
OMICs, Epigenetics, and Genome Editing Techniques for Food and Nutritional Security

 ,
,  , ,
, ,  ,
, 
 ,
,  ,
,  , ,
, ,  ,
,  and
and 
Abstract
:
{kind=link}
{kind=link}
1. Introduction
2. Genomics
2.1. Plant Genome Sequencing and Annotation
2.2. Plant Microbiome Analysis by Metagenomics
2.2.1. Metagenomic Approaches
2.2.2. Application of Metagenomics to the Study of the Plant Microbiome, Breeding, and Food Security
3. Transcriptomics
3.1. Routine Transcriptome Analysis
3.2. Spatial Transcriptomics and In Situ Tissue Profiling
3.3. Single-Cell Transcriptomics
3.4. Metatranscriptomics
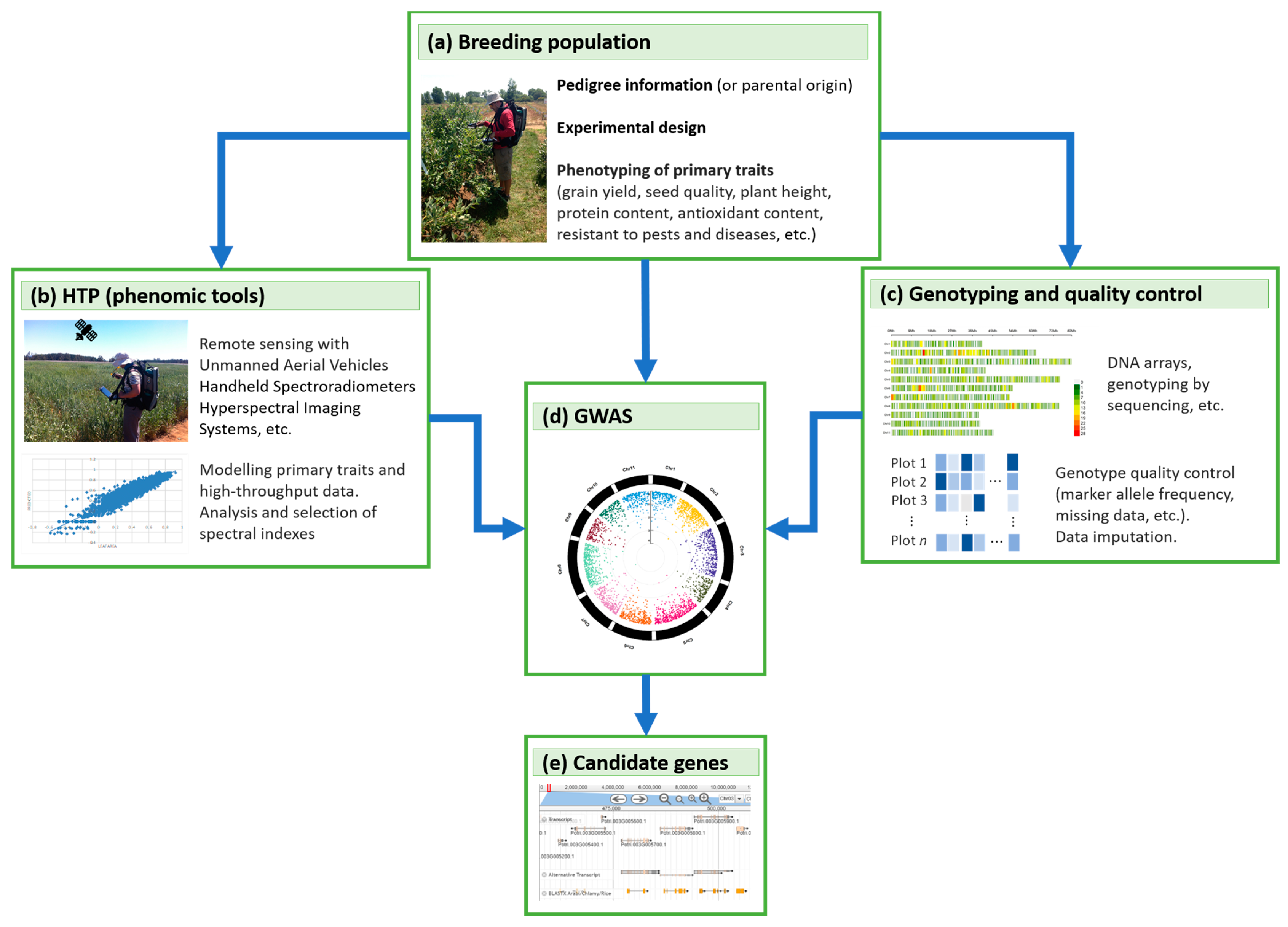
4. GWAS, Genomic, and Phenomic Prediction
5. Plant Epigenetics and Epigenomics: OMICs Studies (Methylome by WGBS and Histone Modifications by ChiP-Seq)
5.1. Regulation of Plant Stress Response by Dynamic Changes in DNA Methylation: Analysis of Global DNA Methylome by WGBS
5.2. Regulation of Plant Stress Response by Dynamic Histone Modifications: Analysis of Genome-Wide Histone Modifications by ChiP-Seq
5.3. Application of Epigenetics and Epigenomics to Improve Crop Germplasm
5.3.1. Epigenetic-Assisted Molecular Breeding
5.3.2. Precise Epigenome Editing Approach
6. Gene Editing Techniques for Food and Nutritional Security
7. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. World Hunger Is Still Not Going Down after Three Years and Obesity Is Still Growing. Available online: https://www.who.int/news/item/15-07-2019-world-hunger-is-still-not-going-down-after-three-years-and-obesity-is-still-growing-un-report (accessed on 25 April 2021).
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Tuppy, H. The amino-acid sequence in the phenylalanyl chain of insulin. I. The identification of lower peptides from partial hydrolysates. Biochem. J. 1951, 49, 463–481. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Tuppy, H. The amino-acid sequence in the phenylalanyl chain of insulin. 2. The investigation of peptides from enzymic hydrolysates. Biochem. J. 1951, 49, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Giani, A.M.; Gallo, G.R.; Gianfranceschi, L.; Formenti, G. Long walk to genomics: History and current approaches to genome sequencing and assembly. Comput. Struct. Biotechnol. J. 2020, 18, 9–19. [Google Scholar] [CrossRef]
- Holley, R.W.; Apgar, J.; Everett, G.A.; Madison, J.T.; Marquisee, M.; Merrill, S.H.; Penswick, J.R.; Zamir, A. Structure of a Ribonucleic Acid. Science 1965, 147, 1462–1465. [Google Scholar] [CrossRef]
- Sanger, F.; Coulson, A.R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 1975, 94, 441–448. [Google Scholar] [CrossRef]
- Maxam, A.M.; Gilbert, W. A new method for sequencing DNA. Proc. Natl. Acad. Sci. USA 1977, 74, 560–564. [Google Scholar] [CrossRef] [Green Version]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, H.; Gesteland, R. Capillary gel electrophoresis for rapid, high resolution DNA sequencing. Nucleic Acids Res. 1990, 18, 1415–1419. [Google Scholar] [CrossRef] [Green Version]
- Gut, I.G. New sequencing technologies. Clin. Transl. Oncol. 2013, 15, 879–881. [Google Scholar] [CrossRef]
- Kumar, K.R.; Cowley, M.J.; Davis, R.L. Next-Generation Sequencing and Emerging Technologies. Semin. Thromb. Hemost. 2019, 45, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 2016, 107, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Gurtowski, J.; Yoo, S.; Nattestad, M.; Marcus, S.; Goodwin, S.; McCombie, W.R.; Schatz, M.C. Third-generation sequencing and the future of genomics. bioRxiv 2016, 048603. [Google Scholar] [CrossRef] [Green Version]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef]
- Jung, H.; Jeon, M.S.; Hodgett, M.; Waterhouse, P.; Eyun, S.I. Comparative Evaluation of Genome Assemblers from Long-Read Sequencing for Plants and Crops. J. Agric. Food Chem. 2020, 68, 7670–7677. [Google Scholar] [CrossRef]
- Wee, Y.; Bhyan, S.B.; Liu, Y.; Lu, J.; Li, X.; Zhao, M. The bioinformatics tools for the genome assembly and analysis based on third-generation sequencing. Brief. Funct. Genom. 2019, 18, 1–12. [Google Scholar] [CrossRef]
- Lappalainen, T.; Scott, A.J.; Brandt, M.; Hall, I.M. Genomic Analysis in the Age of Human Genome Sequencing. Cell 2019, 177, 70–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Kersey, P.J. Plant genome sequences: Past, present, future. Curr. Opin. Plant Biol. 2019, 48, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Blanc, G.; Wolfe, K.H. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 2004, 16, 1667–1678. [Google Scholar] [CrossRef] [Green Version]
- The International Wheat Genome Sequencing Consortium (IWGSC); Appels, R.; Eversole, K.; Stein, N.; Feuillet, C.; Keller, B.; Rogers, J.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361. [Google Scholar] [CrossRef] [Green Version]
- Bolger, M.; Schwacke, R.; Gundlach, H.; Schmutzer, T.; Chen, J.; Arend, D.; Oppermann, M.; Weise, S.; Lange, M.; Fiorani, F.; et al. From plant genomes to phenotypes. J. Biotechnol. 2017, 261, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Akpinar, B.A.; Lucas, S.J.; Vrana, J.; Dolezel, J.; Budak, H. Sequencing chromosome 5D of Aegilops tauschii and comparison with its allopolyploid descendant bread wheat (Triticum aestivum). Plant Biotechnol. J. 2015, 13, 740–752. [Google Scholar] [CrossRef]
- Wendel, J.F.; Jackson, S.A.; Meyers, B.C.; Wing, R.A. Evolution of plant genome architecture. Genome Biol. 2016, 17, 37. [Google Scholar] [CrossRef] [Green Version]
- Zimin, A.V.; Puiu, D.; Hall, R.; Kingan, S.; Clavijo, B.J.; Salzberg, S.L. The first near-complete assembly of the hexaploid bread wheat genome, Triticum aestivum. Gigascience 2017, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- GenBank and WGS Statistics. Available online: https://www.ncbi.nlm.nih.gov/genbank/statistics/ (accessed on 4 February 2021).
