
Variation in Gene Expression between Two Sorghum bicolor Lines Differing in Innate Immunity Response


Abstract
:1. Introduction
2. Results and Discussion
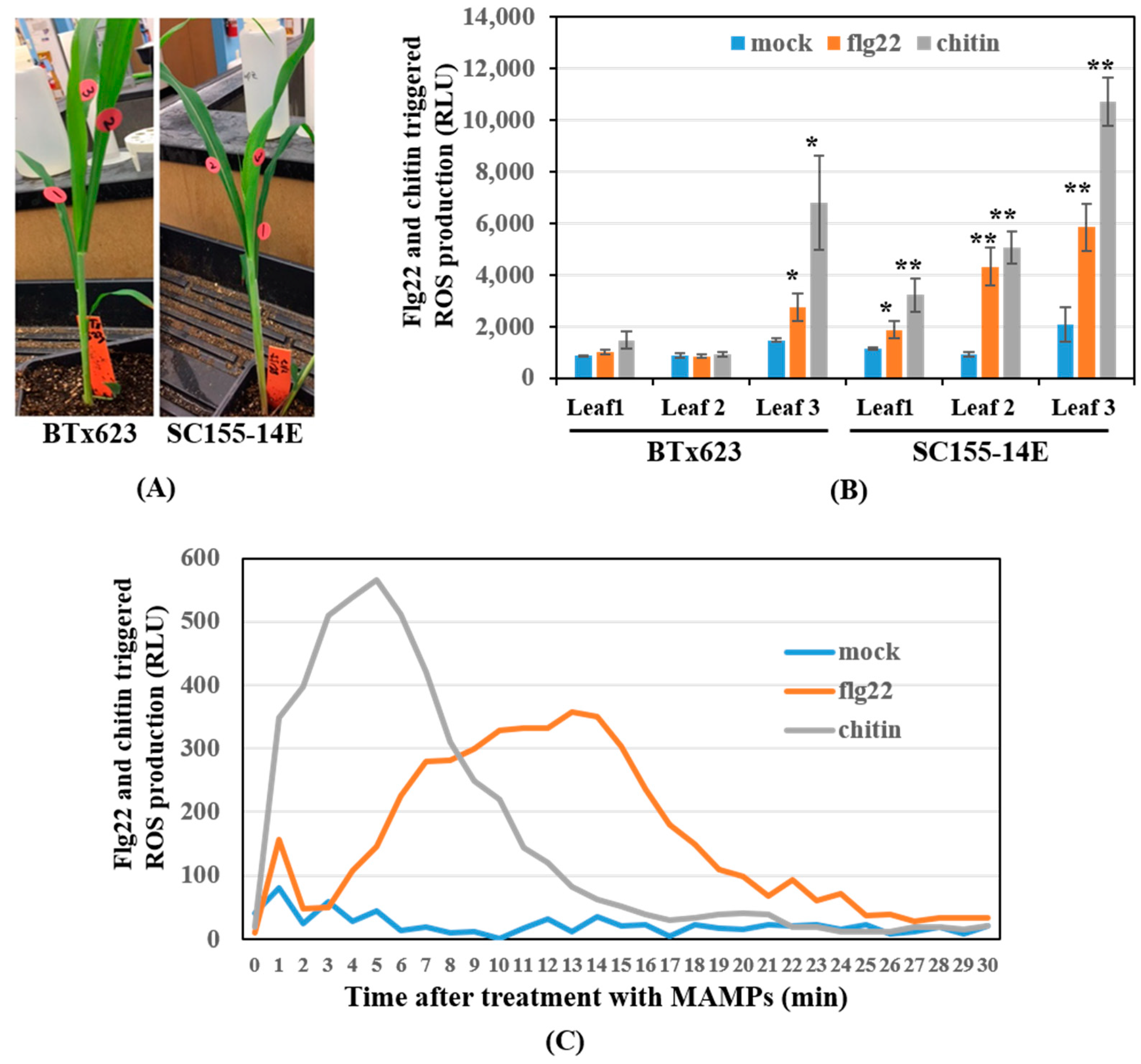
2.1. Variation of the Oxidative