad, Marnes-la-Coquette, France), according to the manufacturer’s protocol. Gene-specific primer pairs for RT-qPCR were designed with Primer 3 software (Whitehead Instutute for Biomedical Researech, Cambridge, MA, USA). Each gene was evaluated at least in two independent runs. Each sample was amplified in biological triplicate by quantitative qRT-PCR using a Roche 2.0 Real-Time PCR Detection System with SYBR Green (Roche, Mannheim, Germany). The cycle program used was as follows: Initial denaturation at 50 °C for 2 min. then 95 °C at 30 s. followed by 2 cycles for 1 min at 95 °C. then 10 min at 95 °C followed by 40 cycles at 95 °C (10 s) and 60 °C (30 s) followed by 65 °C for 15 s. The amplification of the Actin gene was used as the normalization control. The mRNA fold difference was relative to that of untreated samples used as calibrator. The relative quantification value for
MtERF1 was calculated by the 2−ΔΔCT method.
4.7. Promoter Prediction and Analysis for MtERF1
In order to gain insight into the transcriptional regulation of
MtERF1 gene, putative stress- or hormone-responsive cis-acting elements were identified in 1 kb lengths of
M. truncatula DNA sequence located immediately 5′ to the gene coding sequence using the PlantCARE search tool (
http://bioinformatics.psb.ugent.be/webtools/plantcare/html/ (accessed on 15 August 2019)).
4.8. Treatment with Various Abiotic Stress
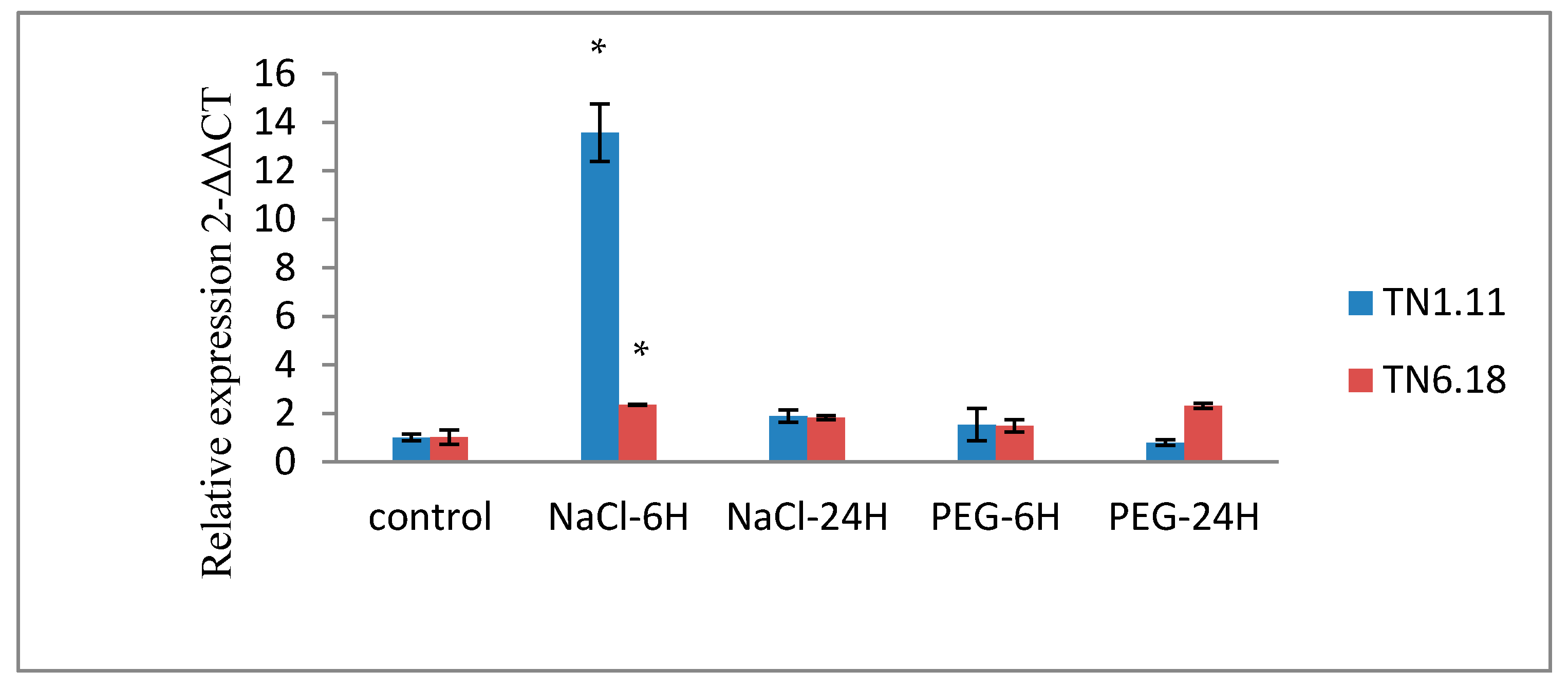
We examined the expression profiles of MTERF1 genes by qRT-PCR as described above under ABA and abiotic stress treatment, including cold, NaCl, and PEG only for the two local lines TN1.11 and TN6.18.
The three additional abiotic elicitors which were used are: osmotic stress (PEG 20%), cold (4 °C) and ABA (10 µM). For the treatment with salt, PEG, and ABA, 21-day-old seedlings of M. truncatula were transferred to Fahräeus nutritive solution containing 200 mM NaCl or 20% PEG and incubated for 6 h and 24 h. For ABA treatment, the solution containing 10 µM ABA and incubated for 24 h. For cold treatment, 21-day-old seedlings of M. truncatula grown in soil were placed in a low temperature chamber at 4 °C for 24 h.
4.9. Statistical Analyses