- Cagirici, H.B.; Sen, T.Z.; Budak, H. mirMachine: A One-Stop Shop for Plant miRNA Annotation. J. Vis. Exp. 2021, 171. [Google Scholar] [CrossRef]
- Leroy, P.; Guilhot, N.; Sakai, H.; Bernard, A.; Choulet, F.; Theil, S.; Reboux, S.; Amano, N.; Flutre, T.; Pelegrin, C.; et al. TriAnnot: A Versatile and High Performance Pipeline for the Automated Annotation of Plant Genomes. Front. Plant Sci. 2012, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coletta, R.D.; Qiu, Y.; Ou, S.; Hufford, M.B.; Hirsch, C.N. How the pan-genome is changing crop genomics and improvement. Genome Biol. 2021, 22, 3. [Google Scholar] [CrossRef] [PubMed]
- Bayer, P.E.; Golicz, A.A.; Scheben, A.; Batley, J.; Edwards, D. Plant pan-genomes are the new reference. Nat. Plants 2020, 6, 914–920. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [Green Version]
- Danilevicz, M.F.; Fernandez, C.G.T.; Marsh, J.I.; Bayer, P.E.; Edwards, D. Plant pangenomics: Approaches, applications and advancements. Curr. Opin. Plant Biol. 2020, 54, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Paux, E.; Faure, S.; Choulet, F.; Roger, D.; Gauthier, V.; Martinant, J.P.; Sourdille, P.; Balfourier, F.; Le Paslier, M.C.; Chauveau, A.; et al. Insertion site-based polymorphism markers open new perspectives for genome saturation and marker-assisted selection in wheat. Plant Biotechnol. J. 2010, 8, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Satya, P. Next generation sequencing technologies for next generation plant breeding. Front. Plant Sci. 2014, 5, 367. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Zhao, X.; Laroche, A.; Lu, Z.X.; Liu, H.; Li, Z. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 2014, 5, 484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pont, C.; Wagner, S.; Kremer, A.; Orlando, L.; Plomion, C.; Salse, J. Paleogenomics: Reconstruction of plant evolutionary trajectories from modern and ancient DNA. Genome Biol. 2019, 20, 29. [Google Scholar] [CrossRef]
- Mendes, R.; Garbeva, P.; Raaijmakers, J.M. The rhizosphere microbiome: Significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 2013, 37, 634–663. [Google Scholar] [CrossRef] [PubMed]
- Daniel, R. The metagenomics of soil. Nat. Rev. Microbiol. 2005, 3, 470–478. [Google Scholar] [CrossRef]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winogradsky, H. Contribution to the study of microflora nitrification of wastewater and; Resistance of germs to unfavorable conditions. Ann. Inst. Pasteur 1949, 76, 35–42. [Google Scholar]
- Sanguin, H.; Remenant, B.; Dechesne, A.; Thioulouse, J.; Vogel, T.M.; Nesme, X.; Moenne-Loccoz, Y.; Grundmann, G.L. Potential of a 16S rRNA-based taxonomic microarray for analyzing the rhizosphere effects of maize on Agrobacterium spp. and bacterial communities. Appl. Environ. Microbiol. 2006, 72, 4302–4312. [Google Scholar] [CrossRef] [Green Version]
- DeAngelis, K.M.; Brodie, E.L.; DeSantis, T.Z.; Andersen, G.L.; Lindow, S.E.; Firestone, M.K. Selective progressive response of soil microbial community to wild oat roots. ISME J. 2009, 3, 168–178. [Google Scholar] [CrossRef] [Green Version]
- Roesch, L.F.; Fulthorpe, R.R.; Riva, A.; Casella, G.; Hadwin, A.K.; Kent, A.D.; Daroub, S.H.; Camargo, F.A.; Farmerie, W.G.; Triplett, E.W. Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 2007, 1, 283–290. [Google Scholar] [CrossRef]
- Fierer, N.; Breitbart, M.; Nulton, J.; Salamon, P.; Lozupone, C.; Jones, R.; Robeson, M.; Edwards, R.A.; Felts, B.; Rayhawk, S.; et al. Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Appl. Environ. Microbiol. 2007, 73, 7059–7066. [Google Scholar] [CrossRef] [Green Version]
- Uroz, S.; Buee, M.; Murat, C.; Frey-Klett, P.; Martin, F. Pyrosequencing reveals a contrasted bacterial diversity between oak rhizosphere and surrounding soil. Environ. Microbiol. Rep. 2010, 2, 281–288. [Google Scholar] [CrossRef]
- Edwards, R.A.; Rodriguez-Brito, B.; Wegley, L.; Haynes, M.; Breitbart, M.; Peterson, D.M.; Saar, M.O.; Alexander, S.; Alexander, E.C., Jr.; Rohwer, F. Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genom. 2006, 7, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rausch, P.; Ruhlemann, M.; Hermes, B.M.; Doms, S.; Dagan, T.; Dierking, K.; Domin, H.; Fraune, S.; von Frieling, J.; Hentschel, U.; et al. Comparative analysis of amplicon and metagenomic sequencing methods reveals key features in the evolution of animal metaorganisms. Microbiome 2019, 7, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, I.C.; Cairney, J.W. Diversity and ecology of soil fungal communities: Increased understanding through the application of molecular techniques. Environ. Microbiol. 2004, 6, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W. Fungal Barcoding Consortium. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [Green Version]
- Blaalid, R.; Kumar, S.; Nilsson, R.H.; Abarenkov, K.; Kirk, P.M.; Kauserud, H. ITS1 versus ITS2 as DNA metabarcodes for fungi. Mol. Ecol. Resour. 2013, 13, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, J.; Singh, K.; Fern, A.; Kirton, E.S.; He, S.; Woyke, T.; Lee, J.; Chen, F.; Dangl, J.L.; Tringe, S.G. Primer and platform effects on 16S rRNA tag sequencing. Front. Microbiol. 2015, 6, 771. [Google Scholar] [CrossRef] [Green Version]
- Bahram, M.; Anslan, S.; Hildebrand, F.; Bork, P.; Tedersoo, L. Newly designed 16S rRNA metabarcoding primers amplify diverse and novel archaeal taxa from the environment. Environ. Microbiol. Rep. 2019, 11, 487–494. [Google Scholar] [CrossRef]
- Shakya, M.; Lo, C.C.; Chain, P.S.G. Advances and Challenges in Metatranscriptomic Analysis. Front. Genet. 2019, 10, 904. [Google Scholar] [CrossRef] [Green Version]
- Escobar-Zepeda, A.; de Leon, A.V.-P.; Sanchez-Flores, A. The Road to Metagenomics: From Microbiology to DNA Sequencing Technologies and Bioinformatics. Front. Genet. 2015, 6, 348. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Handelsman, J. Metagenomics for studying unculturable microorganisms: Cutting the Gordian knot. Genome Biol. 2005, 6, 229. [Google Scholar] [CrossRef] [PubMed]
- Laudadio, I.; Fulci, V.; Palone, F.; Stronati, L.; Cucchiara, S.; Carissimi, C. Quantitative Assessment of Shotgun Metagenomics and 16S rDNA Amplicon Sequencing in the Study of Human Gut Microbiome. OMICS 2018, 22, 248–254. [Google Scholar] [CrossRef]
- Jovel, J.; Patterson, J.; Wang, W.; Hotte, N.; O’Keefe, S.; Mitchel, T.; Perry, T.; Kao, D.; Mason, A.L.; Madsen, K.L.; et al. Characterization of the Gut Microbiome Using 16S or Shotgun Metagenomics. Front. Microbiol. 2016, 7, 459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhang, Y.; Zhang, P.; Trivedi, P.; Riera, N.; Wang, Y.; Liu, X.; Fan, G.; Tang, J.; Coletta-Filho, H.D.; et al. The structure and function of the global citrus rhizosphere microbiome. Nat. Commun. 2018, 9, 4894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Marais, A.; Lefebvre, M.; Theil, S.; Svanella-Dumas, L.; Faure, C.; Candresse, T. Phytovirome Analysis of Wild Plant Populations: Comparison of Double-Stranded RNA and Virion-Associated Nucleic Acid Metagenomic Approaches. J. Virol. 2019, 94. [Google Scholar] [CrossRef]
- Keegan, K.P.; Glass, E.M.; Meyer, F. MG-RAST, a Metagenomics Service for Analysis of Microbial Community Structure and Function. Methods Mol. Biol. 2016, 1399, 207–233. [Google Scholar] [CrossRef] [PubMed]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Wemheuer, B.; Laffy, P.W.; Webster, N.S.; Thomas, T. Taxonomic, functional and expression analysis of viral communities associated with marine sponges. PeerJ 2021, 9, e10715. [Google Scholar] [CrossRef] [PubMed]
- Murali, A.; Bhargava, A.; Wright, E.S. IDTAXA: A novel approach for accurate taxonomic classification of microbiome sequences. Microbiome 2018, 6, 140. [Google Scholar] [CrossRef]
- Young, J.P.W.; Moeskjaer, S.; Afonin, A.; Rahi, P.; Maluk, M.; James, E.K.; Cavassim, M.I.A.; Rashid, M.H.; Aserse, A.A.; Perry, B.J.; et al. Defining the Rhizobium leguminosarum Species Complex. Genes 2021, 12, 111. [Google Scholar] [CrossRef]
- Balvociute, M.; Huson, D.H. SILVA, RDP, Greengenes, NCBI and OTT—How do these taxonomies compare? BMC Genom. 2017, 18, 114. [Google Scholar] [CrossRef] [Green Version]
- Xu, J. Fungal DNA barcoding. Genome 2016, 59, 913–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segata, N.; Waldron, L.; Ballarini, A.; Narasimhan, V.; Jousson, O.; Huttenhower, C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 2012, 9, 811–814. [Google Scholar] [CrossRef]
- Franzosa, E.A.; McIver, L.J.; Rahnavard, G.; Thompson, L.R.; Schirmer, M.; Weingart, G.; Lipson, K.S.; Knight, R.; Caporaso, J.G.; Segata, N.; et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 2018, 15, 962–968. [Google Scholar] [CrossRef]
- Liu, F.; Hewezi, T.; Lebeis, S.L.; Pantalone, V.; Grewal, P.S.; Staton, M.E. Soil indigenous microbiome and plant genotypes cooperatively modify soybean rhizosphere microbiome assembly. BMC Microbiol. 2019, 19, 201. [Google Scholar] [CrossRef]
- Murray, A.E.; Freudenstein, J.; Gribaldo, S.; Hatzenpichler, R.; Hugenholtz, P.; Kampfer, P.; Konstantinidis, K.T.; Lane, C.E.; Papke, R.T.; Parks, D.H.; et al. Roadmap for naming uncultivated Archaea and Bacteria. Nat. Microbiol. 2020, 5, 987–994. [Google Scholar] [CrossRef]
- Schulz, T.; Stoye, J.; Doerr, D. GraphTeams: A method for discovering spatial gene clusters in Hi-C sequencing data. BMC Genom. 2018, 19, 308. [Google Scholar] [CrossRef]
- Berendsen, R.L.; Pieterse, C.M.; Bakker, P.A. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef]
- Ciccazzo, S.; Esposito, A.; Rolli, E.; Zerbe, S.; Daffonchio, D.; Brusetti, L. Different pioneer plant species select specific rhizosphere bacterial communities in a high mountain environment. Springerplus 2014, 3, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Del Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoyo, G.; Moreno-Hagelsieb, G.; Mdel, C.O.-M.; Glick, B.R. Plant growth-promoting bacterial endophytes. Microbiol. Res. 2016, 183, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Turner, T.R.; James, E.K.; Poole, P.S. The plant microbiome. Genome Biol. 2013, 14, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K.; van Themaat, E.V.L.; Ahmadinejad, N.; Assenza, F.; Rauf, P.; Huettel, B.; Reinhardt, R.; Schmelzer, E.; et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef]
- Mendes, R.; Kruijt, M.; de Bruijn, I.; Dekkers, E.; van der Voort, M.; Schneider, J.H.; Piceno, Y.M.; DeSantis, T.Z.; Andersen, G.L.; Bakker, P.A.; et al. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 2011, 332, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Cao, L.; Zeng, J.; Franco, C.M.M.; Yang, Y.; Hu, X.; Liu, Y.; Wang, X.; Gao, Y.; Bu, Z.; et al. The antifungal action mode of the rice endophyte Streptomyces hygroscopicus OsiSh-2 as a potential biocontrol agent against the rice blast pathogen. Pestic. Biochem. Physiol. 2019, 160, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Teplitski, M.; Barak, J.D.; Schneider, K.R. Human enteric pathogens in produce: Un-answered ecological questions with direct implications for food safety. Curr. Opin. Biotechnol. 2009, 20, 166–171. [Google Scholar] [CrossRef]
- Marin, S.; Ramos, A.J.; Cano-Sancho, G.; Sanchis, V. Mycotoxins: Occurrence, toxicology, and exposure assessment. Food Chem. Toxicol. 2013, 60, 218–237. [Google Scholar] [CrossRef]
- Melotto, M.; Brandl, M.T.; Jacob, C.; Jay-Russell, M.T.; Micallef, S.A.; Warburton, M.L.; Van Deynze, A. Breeding Crops for Enhanced Food Safety. Front. Plant Sci. 2020, 11, 428. [Google Scholar] [CrossRef] [Green Version]
- Gorshkov, V.; Osipova, E.; Ponomareva, M.; Ponomarev, S.; Gogoleva, N.; Petrova, O.; Gogoleva, O.; Meshcherov, A.; Balkin, A.; Vetchinkina, E.; et al. Rye Snow Mold-Associated Microdochium nivale Strains Inhabiting a Common Area: Variability in Genetics, Morphotype, Extracellular Enzymatic Activities, and Virulence. J. Fungi 2020, 6, 335. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y.; Miller, S.A. Clinical metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leggett, R.M.; Clark, M.D. A world of opportunities with nanopore sequencing. J. Exp. Bot. 2017, 68, 5419–5429. [Google Scholar] [CrossRef] [PubMed]
- Rang, F.J.; Kloosterman, W.P.; de Ridder, J. From squiggle to basepair: Computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018, 19, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxford Nanopore Technologies. Nanopore Sequencing Accuracy. Available online: https://nanoporetech.com/accuracy (accessed on 10 July 2021).
- Sevim, V.; Lee, J.; Egan, R.; Clum, A.; Hundley, H.; Lee, J.; Everroad, R.C.; Detweiler, A.M.; Bebout, B.M.; Pett-Ridge, J.; et al. Shotgun metagenome data of a defined mock community using Oxford Nanopore, PacBio and Illumina technologies. Sci. Data 2019, 6, 285. [Google Scholar] [CrossRef] [Green Version]
- Jongman, M.; Carmichael, P.C.; Bill, M. Technological Advances in Phytopathogen Detection and Metagenome Profiling Techniques. Curr. Microbiol. 2020, 77, 675–681. [Google Scholar] [CrossRef]
- Llontop, M.E.M.; Sharma, P.; Flores, M.A.; Yang, S.; Pollock, J.; Tian, L.; Huang, C.; Rideout, S.; Heath, L.S.; Li, S.; et al. Strain-Level Identification of Bacterial Tomato Pathogens Directly from Metagenomic Sequences. Phytopathology 2020, 110, 768–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalupowicz, L.; Dombrovsky, A.; Gaba, V.; Luria, N.; Reuven, M.; Beerman, A.; Lachman, O.; Dror, O.; Nissan, G.; Manulis-Sasson, S. Diagnosis of plant diseases using the Nanopore sequencing platform. Plant Pathol. 2019, 68, 229–238. [Google Scholar] [CrossRef]
- Ciuffreda, L.; Rodriguez-Perez, H.; Flores, C. Nanopore sequencing and its application to the study of microbial communities. Comput. Struct. Biotechnol. J. 2021, 19, 1497–1511. [Google Scholar] [CrossRef]
- Schlaeppi, K.; Bulgarelli, D. The plant microbiome at work. Mol. Plant Microbe Interact. 2015, 28, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Bulgarelli, D.; Schlaeppi, K.; Spaepen, S.; van Themaat, E.V.L.; Schulze-Lefert, P. Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 2013, 64, 807–838. [Google Scholar] [CrossRef] [Green Version]
- Dangl, J.L.; Horvath, D.M.; Staskawicz, B.J. Pivoting the plant immune system from dissection to deployment. Science 2013, 341, 746–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Rani, B.; Sharma, V. Transcriptome profiling: Methods and applications—A review. Agric. Rev. 2017, 38, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Velculescu, V.E.; Zhang, L.; Vogelstein, B.; Kinzler, K.W. Serial analysis of gene expression. Science 1995, 270, 484–487. [Google Scholar] [CrossRef] [Green Version]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995, 270, 467–470. [Google Scholar] [CrossRef] [Green Version]
- Klepikova, A.V.; Logacheva, M.D.; Dmitriev, S.E.; Penin, A.A. RNA-seq analysis of an apical meristem time series reveals a critical point in Arabidopsis thaliana flower initiation. BMC Genom. 2015, 16, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorshkov, O.; Mokshina, N.; Gorshkov, V.; Chemikosova, S.; Gogolev, Y.; Gorshkova, T. Transcriptome portrait of cellulose-enriched flax fibres at advanced stage of specialization. Plant Mol. Biol. 2017, 93, 431–449. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Xu, Y.; Yu, C.; He, K.; Tang, Q.; Jia, C.; He, G.; Wang, X.; Kong, Y.; Zhou, G. Transcriptome analysis of genes involved in secondary cell wall biosynthesis in developing internodes of Miscanthus lutarioriparius. Sci. Rep. 2017, 7, 9034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlova, L.V.; Nazipova, A.R.; Gorshkov, O.V.; Petrova, A.A.; Gorshkova, T.A. Elongating maize root: Zone-specific combinations of polysaccharides from type I and type II primary cell walls. Sci. Rep. 2020, 10, 10956. [Google Scholar] [CrossRef]
- Malviya, M.K.; Li, C.N.; Solanki, M.K.; Singh, R.K.; Htun, R.; Singh, P.; Verma, K.K.; Yang, L.T.; Li, Y.R. Comparative analysis of sugarcane root transcriptome in response to the plant growth-promoting Burkholderia anthina MYSP113. PLoS ONE 2020, 15, e0231206. [Google Scholar] [CrossRef] [Green Version]
- Castandet, B.; Hotto, A.M.; Strickler, S.R.; Stern, D.B. ChloroSeq, an Optimized Chloroplast RNA-Seq Bioinformatic Pipeline, Reveals Remodeling of the Organellar Transcriptome Under Heat Stress. G3 Genes Genomes Genet. 2016, 6, 2817–2827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, B.; Qiu, X.; Huang, S.; Wang, X.; Zhang, X.; Dong, T.; Wang, T.; Li, S.; Sun, G.; Zhu, J.; et al. Physiological and transcriptome analyses of photosynthesis and chlorophyll metabolism in variegated Citrus (Shiranuhi and Huangguogan) seedlings. Sci. Rep. 2019, 9, 15670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanowski, A.; Schlaen, R.G.; Perez-Santangelo, S.; Mancini, E.; Yanovsky, M.J. Global transcriptome analysis reveals circadian control of splicing events in Arabidopsis thaliana. Plant J. 2020, 103, 889–902. [Google Scholar] [CrossRef]
- Li, Y.; Guo, G.; Zhou, L.; Chen, Y.; Zong, Y.; Huang, J.; Lu, R.; Liu, C. Transcriptome Analysis Identifies Candidate Genes and Functional Pathways Controlling the Response of Two Contrasting Barley Varieties to Powdery Mildew Infection. Int. J. Mol. Sci. 2019, 21, 151. [Google Scholar] [CrossRef] [Green Version]
- Tsers, I.; Gorshkov, V.; Gogoleva, N.; Parfirova, O.; Petrova, O.; Gogolev, Y. Plant Soft Rot Development and Regulation from the Viewpoint of Transcriptomic Profiling. Plants 2020, 9, 1176. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Duan, S.; Armstrong, M.R.; Xu, J.; Zheng, J.; Hu, J.; Chen, X.; Hein, I.; Li, G.; Jin, L. Comparative Transcriptome Profiling Reveals Compatible and Incompatible Patterns of Potato Toward Phytophthora infestans. G3 Genes Genomes Genet. 2020, 10, 623–634. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liu, Y.; Spetz, C.; Li, L.; Wang, X. Comparative transcriptome analysis in Triticum aestivum infecting wheat dwarf virus reveals the effects of viral infection on phytohormone and photosynthesis metabolism pathways. Phytopathol. Res. 2020, 2, 1–13. [Google Scholar] [CrossRef]
- Kang, W.H.; Sim, Y.M.; Koo, N.; Nam, J.Y.; Lee, J.; Kim, N.; Jang, H.; Kim, Y.M.; Yeom, S.I. Transcriptome profiling of abiotic responses to heat, cold, salt, and osmotic stress of Capsicum annuum L. Sci. Data 2020, 7, 17. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Zhao, X.; Chory, J. The Arabidopsis Transcriptome Responds Specifically and Dynamically to High Light Stress. Cell Rep. 2019, 29, 4186–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, D.; Zhang, Y.; Xiong, X.; Li, M.; Cai, K.; Luo, H.; Zeng, B. Transcriptome analysis on responses of orchardgrass (Dactylis glomerata L.) leaves to a short term flooding. Hereditas 2020, 157, 20. [Google Scholar] [CrossRef] [PubMed]
- Safavi-Rizi, V.; Herde, M.; Stohr, C. RNA-Seq reveals novel genes and pathways associated with hypoxia duration and tolerance in tomato root. Sci. Rep. 2020, 10, 1692. [Google Scholar] [CrossRef]
- Mokshina, N.; Gorshkov, O.; Galinousky, D.; Gorshkova, T. Genes with bast fiber-specific expression in flax plants—Molecular keys for targeted fiber crop improvement. Ind. Crop. Prod. 2020, 152. [Google Scholar] [CrossRef]
- Galinousky, D.; Mokshina, N.; Padvitski, T.; Ageeva, M.; Bogdan, V.; Kilchevsky, A.; Gorshkova, T. The Toolbox for Fiber Flax Breeding: A Pipeline From Gene Expression to Fiber Quality. Front. Genet. 2020, 11, 589881. [Google Scholar] [CrossRef]
- Brandt, R.; Mascher, M.; Thiel, J. Laser Capture Microdissection-Based RNA-Seq of Barley Grain Tissues. Methods Mol. Biol. 2018, 1723, 397–409. [Google Scholar] [CrossRef]
- Gorshkova, T.; Chernova, T.; Mokshina, N.; Gorshkov, V.; Kozlova, L.; Gorshkov, O. Transcriptome Analysis of Intrusively Growing Flax Fibers Isolated by Laser Microdissection. Sci. Rep. 2018, 8, 14570. [Google Scholar] [CrossRef]
- Shulse, C.N.; Cole, B.J.; Ciobanu, D.; Lin, J.; Yoshinaga, Y.; Gouran, M.; Turco, G.M.; Zhu, Y.; O’Malley, R.C.; Brady, S.M.; et al. High-Throughput Single-Cell Transcriptome Profiling of Plant Cell Types. Cell Rep. 2019, 27, 2241–2247. e2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, R.; Tian, X.; Xu, J. Single-Cell Transcriptome Analysis in Plants: Advances and Challenges. Mol. Plant 2021, 14, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, Z.; Wang, F.; Jia, W.; Xu, Z. Combined transcriptomic and metabolomic analyses uncover rearranged gene expression and metabolite metabolism in tobacco during cold acclimation. Sci. Rep. 2020, 10, 5242. [Google Scholar] [CrossRef]
- Gao, W.; Sun, H.X.; Xiao, H.; Cui, G.; Hillwig, M.L.; Jackson, A.; Wang, X.; Shen, Y.; Zhao, N.; Zhang, L.; et al. Combining metabolomics and transcriptomics to characterize tanshinone biosynthesis in Salvia miltiorrhiza. BMC Genom. 2014, 15, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Liu, P.; Fan, J.; Li, L. Comparative transcriptome analysis two genotypes of Acer truncatum Bunge seeds reveals candidate genes that influences seed VLCFAs accumulation. Sci. Rep. 2018, 8, 15504. [Google Scholar] [CrossRef]
- Murat, F.; Van de Peer, Y.; Salse, J. Decoding plant and animal genome plasticity from differential paleo-evolutionary patterns and processes. Genome Biol. Evol. 2012, 4, 917–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; McClain, C.J.; Rai, S.N. Fifteen Years of Gene Set Analysis for High-Throughput Genomic Data: A Review of Statistical Approaches and Future Challenges. Entropy 2020, 22, 427. [Google Scholar] [CrossRef] [Green Version]
- Hill, D.P.; Smith, B.; McAndrews-Hill, M.S.; Blake, J.A. Gene Ontology annotations: What they mean and where they come from. BMC Bioinform. 2008, 9, S2. [Google Scholar] [CrossRef] [Green Version]
- Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernandez-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwacke, R.; Ponce-Soto, G.Y.; Krause, K.; Bolger, A.M.; Arsova, B.; Hallab, A.; Gruden, K.; Stitt, M.; Bolger, M.E.; Usadel, B. MapMan4: A Refined Protein Classification and Annotation Framework Applicable to Multi-Omics Data Analysis. Mol. Plant 2019, 12, 879–892. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Thomas, P.D.; Kejariwal, A.; Campbell, M.J.; Mi, H.; Diemer, K.; Guo, N.; Ladunga, I.; Ulitsky-Lazareva, B.; Muruganujan, A.; Rabkin, S.; et al. PANTHER: A browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 2003, 31, 334–341. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018, 46, D633–D639. [Google Scholar] [CrossRef] [Green Version]
- Asp, M.; Bergenstråhle, J.; Lundeberg, J. Spatially resolved transcriptomes—Next generation tools for tissue exploration. BioEssays 2020, 42, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Moses, L.; Pachter, L. Museum of Spatial Transcriptomics. Available online: https://bookdown.org/lambdamoses/museumst/ (accessed on 1 April 2021).