A three-way analysis of variance was used to test for line, salt treatment differences, and line × treatment interaction effects. Only characters that showed a significant line × treatment interaction were used for further analysis. Means were compared using Duncan’s multiple range test at 5%. To find out whether or not traits are correlated to each other, Pearson coefficients were calculated. The significance level of associations between morphological traits and photosynthetic parameters was set to 0.05. All analyses were performed using SPSS software (SPSS Inc. Released 2007 SPSS for Windows. Version 16.0. Chicago, IL, USA).
Broad-sense heritability (
H2) for each trait was estimated as described by Badri et al. (2016) [
94].
where σ
2 g is the genetic variance observed between the lines and σ
2 e is the environmental variance corresponding to the residual error between the eight replicates of the same genotype (=line).
To investigate the stratification of the lines, salt sensitivity index (SSI) for measured traits in lines of M. truncatula grown under salt stress were subjected to principal component analysis (PCA) and hierarchical cluster analysis (HCA). This analysis was carried out using XLSTAT software (Version 2014.5.03. Addinsoft, Paris, France).
5. Conclusions
Associations were discovered between exhibited phenotypes, lipid peroxidation, soluble proteins, and sugar accumulation, and gene expression. Salt tolerance seems to be related to lower MDA accumulation and a lower biomass reduction. Besides, accumulation of soluble proteins and soluble sugars was linked to salt-tolerance, being much higher for the tolerant genotype.
The expression pattern of MTERF1 supports the fact that this gene is involved in mechanisms associated with salt tolerance and could be considered as a marker to discriminate Medicago genotypes with differing tolerance to salinity. Our expression profiling analysis of the MtERF1 in various tissues and under different abiotic stresses should facilitate the identification of appropriate candidate genes for further functional characterization. The information obtained in this study showed that the MTERF1 gene is strongly induced by salt in root tissues. Thus. MTERF1 could be helpful in selecting candidate genotypes to be used by crop growers in salty areas or as progenitors in breeding programs for salt tolerance.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}