- Kerk, N.M.; Ceserani, T.; Tausta, S.L.; Sussex, I.M.; Nelson, T.M. Laser capture microdissection of cells from plant tissues. Plant Physiol. 2003, 132, 27–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautam, V.; Singh, A.; Singh, S.; Sarkar, A.K. An Efficient LCM-Based Method for Tissue Specific Expression Analysis of Genes and miRNAs. Sci. Rep. 2016, 6, 21577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuper, H.; Amari, K.; Krenz, B. Analyzing the G3BP-like gene family of Arabidopsis thaliana in early turnip mosaic virus infection. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Nelson, T.; Tausta, S.L.; Gandotra, N.; Liu, T. Laser microdissection of plant tissue: What you see is what you get. Annu. Rev. Plant Biol. 2006, 57, 181–201. [Google Scholar] [CrossRef]
- Liew, L.C.; Wang, Y.; Peirats-Llobet, M.; Berkowitz, O.; Whelan, J.; Lewsey, M.G. Laser-Capture Microdissection RNA-sequencing for spatial and temporal tissue-specific gene expression analysis in plants. J. Vis. Exp. 2020, 162. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, M.; Uneyama, C.; Miyazaki, K.; Toyoda, K.; Hirose, M. Methacarn fixation: A novel tool for analysis of gene expressions in paraffin-embedded tissue specimens. Lab. Investig. 2000, 80, 199–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serova, T.A.; Tikhonovich, I.A.; Tsyganov, V.E. Analysis of nodule senescence in pea (Pisum sativum L.) using laser microdissection, real-time PCR, and ACC immunolocalization. J. Plant. Physiol. 2017, 212, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Schrader, J.; Nilsson, J.; Mellerowicz, E.; Berglund, A.; Nilsson, P.; Hertzberg, M.; Sandberg, G. A high-resolution transcript profile across the wood-forming meristem of poplar identifies potential regulators of cambial stem cell identity. Plant Cell 2004, 16, 2278–2292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Immanen, J.; Nieminen, K.; Smolander, O.-P.; Kojima, M.; Alonso Serra, J.; Koskinen, P.; Zhang, J.; Elo, A.; Mähönen, A.P.; Street, N.; et al. Cytokinin and auxin display distinct but interconnected distribution and signaling profiles to stimulate cambial activity. Curr. Biol. 2016, 26, 1990–1997. [Google Scholar] [CrossRef] [Green Version]
- Sundell, D.; Street, N.R.; Kumar, M.; Mellerowicz, E.J.; Kucukoglu, M.; Johnsson, C.; Kumar, V.; Mannapperuma, C.; Delhomme, N.; Nilsson, O.; et al. AspWood: High-spatial-resolution transcriptome profiles reveal uncharacterized modularity of wood formation in Populus tremula. Plant Cell 2017, 29, 1585–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angerer, L.M.; Angerer, R.C. Detection of poly A+ RNA in sea urchin eggs and embryos by quantitative in situ hybridization. Nucleic Acids Res. 1981, 9, 2819–2840. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, R.A.; Maslyar, D.J.; Heupel, R.C.; Harada, J.J. Spatial patterns of gene expression in Brassica napus seedlings: Identification of a cortex-specific gene and localization of mRNAs encoding isocitrate lyase and a polypeptide homologous to proteinases. Plant Cell 1989, 1, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Young, A.P.; Jackson, D.J.; Wyeth, R.C. A technical review and guide to RNA fluorescence in situ hybridization. PeerJ 2020, 8, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Singer, R.H.; Ward, D.C. Actin gene expression visualized in chicken muscle tissue culture by using in situ hybridization with a biotinated nucleotide analog. Proc. Natl. Acad. Sci. USA 1982, 79, 7331–7335. [Google Scholar] [CrossRef] [Green Version]
- Kitomi, Y.; Hanzawa, E.; Kuya, N.; Inoue, H.; Hara, N.; Kawai, S.; Kanno, N.; Endo, M.; Sugimoto, K.; Yamazaki, T.; et al. Root angle modifications by the DRO1 homolog improve rice yields in saline paddy fields. Proc. Natl. Acad. Sci. USA 2020, 117, 21242–21250. [Google Scholar] [CrossRef]
- Yang, W.; Cortijo, S.; Korsbo, N.; Roszak, P.; Schiessl, K.; Gurzadyan, A.; Wightman, R.; Jonsson, H.; Meyerowitz, E. Molecular mechanism of cytokinin-activated cell division in Arabidopsis. Science 2021, 371, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.; Rosa, S. Gaining insight into plant gene transcription using smFISH. Transcription 2018, 9, 166–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Femino, A.M.; Fay, F.S.; Fogarty, K.; Singer, R.H. Visualization of single RNA transcripts in situ. Science 1998, 280, 585–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, S.; Duncan, S.; Dean, C. Mutually exclusive sense-antisense transcription at FLC facilitates environmentally induced gene repression. Nat. Commun. 2016, 7, 13031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, S.; Olsson, T.S.G.; Hartley, M.; Dean, C.; Rosa, S. A method for detecting single mRNA molecules in Arabidopsis thaliana. Plant Methods 2016, 12, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Duncan, S.; Olsson, T.S.G.; Hartley, M.; Dean, C.; Rosa, S. Single molecule RNA FISH in Arabidopsis root cells. Bio Protocol 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Huang, K.; Demirci, F.; Batish, M.; Treible, W.; Meyers, B.C.; Caplan, J.L. Quantitative, super-resolution localization of small RNAs with sRNA-PAINT. Nucleic Acids Res. 2020, 48, 1–13. [Google Scholar] [CrossRef]
- Huang, K.; Batish, M.; Teng, C.; Harkess, A.; Meyers, B.C.; Caplan, J.L. Quantitative fluorescence in situ hybridization detection of plant mRNAs with single-molecule resolution. In RNA Tagging: Methods and Protocols; Heinlein, M., Ed.; Springer: New York, NY, USA, 2020; Volume 2166, pp. 23–33. [Google Scholar]
- Wang, F.; Flanagan, J.; Su, N.; Wang, L.-C.; Bui, S.; Nielson, A.; Wu, X.; Vo, H.-T.; Ma, X.-J.; Luo, Y. RNAscope: A novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 2012, 14, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Bowling, A.J.; Pence, H.E.; Church, J.B. Application of a novel and automated branched DNA in situ hybridization method for the rapid and sensitive localization of mRNA molecules in plant tissues. Appl. Plant Sci. 2014, 2, 1–5. [Google Scholar] [CrossRef]
- Bergua, M.; Phelan, D.M.; Bak, A.; Bloom, D.C.; Folimonova, S.Y. Simultaneous visualization of two Citrus tristeza virus genotypes provides new insights into the structure of multi-component virus populations in a host. Virology 2016, 491, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Munganyinka, E.; Margaria, P.; Sheat, S.; Ateka, E.M.; Tairo, F.; Ndunguru, J.; Winter, S. Localization of cassava brown streak virus in Nicotiana rustica and cassava Manihot esculenta (Crantz) using RNAscope® in situ hybridization. Virol. J. 2018, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sheat, S.; Winter, S.; Margaria, P. Duplex in situ hybridization of virus nucleic acids in plant tissues using RNAscope®. In In Situ Hybridization Protocols, 5th ed.; Nielsen, B.S., Jones, J., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2020; Volume 2148, pp. 203–215. [Google Scholar]
- Solanki, S.; Ameen, G.; Zhao, J.; Flaten, J.; Borowicz, P.; Brueggeman, R.S. Visualization of spatial gene expression in plants by modified RNAscope fluorescent in situ hybridization. Plant Methods 2020, 16, 1–9. [Google Scholar] [CrossRef]
- plaBiPD. Available online: https://www.plabipd.de/index.ep (accessed on 1 April 2021).
- Wang, K.N. (Ed.) Agrobacterium Protocols, 3rd ed.; Springer: New York, NY, USA, 2015; Volume 1–2, p. 365. [Google Scholar]
- Kumar, S.; Barone, P.; Smith, M. (Eds.) Transgenic Plants: Methods and Protocols; Springer: New York, NY, USA, 2020; Volume 1864, p. 438. [Google Scholar]
- Valla, S.; Lale, R. (Eds.) DNA Cloning and Assembly Methods; Springer: New York, NY, USA, 2014; Volume 1116, p. 308. [Google Scholar]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, V. Method of the year: Spatially resolved transcriptomics. Nat. Methods 2021, 18, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, S.; Salmén, F.; Terebieniec, B.K.; Vickovic, S.; Navarro, J.F.; Alexeyenko, A.; Reimegård, J.; McKee, L.S.; Mannapperuma, C.; Bulone, V.; et al. Spatially resolved transcriptome profiling in model plant species. Nat. Plants 2017, 3, 1–11. [Google Scholar] [CrossRef]
- Giacomello, S.; Lundeberg, J. Preparation of plant tissue to enable spatial transcriptomics profiling using barcoded microarrays. Nat. Protoc. 2018, 13, 2425–2446. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, K.; Shasha, D.E.; Wang, J.Y.; Jung, J.W.; Lambert, G.M.; Galbraith, D.W.; Benfey, P.N. A gene expression map of the Arabidopsis root. Science 2003, 302, 1956–1960. [Google Scholar] [CrossRef] [Green Version]
- Birnbaum, K.; Jung, J.W.; Wang, J.Y.; Lambert, G.M.; Hirst, J.A.; Galbraith, D.W.; Benfey, P.N. Cell type–specific expression profiling in plants via cell sorting of protoplasts from fluorescent reporter lines. Nat. Methods 2005, 2, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.M.; Orlando, D.A.; Lee, J.-Y.; Wang, J.Y.; Koch, J.; Dinneny, J.R.; Mace, D.; Ohler, U.; Benfey, P.N. A high-resolution root spatiotemporal map reveals dominant expression patterns. Science 2007, 318, 801–806. [Google Scholar] [CrossRef]
- Zanetti, M.E.; Chang, I.-F.; Gong, F.; Galbraith, D.W.; Bailey-Serres, J. Immunopurification of polyribosomal complexes of arabidopsis for global analysis of gene expression. Plant Physiol. 2005, 138, 624–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thellmann, M.; Andersen, T.G.; Vermeer, J.E.M. Translating Ribosome Affinity Purification (TRAP) to investigate Arabidopsis thaliana root development at a cell type-specific scale. J. Vis. Exp. 2020, 159, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Barthelson, R.A.; Lambert, G.M.; Galbraith, D.W. Global characterization of cell-specific gene expression through fluorescence-activated sorting of nuclei. Plant Physiol. 2008, 147, 30–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deal, R.B.; Henikoff, S. A simple method for gene expression and chromatin profiling of individual cell types within a tissue. Dev. Cell 2010, 18, 1030–1040. [Google Scholar] [CrossRef] [Green Version]
- Deal, R.B.; Henikoff, S. The INTACT method for cell type–specific gene expression and chromatin profiling in Arabidopsis thaliana. Nat. Protoc. 2011, 6, 56–68. [Google Scholar] [CrossRef]
- Palovaara, J.; Saiga, S.; Wendrich, J.R.; van‘t Wout Hofland, N.; van Schayck, J.P.; Hater, F.; Mutte, S.; Sjollema, J.; Boekschoten, M.; Hooiveld, G.J.; et al. Transcriptome dynamics revealed by a gene expression atlas of the early Arabidopsis embryo. Nat. Plants 2017, 3, 894–904. [Google Scholar] [CrossRef] [Green Version]
- Bobrovskikh, A.; Doroshkov, A.; Mazzoleni, S.; Carteni, F.; Giannino, F.; Zubairova, U. A Sight on Single-Cell Transcriptomics in Plants Through the Prism of Cell-Based Computational Modeling Approaches: Benefits and Challenges for Data Analysis. Front Genet 2021, 12, 652974. [Google Scholar] [CrossRef] [PubMed]
- Thibivilliers, S.; Libault, M. Plant single-cell multiomics: Cracking the molecular profiles of plant cells. Trends Plant Sci. 2021, 26, 662–663. [Google Scholar] [CrossRef] [PubMed]
- Seyfferth, C.; Renema, J.; Wendrich, J.R.; Eekhout, T.; Seurinck, R.; Vandamme, N.; Blob, B.; Saeys, Y.; Helariutta, Y.; Birnbaum, K.D.; et al. Advances and opportunities of single-cell transcriptomics for plant research. Annu. Rev. Plant Biol. 2021, 72, 1–20. [Google Scholar] [CrossRef]
- Brennecke, P.; Anders, S.; Kim, J.K.; Kołodziejczyk, A.A.; Zhang, X.; Proserpio, V.; Baying, B.; Benes, V.; Teichmann, S.A.; Marioni, J.C.; et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat. Methods 2013, 10, 1093–1095. [Google Scholar] [CrossRef]
- Efroni, I.; Ip, P.-L.; Nawy, T.; Mello, A.; Birnbaum, K.D. Quantification of cell identity from single-cell gene expression profiles. Genome Biol. 2015, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Efroni, I.; Mello, A.; Nawy, T.; Ip, P.-L.; Rahni, R.; DelRose, N.; Powers, A.; Satija, R.; Birnbaum, K.D. Root regeneration triggers an embryo-like sequence guided by hormonal interactions. Cell 2016, 165, 1721–1733. [Google Scholar] [CrossRef] [Green Version]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Wendrich, J.R.; Yang, B.; Vandamme, N.; Verstaen, K.; Smet, W.; Van de Velde, C.; Minne, M.; Wybouw, B.; Mor, E.; Arents, H.E.; et al. Vascular transcription factors guide plant epidermal responses to limiting phosphate conditions. Science 2020, 370, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Farmer, A.; Thibivilliers, S.; Ryu, K.H.; Schiefelbein, J.; Libault, M. Single-nucleus RNA and ATAC sequencing reveals the impact of chromatin accessibility on gene expression in Arabidopsis roots at the single-cell level. Mol. Plant 2021, 14, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Liu, Z.; Jia, J.; Mo, W.; Fang, L.; Lu, D.; Liu, B.; Zhang, H.; Chen, W.; Zhai, J. FlsnRNA-seq: Protoplasting-free full-length single-nucleus RNA profiling in plants. Genome Biol. 2021, 22, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dorrity, M.W.; Alexandre, C.M.; Hamm, M.; Vigil, A.-L.; Fields, S.; Queitsch, C.; Cuperus, J. The regulatory landscape of Arabidopsis thaliana roots at single-cell resolution. bioRxiv 2021. [Google Scholar] [CrossRef]
- Rich-Griffin, C.; Stechemesser, A.; Finch, J.; Lucas, E.; Ott, S.; Schäfer, P. Single-cell transcriptomics: A high-resolution avenue for plant functional genomics. Trends Plant Sci. 2020, 25, 186–197. [Google Scholar] [CrossRef]
- Valihrach, L.; Androvic, P.; Kubista, M. Platforms for single-cell collection and analysis. Int. J. Mol. Sci. 2018, 19, 807. [Google Scholar] [CrossRef] [Green Version]
- McGinnis, C.S.; Murrow, L.M.; Gartner, Z.J. DoubletFinder: Doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019, 8, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Wolock, S.L.; Lopez, R.; Klein, A.M. Scrublet: Computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 2019, 8, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DePasquale, E.A.K.; Schnell, D.J.; Van Camp, P.-J.; Valiente-Alandí, Í.; Blaxall, B.C.; Grimes, H.L.; Singh, H.; Salomonis, N. DoubletDecon: Deconvoluting doublets from single-cell RNA-sequencing data. Cell Rep. 2019, 29, 1718–1727. [Google Scholar] [CrossRef]
- DePasquale, E.A.K.; Schnell, D.; Chetal, K.; Salomonis, N. Protocol for identification and removal of doublets with DoubletDecon. STAR Protoc. 2020, 1, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Zappia, L.; Phipson, B.; Oshlack, A. Exploring the single-cell RNA-seq analysis landscape with the scRNA-tools database. PLoS Comput. Biol. 2018, 14, e1006245. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Denyer, T.; Timmermans, M.C.P. PscB: A browser to explore plant single cell RNA-sequencing data sets. Plant Physiol. 2020, 183, 464–467. [Google Scholar] [CrossRef] [Green Version]
- Baldrian, P. The known and the unknown in soil microbial ecology. FEMS Microbiol. Ecol. 2019, 95. [Google Scholar] [CrossRef]
- Zifcakova, L.; Vetrovsky, T.; Lombard, V.; Henrissat, B.; Howe, A.; Baldrian, P. Feed in summer, rest in winter: Microbial carbon utilization in forest topsoil. Microbiome 2017, 5, 122. [Google Scholar] [CrossRef] [Green Version]
- Damon, C.; Lehembre, F.; Oger-Desfeux, C.; Luis, P.; Ranger, J.; Fraissinet-Tachet, L.; Marmeisse, R. Metatranscriptomics reveals the diversity of genes expressed by eukaryotes in forest soils. PLoS ONE 2012, 7, e28967. [Google Scholar] [CrossRef]
- Geisen, S.; Tveit, A.T.; Clark, I.M.; Richter, A.; Svenning, M.M.; Bonkowski, M.; Urich, T. Metatranscriptomic census of active protists in soils. ISME J. 2015, 9, 2178–2190. [Google Scholar] [CrossRef]
- White, R.A., 3rd; Bottos, E.M.; Chowdhury, T.R.; Zucker, J.D.; Brislawn, C.J.; Nicora, C.D.; Fansler, S.J.; Glaesemann, K.R.; Glass, K.; Jansson, J.K. Moleculo Long-Read Sequencing Facilitates Assembly and Genomic Binning from Complex Soil Metagenomes. mSystems 2016, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, H.L.; Savin, K.W.; Wadeson, J.; Gupta, V.; Mele, P.M. Comparative Metatranscriptomics of Wheat Rhizosphere Microbiomes in Disease Suppressive and Non-suppressive Soils for Rhizoctonia solani AG8. Front. Microbiol. 2018, 9, 859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marti, J.M.; Arias-Giraldo, L.F.; Diaz-Villanueva, W.; Arnau, V.; Rodriguez-Franco, A.; Garay, C.P. Metatranscriptomic dynamics after Verticillium dahliae infection and root damage in Olea europaea. BMC Plant Biol. 2020, 20, 79. [Google Scholar] [CrossRef] [Green Version]
- Jo, Y.; Back, C.G.; Choi, H.; Cho, W.K. Comparative Microbiome Study of Mummified Peach Fruits by Metagenomics and Metatranscriptomics. Plants 2020, 9, 1052. [Google Scholar] [CrossRef] [PubMed]
- Westermann, A.J.; Gorski, S.A.; Vogel, J. Dual RNA-seq of pathogen and host. Nat. Rev. Microbiol. 2012, 10, 618–630. [Google Scholar] [CrossRef]
- Ettwiller, L.; Buswell, J.; Yigit, E.; Schildkraut, I. A novel enrichment strategy reveals unprecedented number of novel transcription start sites at single base resolution in a model prokaryote and the gut microbiome. BMC Genom. 2016, 17, 199. [Google Scholar] [CrossRef] [Green Version]
- Sharma, C.M.; Hoffmann, S.; Darfeuille, F.; Reignier, J.; Findeiss, S.; Sittka, A.; Chabas, S.; Reiche, K.; Hackermuller, J.; Reinhardt, R.; et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 2010, 464, 250–255. [Google Scholar] [CrossRef]
- Gorshkov, V.; Gubaev, R.; Petrova, O.; Daminova, A.; Gogoleva, N.; Ageeva, M.; Parfirova, O.; Prokchorchik, M.; Nikolaichik, Y.; Gogolev, Y. Transcriptome profiling helps to identify potential and true molecular switches of stealth to brute force behavior in Pectobacterium atrosepticum during systemic colonization of tobacco plants. Eur. J. Plant. Pathol. 2018, 152, 957–976. [Google Scholar] [CrossRef]
- Crump, B.C.; Wojahn, J.M.; Tomas, F.; Mueller, R.S. Metatranscriptomics and Amplicon Sequencing Reveal Mutualisms in Seagrass Microbiomes. Front. Microbiol. 2018, 9, 388. [Google Scholar] [CrossRef] [PubMed]
- Saminathan, T.; Garcia, M.; Ghimire, B.; Lopez, C.; Bodunrin, A.; Nimmakayala, P.; Abburi, V.L.; Levi, A.; Balagurusamy, N.; Reddy, U.K. Metagenomic and Metatranscriptomic Analyses of Diverse Watermelon Cultivars Reveal the Role of Fruit Associated Microbiome in Carbohydrate Metabolism and Ripening of Mature Fruits. Front. Plant Sci. 2018, 9, 4. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Cabrero, D.; Abugessaisa, I.; Maier, D.; Teschendorff, A.; Merkenschlager, M.; Gisel, A.; Ballestar, E.; Bongcam-Rudloff, E.; Conesa, A.; Tegner, J. Data integration in the era of omics: Current and future challenges. BMC Syst. Biol. 2014, 8, I1. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.Y.; Kim, H.U.; Lee, S.Y. Data integration and analysis of biological networks. Curr. Opin. Biotechnol. 2010, 21, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, A.; Kanaya, S.; Nishida, K. Integrated network analysis and effective tools in plant systems biology. Front. Plant Sci. 2014, 5, 598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Risueno, M.A.; Busch, W.; Benfey, P.N. Omics meet networks—Using systems approaches to infer regulatory networks in plants. Curr. Opin. Plant Biol. 2010, 13, 126–131. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Bai, Y. Analyzing LC/MS metabolic profiling data in the context of existing metabolic networks. Curr. Metab. 2013, 1, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Tagkopoulos, I. Data integration and predictive modeling methods for multi-omics datasets. Mol. Omics 2018, 14, 8–25. [Google Scholar] [CrossRef]
- Plomion, C.; Bastien, C.; Bogeat-Triboulot, M.-B.; Bouffier, L.; Déjardin, A.; Duplessis, S.; Fady, B.; Heuertz, M.; Le Gac, A.-L.; Provost, S.; et al. Forest tree genomics: 10 achievements from the past 10 years and future prospects. Ann. For. Sci. 2016, 73, 77–103. [Google Scholar] [CrossRef] [Green Version]
- Ballesta, P.; Maldonado, C.; Perez-Rodriguez, P.; Mora, F. SNP and Haplotype-Based Genomic Selection of Quantitative Traits in Eucalyptus globulus. Plants 2019, 8, 331. [Google Scholar] [CrossRef] [Green Version]
- Tsai, H.Y.; Cericola, F.; Edriss, V.; Andersen, J.R.; Orabi, J.; Jensen, J.D.; Jahoor, A.; Janss, L.; Jensen, J. Use of multiple traits genomic prediction, genotype by environment interactions and spatial effect to improve prediction accuracy in yield data. PLoS ONE 2020, 15, e0232665. [Google Scholar] [CrossRef]
- Fikere, M.; Barbulescu, D.M.; Malmberg, M.M.; Maharjan, P.; Salisbury, P.A.; Kant, S.; Panozzo, J.; Norton, S.; Spangenberg, G.C.; Cogan, N.O.I.; et al. Genomic Prediction and Genetic Correlation of Agronomic, Blackleg Disease, and Seed Quality Traits in Canola (Brassica napus L.). Plants 2020, 9, 719. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, C.; Mora-Poblete, F.; Contreras-Soto, R.I.; Ahmar, S.; Chen, J.T.; Junior, A.T.D.T.; Scapim, C.A. Genome-Wide Prediction of Complex Traits in Two Outcrossing Plant Species Through Deep Learning and Bayesian Regularized Neural Network. Front. Plant Sci. 2020, 11, 593897. [Google Scholar] [CrossRef]
- Haberer, G.; Kamal, N.; Bauer, E.; Gundlach, H.; Fischer, I.; Seidel, M.A.; Spannagl, M.; Marcon, C.; Ruban, A.; Urbany, C.; et al. European maize genomes highlight intraspecies variation in repeat and gene content. Nat. Genet. 2020, 52, 950–957. [Google Scholar] [CrossRef]
- Lopez-Cortes, X.A.; Matamala, F.; Maldonado, C.; Mora-Poblete, F.; Scapim, C.A. A Deep Learning Approach to Population Structure Inference in Inbred Lines of Maize. Front. Genet. 2020, 11, 543459. [Google Scholar] [CrossRef] [PubMed]
- Cappetta, E.; Andolfo, G.; Di Matteo, A.; Barone, A.; Frusciante, L.; Ercolano, M.R. Accelerating Tomato Breeding by Exploiting Genomic Selection Approaches. Plants 2020, 9, 1236. [Google Scholar] [CrossRef]
- Wang, X.; Gao, L.; Jiao, C.; Stravoravdis, S.; Hosmani, P.S.; Saha, S.; Zhang, J.; Mainiero, S.; Strickler, S.R.; Catala, C.; et al. Genome of Solanum pimpinellifolium provides insights into structural variants during tomato breeding. Nat. Commun. 2020, 11, 5817. [Google Scholar] [CrossRef]
- Sun, C.; Dong, Z.; Zhao, L.; Ren, Y.; Zhang, N.; Chen, F. The Wheat 660K SNP array demonstrates great potential for marker-assisted selection in polyploid wheat. Plant Biotechnol. J. 2020, 18, 1354–1360. [Google Scholar] [CrossRef]
- Babu, P.; Baranwal, D.K.; Harikrishna; Pal, D.; Bharti, H.; Joshi, P.; Thiyagarajan, B.; Gaikwad, K.B.; Bhardwaj, S.C.; Singh, G.P.; et al. Application of Genomics Tools in Wheat Breeding to Attain Durable Rust Resistance. Front. Plant Sci. 2020, 11, 567147. [Google Scholar] [CrossRef]
- Liu, C.; Song, J.; Wang, Y.; Huang, X.; Zhang, F.; Wang, W.; Xu, J.; Zhang, Y.; Yu, H.; Pang, Y.; et al. Rapid prediction of head rice yield and grain shape for genome-wide association study in indica rice. J. Cereal Sci. 2020, 96. [Google Scholar] [CrossRef]
- Morales, K.Y.; Singh, N.; Perez, F.A.; Ignacio, J.C.; Thapa, R.; Arbelaez, J.D.; Tabien, R.E.; Famoso, A.; Wang, D.R.; Septiningsih, E.M.; et al. An improved 7K SNP array, the C7AIR, provides a wealth of validated SNP markers for rice breeding and genetics studies. PLoS ONE 2020, 15, e0232479. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, C.; Mora, F.; Scapim, C.A.; Coan, M. Genome-wide haplotype-based association analysis of key traits of plant lodging and architecture of maize identifies major determinants for leaf angle: hapLA4. PLoS ONE 2019, 14, e0212925. [Google Scholar] [CrossRef] [Green Version]
- Mora-Poblete, F.; Ballesta, P.; Lobos, G.A.; Molina-Montenegro, M.; Gleadow, R.; Ahmar, S.; Jimenez-Aspee, F. Genome-wide association study of cyanogenic glycosides, proline, sugars, and pigments in Eucalyptus cladocalyx after 18 consecutive dry summers. Physiol. Plant. 2021. [Google Scholar] [CrossRef]
- Allier, A.; Teyssedre, S.; Lehermeier, C.; Moreau, L.; Charcosset, A. Optimized breeding strategies to harness genetic resources with different performance levels. BMC Genom. 2020, 21, 349. [Google Scholar] [CrossRef]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef]
- Daetwyler, H.D.; Calus, M.P.; Pong-Wong, R.; de Los Campos, G.; Hickey, J.M. Genomic prediction in animals and plants: Simulation of data, validation, reporting, and benchmarking. Genetics 2013, 193, 347–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supple, M.A.; Bragg, J.G.; Broadhurst, L.M.; Nicotra, A.B.; Byrne, M.; Andrew, R.L.; Widdup, A.; Aitken, N.C.; Borevitz, J.O. Landscape genomic prediction for restoration of a Eucalyptus foundation species under climate change. eLife 2018, 7. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression Shrinkage and Selection via the Lasso. J. R. Stat. Soc. Ser. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Habier, D.; Fernando, R.L.; Kizilkaya, K.; Garrick, D.J. Extension of the bayesian alphabet for genomic selection. BMC Bioinform. 2011, 12, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- Heslot, N.; Yang, H.-P.; Sorrells, M.E.; Jannik, J.-L. Genomic Selection in Plant Breeding: A Comparison of Models. Crop Breed. Genet. 2012, 52, 146–160. [Google Scholar] [CrossRef]
- Du, Q.; Lu, W.; Quan, M.; Xiao, L.; Song, F.; Li, P.; Zhou, D.; Xie, J.; Wang, L.; Zhang, D. Genome-Wide Association Studies to Improve Wood Properties: Challenges and Prospects. Front. Plant Sci. 2018, 9, 1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Su, G.; Janss, L.; Zhang, Y.; Lund, M.S. Model comparison on genomic predictions using high-density markers for different groups of bulls in the Nordic Holstein population. J. Dairy Sci. 2013, 96, 4678–4687. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Lund, M.S.; Sun, D.; Zhang, Q.; Su, G. Impact of relationships between test and training animals and among training animals on reliability of genomic prediction. J. Anim. Breed. Genet. 2015, 132, 366–375. [Google Scholar] [CrossRef]
- Rutkoski, J.; Poland, J.; Mondal, S.; Autrique, E.; Perez, L.G.; Crossa, J.; Reynolds, M.; Singh, R. Canopy Temperature and Vegetation Indices from High-Throughput Phenotyping Improve Accuracy of Pedigree and Genomic Selection for Grain Yield in Wheat. G3 Genes Genomes Genet. 2016, 6, 2799–2808. [Google Scholar] [CrossRef] [Green Version]
- Crain, J.; Mondal, S.; Rutkoski, J.; Singh, R.P.; Poland, J. Combining High-Throughput Phenotyping and Genomic Information to Increase Prediction and Selection Accuracy in Wheat Breeding. Plant Genome 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrera-Bosquet, L.; Crossa, J.; von Zitzewitz, J.; Serret, M.D.; Araus, J.L. High-throughput phenotyping and genomic selection: The frontiers of crop breeding converge. J. Integr. Plant Biol. 2012, 54, 312–320. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.; Wang, X.; Kumar, U.; Gao, L.; Noor, M.; Imtiaz, M.; Singh, R.P.; Poland, J. High-Throughput Phenotyping Enabled Genetic Dissection of Crop Lodging in Wheat. Front. Plant Sci. 2019, 10, 394. [Google Scholar] [CrossRef] [Green Version]
- Mackay, I.; Ober, E.; Hickey, J. GplusE: Beyond genomic selection. Food Energy Secur. 2015, 4, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Rutkoski, J.E.; Poland, J.A.; Crossa, J.; Jannink, J.L.; Sorrells, M.E. Multitrait, Random Regression, or Simple Repeatability Model in High-Throughput Phenotyping Data Improve Genomic Prediction for Wheat Grain Yield. Plant Genome 2017, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leng, P.; Lubberstedt, T.; Xu, M.L. Genomics-assisted breeding—A revolutionary strategy for crop improvement. J. Integr. Agric. 2017, 16, 2674–2685. [Google Scholar] [CrossRef]
- Feng, H.; Guo, Z.; Yang, W.; Huang, C.; Chen, G.; Fang, W.; Xiong, X.; Zhang, H.; Wang, G.; Xiong, L.; et al. An integrated hyperspectral imaging and genome-wide association analysis platform provides spectral and genetic insights into the natural variation in rice. Sci. Rep. 2017, 7, 4401. [Google Scholar] [CrossRef] [Green Version]
- Rincent, R.; Charpentier, J.P.; Faivre-Rampant, P.; Paux, E.; Le Gouis, J.; Bastien, C.; Segura, V. Phenomic Selection Is a Low-Cost and High-Throughput Method Based on Indirect Predictions: Proof of Concept on Wheat and Poplar. G3 Genes Genomes Genet. 2018, 8, 3961–3972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, M.R.; Gonzalez-Perez, L.; Crossa, J.; Perez-Rodriguez, P.; Montesinos-Lopez, O.; Singh, R.P.; Dreisigacker, S.; Poland, J.; Rutkoski, J.; Sorrells, M.; et al. Hyperspectral Reflectance-Derived Relationship Matrices for Genomic Prediction of Grain Yield in Wheat. G3 Genes Genomes Genet. 2019, 9, 1231–1247. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, M.; Paszkowski, J. Epigenetic memory in plants. EMBO J. 2014, 33, 1987–1998. [Google Scholar] [CrossRef] [Green Version]
- Roudier, F.; Ahmed, I.; Berard, C.; Sarazin, A.; Mary-Huard, T.; Cortijo, S.; Bouyer, D.; Caillieux, E.; Duvernois-Berthet, E.; Al-Shikhley, L.; et al. Integrative epigenomic mapping defines four main chromatin states in Arabidopsis. EMBO J. 2011, 30, 1928–1938. [Google Scholar] [CrossRef] [Green Version]
- Rigal, M.; Mathieu, O. A “mille-feuille” of silencing: Epigenetic control of transposable elements. Biochim. Biophys. Acta 2011, 1809, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Haag, J.R.; Pikaard, C.S. Multisubunit RNA polymerases IV and V: Purveyors of non-coding RNA for plant gene silencing. Nat. Rev. Mol. Cell Biol. 2011, 12, 483–492. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Stroud, H.; Greenberg, M.V.; Feng, S.; Bernatavichute, Y.V.; Jacobsen, S.E. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 2013, 152, 352–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014, 21, 64–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebbs, M.L.; Bender, J. Locus-specific control of DNA methylation by the Arabidopsis SUVH5 histone methyltransferase. Plant Cell 2006, 18, 1166–1176. [Google Scholar] [CrossRef] [Green Version]
- Kakutani, T.; Jeddeloh, J.A.; Richards, E.J. Characterization of an Arabidopsis thaliana DNA hypomethylation mutant. Nucleic Acids Res. 1995, 23, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Zemach, A.; Kim, M.Y.; Hsieh, P.H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Yadav, N.S.; Morgenstern, Y.; Zemach, A.; Grafi, G. Activation of Tag1 transposable elements in Arabidopsis dedifferentiating cells and their regulation by CHROMOMETHYLASE 3-mediated CHG methylation. Biochim. Biophys. Acta 2016, 1859, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.S.; Khadka, J.; Domb, K.; Zemach, A.; Grafi, G. CMT3 and SUVH4/KYP silence the exonic Evelknievel retroelement to allow for reconstitution of CMT1 mRNA. Epigenetics Chromatin 2018, 11, 69. [Google Scholar] [CrossRef] [Green Version]
- Gehring, M.; Henikoff, S. DNA methylation and demethylation in Arabidopsis. Arab. Book 2008, 6, e0102. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Lee, U.S.; Wagner, D. Tug of war: Adding and removing histone lysine methylation in Arabidopsis. Curr. Opin. Plant Biol. 2016, 34, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Lu, F.; Cui, X.; Cao, X. Histone methylation in higher plants. Annu. Rev. Plant Biol. 2010, 61, 395–420. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, J.P.; Johnson, L.; Jasencakova, Z.; Zhang, X.; PerezBurgos, L.; Singh, P.B.; Cheng, X.; Schubert, I.; Jenuwein, T.; Jacobsen, S.E. Dimethylation of histone H3 lysine 9 is a critical mark for DNA methylation and gene silencing in Arabidopsis thaliana. Chromosoma 2004, 112, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Du, X.; Tang, K.; Yang, Z.; Pan, L.; Zhu, P.; Luo, J.; Jiang, Y.; Zhang, H.; Wan, H.; et al. Arabidopsis AGDP1 links H3K9me2 to DNA methylation in heterochromatin. Nat. Commun. 2018, 9, 4547. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sung, S. Polycomb-mediated gene silencing in Arabidopsis thaliana. Mol. Cells 2014, 37, 841–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Hu, Y.; Zhou, D.X. Epigenetic gene regulation by plant Jumonji group of histone demethylase. Biochim. Biophys. Acta 2011, 1809, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.S.; Titov, V.; Ayemere, I.; Byeon, B.; Ilnytskyy, Y.; Kovalchuk, I. Multigenerational exposure to heat stress induces phenotypic resilience, and genetic and epigenetic variations in Arabidopsis thaliana offspring. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wong, M.M.; Chong, G.L.; Verslues, P.E. Epigenetics and RNA Processing: Connections to Drought, Salt, and ABA? Methods Mol. Biol. 2017, 1631, 3–21. [Google Scholar] [CrossRef]
- Wang, G.; Li, H.; Meng, S.; Yang, J.; Ye, N.; Zhang, J. Analysis of Global Methylome and Gene Expression during Carbon Reserve Mobilization in Stems under Soil Drying. Plant Physiol. 2020, 183, 1809–1824. [Google Scholar] [CrossRef]
- Al-Harrasi, I.; Al-Yahyai, R.; Yaish, M.W. Differential DNA methylation and transcription profiles in date palm roots exposed to salinity. PLoS ONE 2018, 13, e0191492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaish, M.W.; Al-Lawati, A.; Al-Harrasi, I.; Patankar, H.V. Genome-wide DNA Methylation analysis in response to salinity in the model plant caliph medic (Medicago truncatula). BMC Genom. 2018, 19, 78. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Min, L.; Wang, M.; Wang, C.; Zhao, Y.; Li, Y.; Fang, Q.; Wu, Y.; Xie, S.; Ding, Y.; et al. Disrupted Genome Methylation in Response to High Temperature Has Distinct Affects on Microspore Abortion and Anther Indehiscence. Plant Cell 2018, 30, 1387–1403. [Google Scholar] [CrossRef] [Green Version]
- Hossain, M.S.; Kawakatsu, T.; Kim, K.D.; Zhang, N.; Nguyen, C.T.; Khan, S.M.; Batek, J.M.; Joshi, T.; Schmutz, J.; Grimwood, J.; et al. Divergent cytosine DNA methylation patterns in single-cell, soybean root hairs. New Phytol. 2017, 214, 808–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Huang, Q.; Sun, M.; Zhang, T.; Li, H.; Chen, B.; Xu, K.; Gao, G.; Li, F.; Yan, G.; et al. Global DNA methylation variations after short-term heat shock treatment in cultured microspores of Brassica napus cv. Topas. Sci. Rep. 2016, 6, 38401. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Li, J.; Li, H.; Li, F.; Xu, K.; Yan, G.; Chen, B.; Qiao, J.; Wu, X. Comparison of the heat stress induced variations in DNA methylation between heat-tolerant and heat-sensitive rapeseed seedlings. Breed. Sci. 2014, 64, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villagomez-Aranda, A.L.; Garcia-Ortega, L.F.; Torres-Pacheco, I.; Guevara-Gonzalez, R.G. Whole-Genome DNA Methylation Analysis in Hydrogen Peroxide Overproducing Transgenic Tobacco Resistant to Biotic and Abiotic Stresses. Plants 2021, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhou, S.; Gong, X.; Song, Y.; van Nocker, S.; Ma, F.; Guan, Q. Single-base methylome analysis reveals dynamic epigenomic differences associated with water deficit in apple. Plant. Biotechnol. J. 2018, 16, 672–687. [Google Scholar] [CrossRef]
- Rajkumar, M.S.; Shankar, R.; Garg, R.; Jain, M. Bisulphite sequencing reveals dynamic DNA methylation under desiccation and salinity stresses in rice cultivars. Genomics 2020, 112, 3537–3548. [Google Scholar] [CrossRef]
- Li, R.; Hu, F.; Li, B.; Zhang, Y.; Chen, M.; Fan, T.; Wang, T. Whole genome bisulfite sequencing methylome analysis of mulberry (Morus alba) reveals epigenome modifications in response to drought stress. Sci. Rep. 2020, 10, 8013. [Google Scholar] [CrossRef]
- Qian, Y.; Hu, W.; Liao, J.; Zhang, J.; Ren, Q. The Dynamics of DNA methylation in the maize (Zea mays L.) inbred line B73 response to heat stress at the seedling stage. Biochem. Biophys. Res. Commun. 2019, 512, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Miao, X.; Cui, J.; Deng, J.; Wang, X.; Wang, Y.; Zhang, Y.; Gao, S.; Yang, K. Genome-wide high-resolution mapping of DNA methylation identifies epigenetic variation across different salt stress in Maize (Zea mays L.). Euphytica 2018, 214. [Google Scholar] [CrossRef]
- An, Y.C.; Goettel, W.; Han, Q.; Bartels, A.; Liu, Z.; Xiao, W. Dynamic Changes of Genome-Wide DNA Methylation during Soybean Seed Development. Sci. Rep. 2017, 7, 12263. [Google Scholar] [CrossRef] [PubMed]
- Atighi, M.R.; Verstraeten, B.; De Meyer, T.; Kyndt, T. Genome-wide DNA hypomethylation shapes nematode pattern-triggered immunity in plants. New Phytol. 2020, 227, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Lamke, J.; Baurle, I. Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol. 2017, 18, 124. [Google Scholar] [CrossRef]
- Boyko, A.; Kovalchuk, I. Transgenerational response to stress in Arabidopsis thaliana. Plant Signal. Behav. 2010, 5, 995–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suter, L.; Widmer, A. Phenotypic effects of salt and heat stress over three generations in Arabidopsis thaliana. PLoS ONE 2013, 8, e80819. [Google Scholar] [CrossRef]
- Ramirez-Carrasco, G.; Martinez-Aguilar, K.; Alvarez-Venegas, R. Transgenerational Defense Priming for Crop Protection against Plant Pathogens: A Hypothesis. Front. Plant Sci. 2017, 8, 696. [Google Scholar] [CrossRef] [Green Version]
- Wibowo, A.; Becker, C.; Marconi, G.; Durr, J.; Price, J.; Hagmann, J.; Papareddy, R.; Putra, H.; Kageyama, J.; Becker, J.; et al. Hyperosmotic stress memory in Arabidopsis is mediated by distinct epigenetically labile sites in the genome and is restricted in the male germline by DNA glycosylase activity. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Chen, L.; Xia, H.; Wei, H.; Lou, Q.; Li, M.; Li, T.; Luo, L. Transgenerational epimutations induced by multi-generation drought imposition mediate rice plant’s adaptation to drought condition. Sci. Rep. 2017, 7, 39843. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Zhang, Y.; Xu, C.; Lin, X.; Zang, Q.; Zhuang, T.; Jiang, L.; von Wettstein, D.; Liu, B. Transgenerational inheritance of modified DNA methylation patterns and enhanced tolerance induced by heavy metal stress in rice (Oryza sativa L.). PLoS ONE 2012, 7, e41143. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.M.; Sasaki, T.; Ueda, M.; Sako, K.; Seki, M. Chromatin changes in response to drought, salinity, heat, and cold stresses in plants. Front. Plant Sci. 2015, 6, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dijk, K.; Ding, Y.; Malkaram, S.; Riethoven, J.J.; Liu, R.; Yang, J.; Laczko, P.; Chen, H.; Xia, Y.; Ladunga, I.; et al. Dynamic changes in genome-wide histone H3 lysine 4 methylation patterns in response to dehydration stress in Arabidopsis thaliana. BMC Plant Biol. 2010, 10, 238. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Fan, G.; Li, X. Genome-wide analysis of three histone marks and gene expression in Paulownia fortunei with phytoplasma infection. BMC Genom. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Zhai, X.; Zhao, Z.; Fan, G. Whole-genome landscape of H3K4me3, H3K36me3 and H3K9ac and their association with gene expression during Paulownia witches’ broom disease infection and recovery processes. 3 Biotech 2020, 10, 336. [Google Scholar] [CrossRef]
- Hussey, S.G.; Loots, M.T.; van der Merwe, K.; Mizrachi, E.; Myburg, A.A. Integrated analysis and transcript abundance modelling of H3K4me3 and H3K27me3 in developing secondary xylem. Sci. Rep. 2017, 7, 3370. [Google Scholar] [CrossRef]
- Zeng, Z.; Zhang, W.; Marand, A.P.; Zhu, B.; Buell, C.R.; Jiang, J. Cold stress induces enhanced chromatin accessibility and bivalent histone modifications H3K4me3 and H3K27me3 of active genes in potato. Genome Biol. 2019, 20, 123. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liang, Y.; Dong, Y.; Gao, Y.; Yang, X.; Yuan, J.; Qiu, D. The Magnaporthe oryzae Alt A 1-like protein MoHrip1 binds to the plant plasma membrane. Biochem. Biophys. Res. Commun. 2017, 492, 55–60. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, G.; Liu, X.; Ding, X.; Zhang, D.; Wang, X.; Zhou, Y.; Yan, H.; Li, T.; Wu, K.; et al. Histone demethylase SlJMJ6 promotes fruit ripening by removing H3K27 methylation of ripening-related genes in tomato. New Phytol. 2020, 227, 1138–1156. [Google Scholar] [CrossRef]
- Liu, B.; Wendel, J.F. Epigenetic phenomena and the evolution of plant allopolyploids. Mol. Phylogenet Evol. 2003, 29, 365–379. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Fischer, M.; Colot, V.; Bossdorf, O. Epigenetic variation creates potential for evolution of plant phenotypic plasticity. New Phytol. 2013, 197, 314–322. [Google Scholar] [CrossRef]
- Varotto, S.; Tani, E.; Abraham, E.; Krugman, T.; Kapazoglou, A.; Melzer, R.; Radanovic, A.; Miladinovic, D. Epigenetics: Possible applications in climate-smart crop breeding. J. Exp. Bot. 2020, 71, 5223–5236. [Google Scholar] [CrossRef]
- Yang, X.; Kundariya, H.; Xu, Y.Z.; Sandhu, A.; Yu, J.; Hutton, S.F.; Zhang, M.; Mackenzie, S.A. MutS HOMOLOG1-derived epigenetic breeding potential in tomato. Plant Physiol. 2015, 168, 222–232. [Google Scholar] [CrossRef] [Green Version]
- Raju, S.K.K.; Shao, M.R.; Sanchez, R.; Xu, Y.Z.; Sandhu, A.; Graef, G.; Mackenzie, S. An epigenetic breeding system in soybean for increased yield and stability. Plant Biotechnol. J. 2018, 16, 1836–1847. [Google Scholar] [CrossRef] [Green Version]
- Hauben, M.; Haesendonckx, B.; Standaert, E.; Van Der Kelen, K.; Azmi, A.; Akpo, H.; Van Breusegem, F.; Guisez, Y.; Bots, M.; Lambert, B.; et al. Energy use efficiency is characterized by an epigenetic component that can be directed through artificial selection to increase yield. Proc. Natl. Acad. Sci. USA 2009, 106, 20109–20114. [Google Scholar] [CrossRef] [Green Version]
- Greaves, I.K.; Groszmann, M.; Wang, A.; Peacock, W.J.; Dennis, E.S. Inheritance of Trans Chromosomal Methylation patterns from Arabidopsis F1 hybrids. Proc. Natl. Acad. Sci. USA 2014, 111, 2017–2022. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Greaves, I.K.; Groszmann, M.; Wu, L.M.; Dennis, E.S.; Peacock, W.J. Hybrid mimics and hybrid vigor in Arabidopsis. Proc. Natl. Acad. Sci. USA 2015, 112, E4959–E4967. [Google Scholar] [CrossRef] [Green Version]
- Jonas, E.; de Koning, D.J. Does genomic selection have a future in plant breeding? Trends Biotechnol. 2013, 31, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Oakey, H.; Cullis, B.; Thompson, R.; Comadran, J.; Halpin, C.; Waugh, R. Genomic Selection in Multi-environment Crop Trials. G3 Genes Genomes Genet. 2016, 6, 1313–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannes, F.; Porcher, E.; Teixeira, F.K.; Saliba-Colombani, V.; Simon, M.; Agier, N.; Bulski, A.; Albuisson, J.; Heredia, F.; Audigier, P.; et al. Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet. 2009, 5, e1000530. [Google Scholar] [CrossRef]
- Reinders, J.; Wulff, B.B.; Mirouze, M.; Mari-Ordonez, A.; Dapp, M.; Rozhon, W.; Bucher, E.; Theiler, G.; Paszkowski, J. Compromised stability of DNA methylation and transposon immobilization in mosaic Arabidopsis epigenomes. Genes Dev. 2009, 23, 939–950. [Google Scholar] [CrossRef] [Green Version]
- Roux, F.; Colome-Tatche, M.; Edelist, C.; Wardenaar, R.; Guerche, P.; Hospital, F.; Colot, V.; Jansen, R.C.; Johannes, F. Genome-wide epigenetic perturbation jump-starts patterns of heritable variation found in nature. Genetics 2011, 188, 1015–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortijo, S.; Wardenaar, R.; Colome-Tatche, M.; Gilly, A.; Etcheverry, M.; Labadie, K.; Caillieux, E.; Hospital, F.; Aury, J.M.; Wincker, P.; et al. Mapping the epigenetic basis of complex traits. Science 2014, 343, 1145–1148. [Google Scholar] [CrossRef]
- Bond, D.M.; Baulcombe, D.C. Small RNAs and heritable epigenetic variation in plants. Trends Cell Biol. 2014, 24, 100–107. [Google Scholar] [CrossRef]
- Latzel, V.; Allan, E.; Bortolini Silveira, A.; Colot, V.; Fischer, M.; Bossdorf, O. Epigenetic diversity increases the productivity and stability of plant populations. Nat. Commun. 2013, 4, 2875. [Google Scholar] [CrossRef]
- Abdelnoor, R.V.; Yule, R.; Elo, A.; Christensen, A.C.; Meyer-Gauen, G.; Mackenzie, S.A. Substoichiometric shifting in the plant mitochondrial genome is influenced by a gene homologous to MutS. Proc. Natl. Acad. Sci. USA 2003, 100, 5968–5973. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.Z.; Arrieta-Montiel, M.P.; Virdi, K.S.; de Paula, W.B.; Widhalm, J.R.; Basset, G.J.; Davila, J.I.; Elthon, T.E.; Elowsky, C.G.; Sato, S.J.; et al. MutS HOMOLOG1 is a nucleoid protein that alters mitochondrial and plastid properties and plant response to high light. Plant Cell 2011, 23, 3428–3441. [Google Scholar] [CrossRef] [Green Version]
- Shedge, V.; Davila, J.; Arrieta-Montiel, M.P.; Mohammed, S.; Mackenzie, S.A. Extensive rearrangement of the Arabidopsis mitochondrial genome elicits cellular conditions for thermotolerance. Plant Physiol. 2010, 152, 1960–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.Z.; Rde, L.S.; Virdi, K.S.; Arrieta-Montiel, M.P.; Razvi, F.; Li, S.; Ren, G.; Yu, B.; Alexander, D.; Guo, L.; et al. The chloroplast triggers developmental reprogramming when mutS HOMOLOG1 is suppressed in plants. Plant Physiol. 2012, 159, 710–720. [Google Scholar] [CrossRef] [Green Version]
- Virdi, K.S.; Wamboldt, Y.; Kundariya, H.; Laurie, J.D.; Keren, I.; Kumar, K.R.S.; Block, A.; Basset, G.; Luebker, S.; Elowsky, C.; et al. MSH1 Is a Plant Organellar DNA Binding and Thylakoid Protein under Precise Spatial Regulation to Alter Development. Mol. Plant 2016, 9, 245–260. [Google Scholar] [CrossRef] [Green Version]
- Kalisz, S.; Kramer, E.M. Variation and constraint in plant evolution and development. Heredity 2008, 100, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Virdi, K.S.; Laurie, J.D.; Xu, Y.Z.; Yu, J.; Shao, M.R.; Sanchez, R.; Kundariya, H.; Wang, D.; Riethoven, J.J.; Wamboldt, Y.; et al. Arabidopsis MSH1 mutation alters the epigenome and produces heritable changes in plant growth. Nat. Commun. 2015, 6, 6386. [Google Scholar] [CrossRef] [Green Version]
- Shao, M.R.; Raju, S.K.K.; Laurie, J.D.; Sanchez, R.; Mackenzie, S.A. Stress-responsive pathways and small RNA changes distinguish variable developmental phenotypes caused by MSH1 loss. BMC Plant Biol. 2017, 17, 47. [Google Scholar] [CrossRef] [Green Version]
- de la Rosa Santamaria, R.; Shao, M.R.; Wang, G.; Nino-Liu, D.O.; Kundariya, H.; Wamboldt, Y.; Dweikat, I.; Mackenzie, S.A. MSH1-induced non-genetic variation provides a source of phenotypic diversity in Sorghum bicolor. PLoS ONE 2014, 9, e108407. [Google Scholar] [CrossRef]
- Manning, K.; Tor, M.; Poole, M.; Hong, Y.; Thompson, A.J.; King, G.J.; Giovannoni, J.J.; Seymour, G.B. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 2006, 38, 948–952. [Google Scholar] [CrossRef]
- Bilichak, A.; Kovalchuk, I. Transgenerational response to stress in plants and its application for breeding. J. Exp. Bot. 2016, 67, 2081–2092. [Google Scholar] [CrossRef]
- Johnson, L.M.; Du, J.; Hale, C.J.; Bischof, S.; Feng, S.; Chodavarapu, R.K.; Zhong, X.; Marson, G.; Pellegrini, M.; Segal, D.J.; et al. SRA- and SET-domain-containing proteins link RNA polymerase V occupancy to DNA methylation. Nature 2014, 507, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallego-Bartolome, J.; Gardiner, J.; Liu, W.; Papikian, A.; Ghoshal, B.; Kuo, H.Y.; Zhao, J.M.; Segal, D.J.; Jacobsen, S.E. Targeted DNA demethylation of the Arabidopsis genome using the human TET1 catalytic domain. Proc. Natl. Acad. Sci. USA 2018, 115, E2125–E2134. [Google Scholar] [CrossRef] [Green Version]
- Vojta, A.; Dobrinic, P.; Tadic, V.; Bockor, L.; Korac, P.; Julg, B.; Klasic, M.; Zoldos, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, T.; Meister, G.E.; Workman, R.E.; Kato, N.C.; Spellberg, M.J.; Turker, F.; Timp, W.; Ostermeier, M.; Novina, C.D. Targeted DNA methylation in human cells using engineered dCas9-methyltransferases. Sci. Rep. 2017, 7, 6732. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.I.; Celik, H.; Rois, L.E.; Fishberger, G.; Fowler, T.; Rees, R.; Kramer, A.; Martens, A.; Edwards, J.R.; Challen, G.A. Reprogrammable CRISPR/Cas9-based system for inducing site-specific DNA methylation. Biol. Open 2016, 5, 866–874. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Tao, Y.; Gao, X.; Zhang, L.; Li, X.; Zou, W.; Ruan, K.; Wang, F.; Xu, G.L.; Hu, R. A CRISPR-based approach for targeted DNA demethylation. Cell Discov. 2016, 2, 16009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, S.R.; Cui, Y.; Lubecka, K.; Stefanska, B.; Irudayaraj, J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget 2016, 7, 46545–46556. [Google Scholar] [CrossRef] [Green Version]
- Papikian, A.; Liu, W.; Gallego-Bartolome, J.; Jacobsen, S.E. Site-specific manipulation of Arabidopsis loci using CRISPR-Cas9 SunTag systems. Nat. Commun. 2019, 10, 729. [Google Scholar] [CrossRef]
- Nunez, J.K.; Chen, J.; Pommier, G.C.; Cogan, J.Z.; Replogle, J.M.; Adriaens, C.; Ramadoss, G.N.; Shi, Q.; Hung, K.L.; Samelson, A.J.; et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell 2021, 184, 2503–2519. e2517. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Qi, L.S. A CRISPR-dCas Toolbox for Genetic Engineering and Synthetic Biology. J. Mol. Biol. 2019, 431, 34–47. [Google Scholar] [CrossRef]
- Quenneville, S.; Turelli, P.; Bojkowska, K.; Raclot, C.; Offner, S.; Kapopoulou, A.; Trono, D. The KRAB-ZFP/KAP1 system contributes to the early embryonic establishment of site-specific DNA methylation patterns maintained during development. Cell Rep. 2012, 2, 766–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernie, A.R.; Yan, J. De Novo Domestication: An Alternative Route toward New Crops for the Future. Mol. Plant 2019, 12, 615–631. [Google Scholar] [CrossRef] [Green Version]
- Zsogon, A.; Cermak, T.; Naves, E.R.; Notini, M.M.; Edel, K.H.; Weinl, S.; Freschi, L.; Voytas, D.F.; Kudla, J.; Peres, L.E.P. De novo domestication of wild tomato using genome editing. Nat. Biotechnol. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Cui, Y.; Dong, G.; Feng, A.; Wang, D.; Zhao, C.; Zhang, Y.; Hu, J.; Zeng, D.; Guo, L.; et al. Using CRISPR-Cas9 to generate semi-dwarf rice lines in elite landraces. Sci. Rep. 2019, 9, 19096. [Google Scholar] [CrossRef] [Green Version]
- Lacchini, E.; Kiegle, E.; Castellani, M.; Adam, H.; Jouannic, S.; Gregis, V.; Kater, M.M. CRISPR-mediated accelerated domestication of African rice landraces. PLoS ONE 2020, 15, e0229782. [Google Scholar] [CrossRef] [Green Version]
- Okuzaki, A.; Ogawa, T.; Koizuka, C.; Kaneko, K.; Inaba, M.; Imamura, J.; Koizuka, N. CRISPR/Cas9-mediated genome editing of the fatty acid desaturase 2 gene in Brassica napus. Plant Physiol. Biochem. 2018, 131, 63–69. [Google Scholar] [CrossRef]
- Cermak, T.; Baltes, N.J.; Cegan, R.; Zhang, Y.; Voytas, D.F. High-frequency, precise modification of the tomato genome. Genome Biol. 2015, 16, 232. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Sanchez-Leon, S.; Gil-Humanes, J.; Ozuna, C.V.; Gimenez, M.J.; Sousa, C.; Voytas, D.F.; Barro, F. Low-gluten, nontransgenic wheat engineered with CRISPR/Cas9. Plant Biotechnol. J. 2018, 16, 902–910. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Nishimasu, H.; Shi, X.; Ishiguro, S.; Gao, L.; Hirano, S.; Okazaki, S.; Noda, T.; Abudayyeh, O.O.; Gootenberg, J.S.; Mori, H.; et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 2018, 361, 1259–1262. [Google Scholar] [CrossRef]
- Gorbunova, V.; Levy, A.A. Non-homologous DNA end joining in plant cells is associated with deletions and filler DNA insertions. Nucleic Acids Res. 1997, 25, 4650–4657. [Google Scholar] [CrossRef] [Green Version]
- Lindbo, J.A. A historical overview of RNAi in plants. Methods Mol. Biol. 2012, 894, 1–16. [Google Scholar] [CrossRef]
- Budhagatapalli, N.; Rutten, T.; Gurushidze, M.; Kumlehn, J.; Hensel, G. Targeted Modification of Gene Function Exploiting Homology-Directed Repair of TALEN-Mediated Double-Strand Breaks in Barley. G3 Genes Genomes Genet. 2015, 5, 1857–1863. [Google Scholar] [CrossRef] [Green Version]
- Svitashev, S.; Young, J.K.; Schwartz, C.; Gao, H.; Falco, S.C.; Cigan, A.M. Targeted Mutagenesis, Precise Gene Editing, and Site-Specific Gene Insertion in Maize Using Cas9 and Guide RNA. Plant Physiol. 2015, 169, 931–945. [Google Scholar] [CrossRef] [Green Version]
- Najera, V.A.; Twyman, R.M.; Christou, P.; Zhu, C. Applications of multiplex genome editing in higher plants. Curr. Opin. Biotechnol. 2019, 59, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Kannan, B.; Jung, J.H.; Moxley, G.W.; Lee, S.M.; Altpeter, F. TALEN-mediated targeted mutagenesis of more than 100 COMT copies/alleles in highly polyploid sugarcane improves saccharification efficiency without compromising biomass yield. Plant. Biotechnol. J. 2018, 16, 856–866. [Google Scholar] [CrossRef]
- Stuttmann, J.; Barthel, K.; Martin, P.; Ordon, J.; Erickson, J.L.; Herr, R.; Ferik, F.; Kretschmer, C.; Berner, T.; Keilwagen, J.; et al. Highly efficient multiplex editing: One-shot generation of 8x Nicotiana benthamiana and 12x Arabidopsis mutants. Plant J. 2021, 106, 8–22. [Google Scholar] [CrossRef]
- Wang, Y.; Cheng, X.; Shan, Q.; Zhang, Y.; Liu, J.; Gao, C.; Qiu, J.L. Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat. Biotechnol. 2014, 32, 947–951. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Zhang, K.; Chen, K.; Gao, C. Targeted mutagenesis in Zea mays using TALENs and the CRISPR/Cas system. J. Genet. Genom. 2014, 41, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Ku, H.K.; Ha, S.H. Improving Nutritional and Functional Quality by Genome Editing of Crops: Status and Perspectives. Front. Plant Sci. 2020, 11, 577313. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Schmidt, C.; Fransz, P.; Ronspies, M.; Dreissig, S.; Fuchs, J.; Heckmann, S.; Houben, A.; Puchta, H. Changing local recombination patterns in Arabidopsis by CRISPR/Cas mediated chromosome engineering. Nat. Commun. 2020, 11, 4418. [Google Scholar] [CrossRef]
- Kumlehn, J.; Pietralla, J.; Hensel, G.; Pacher, M.; Puchta, H. The CRISPR/Cas revolution continues: From efficient gene editing for crop breeding to plant synthetic biology. J. Integr. Plant Biol. 2018, 60, 1127–1153. [Google Scholar] [CrossRef]
- Schindele, A.; Dorn, A.; Puchta, H. CRISPR/Cas brings plant biology and breeding into the fast lane. Curr. Opin. Biotechnol. 2020, 61, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Ganie, S.A.; Wani, S.H.; Henry, R.; Hensel, G. Improving rice salt tolerance by precision breeding in a new era. Curr. Opin. Plant Biol. 2021, 60, 101996. [Google Scholar] [CrossRef]
- Chandrasekaran, J.; Brumin, M.; Wolf, D.; Leibman, D.; Klap, C.; Pearlsman, M.; Sherman, A.; Arazi, T.; Gal-On, A. Development of broad virus resistance in non-transgenic cucumber using CRISPR/Cas9 technology. Mol. Plant Pathol. 2016, 17, 1140–1153. [Google Scholar] [CrossRef] [Green Version]
- Waltz, E. Gene-edited CRISPR mushroom escapes US regulation. Nature 2016, 532, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osakabe, Y.; Liang, Z.; Ren, C.; Nishitani, C.; Osakabe, K.; Wada, M.; Komori, S.; Malnoy, M.; Velasco, R.; Poli, M.; et al. CRISPR-Cas9-mediated genome editing in apple and grapevine. Nat. Protoc. 2018, 13, 2844–2863. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.M.; Belisle, M.; Frommer, W.B. The evolving landscape around genome editing in agriculture: Many countries have exempted or move to exempt forms of genome editing from GMO regulation of crop plants. EMBO Rep. 2020, 21, e50680. [Google Scholar] [CrossRef]
- Evangelatos, N.; Upadya, S.P.; Venne, J.; Satyamoorthy, K.; Brand, H.; Ramashesha, C.S.; Brand, A. Digital Transformation and Governance Innovation for Public Biobanks and Free/Libre Open Source Software Using a Blockchain Technology. OMICS 2020, 24, 278–285. [Google Scholar] [CrossRef]
- Liu, J.; Huang, L.; Wang, C.; Liu, Y.; Yan, Z.; Wang, Z.; Xiang, L.; Zhong, X.; Gong, F.; Zheng, Y.; et al. Genome-Wide Association Study Reveals Novel Genomic Regions Associated With High Grain Protein Content in Wheat Lines Derived From Wild Emmer Wheat. Front. Plant Sci. 2019, 10, 464. [Google Scholar] [CrossRef]
- Galan, R.J.; Bernal-Vasquez, A.M.; Jebsen, C.; Piepho, H.P.; Thorwarth, P.; Steffan, P.; Gordillo, A.; Miedaner, T. Integration of genotypic, hyperspectral, and phenotypic data to improve biomass yield prediction in hybrid rye. Theor. Appl. Genet. 2020, 133, 3001–3015. [Google Scholar] [CrossRef]
- Marzec, M.; Braszewska-Zalewska, A.; Hensel, G. Prime Editing: A New Way for Genome Editing. Trends Cell Biol. 2020, 30, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Khatri, S.; Sharma, S. How does organic farming shape the soil- and plant-associated microbiota? Symbiosis 2021, 1–8. [Google Scholar] [CrossRef]
- Belimov, A.A.; Shaposhnikov, A.I.; Azarova, T.S.; Makarova, N.M.; Safronova, V.I.; Litvinskiy, V.A.; Nosikov, V.V.; Zavalin, A.A.; Tikhonovich, I.A. Microbial Consortium of PGPR, Rhizobia and Arbuscular Mycorrhizal Fungus Makes Pea Mutant SGECd(t) Comparable with Indian Mustard in Cadmium Tolesrance and Accumulation. Plants 2020, 9, 975. [Google Scholar] [CrossRef]
- Reynolds, M.; Atkin, O.K.; Bennett, M.; Cooper, M.; Dodd, I.C.; Foulkes, M.J.; Frohberg, C.; Hammer, G.; Henderson, I.R.; Huang, B.; et al. Addressing Research Bottlenecks to Crop Productivity. Trends Plant Sci. 2021, 26, 607–630. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gogolev, Y.V.; Ahmar, S.; Akpinar, B.A.; Budak, H.; Kiryushkin, A.S.; Gorshkov, V.Y.; Hensel, G.; Demchenko, K.N.; Kovalchuk, I.; Mora-Poblete, F.; et al. OMICs, Epigenetics, and Genome Editing Techniques for Food and Nutritional Security. Plants 2021, 10, 1423. https://doi.org/10.3390/plants10071423
Gogolev YV, Ahmar S, Akpinar BA, Budak H, Kiryushkin AS, Gorshkov VY, Hensel G, Demchenko KN, Kovalchuk I, Mora-Poblete F, et al. OMICs, Epigenetics, and Genome Editing Techniques for Food and Nutritional Security. Plants. 2021; 10(7):1423. https://doi.org/10.3390/plants10071423
Chicago/Turabian StyleGogolev, Yuri V., Sunny Ahmar, Bala Ani Akpinar, Hikmet Budak, Alexey S. Kiryushkin, Vladimir Y. Gorshkov, Goetz Hensel, Kirill N. Demchenko, Igor Kovalchuk, Freddy Mora-Poblete, and et al. 2021. "OMICs, Epigenetics, and Genome Editing Techniques for Food and Nutritional Security" Plants 10, no. 7: 1423. https://doi.org/10.3390/plants10071423
APA StyleGogolev, Y. V., Ahmar, S., Akpinar, B. A., Budak, H., Kiryushkin, A. S., Gorshkov, V. Y., Hensel, G., Demchenko, K. N., Kovalchuk, I., Mora-Poblete, F., Muslu, T., Tsers, I. D., Yadav, N. S., & Korzun, V. (2021). OMICs, Epigenetics, and Genome Editing Techniques for Food and Nutritional Security. Plants, 10(7), 1423. https://doi.org/10.3390/plants10071423