Burst in Different Sorghum Leaves
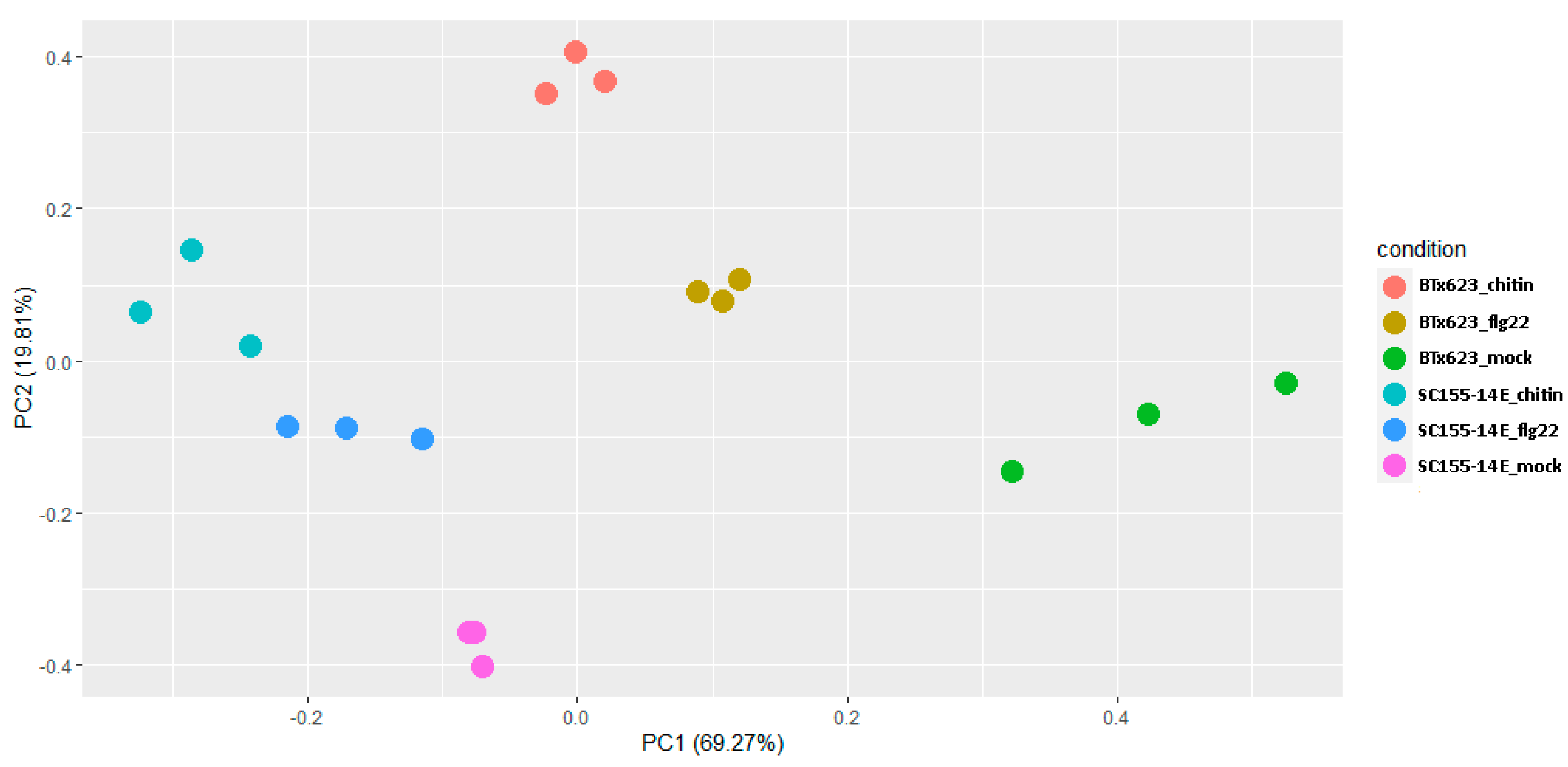
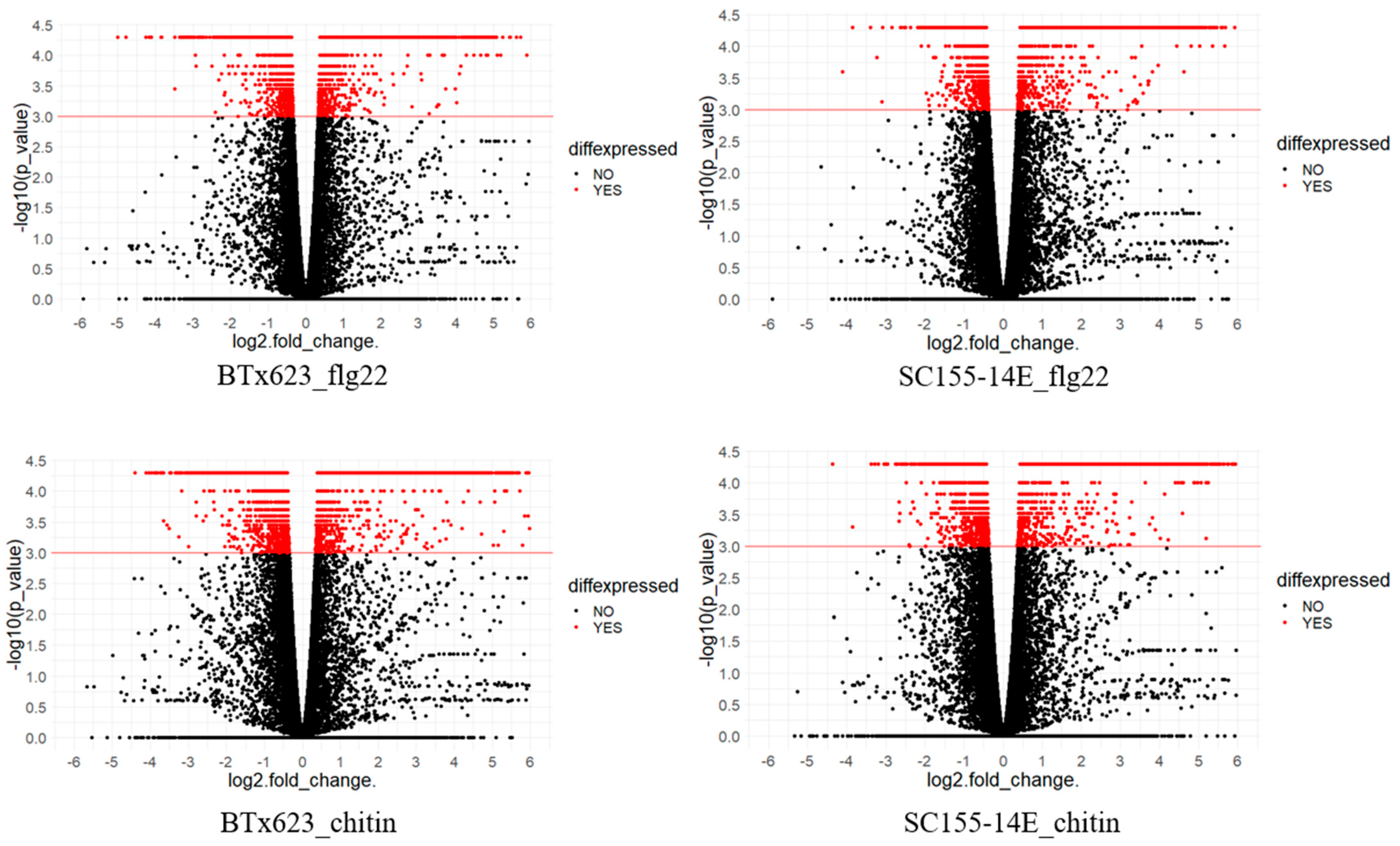
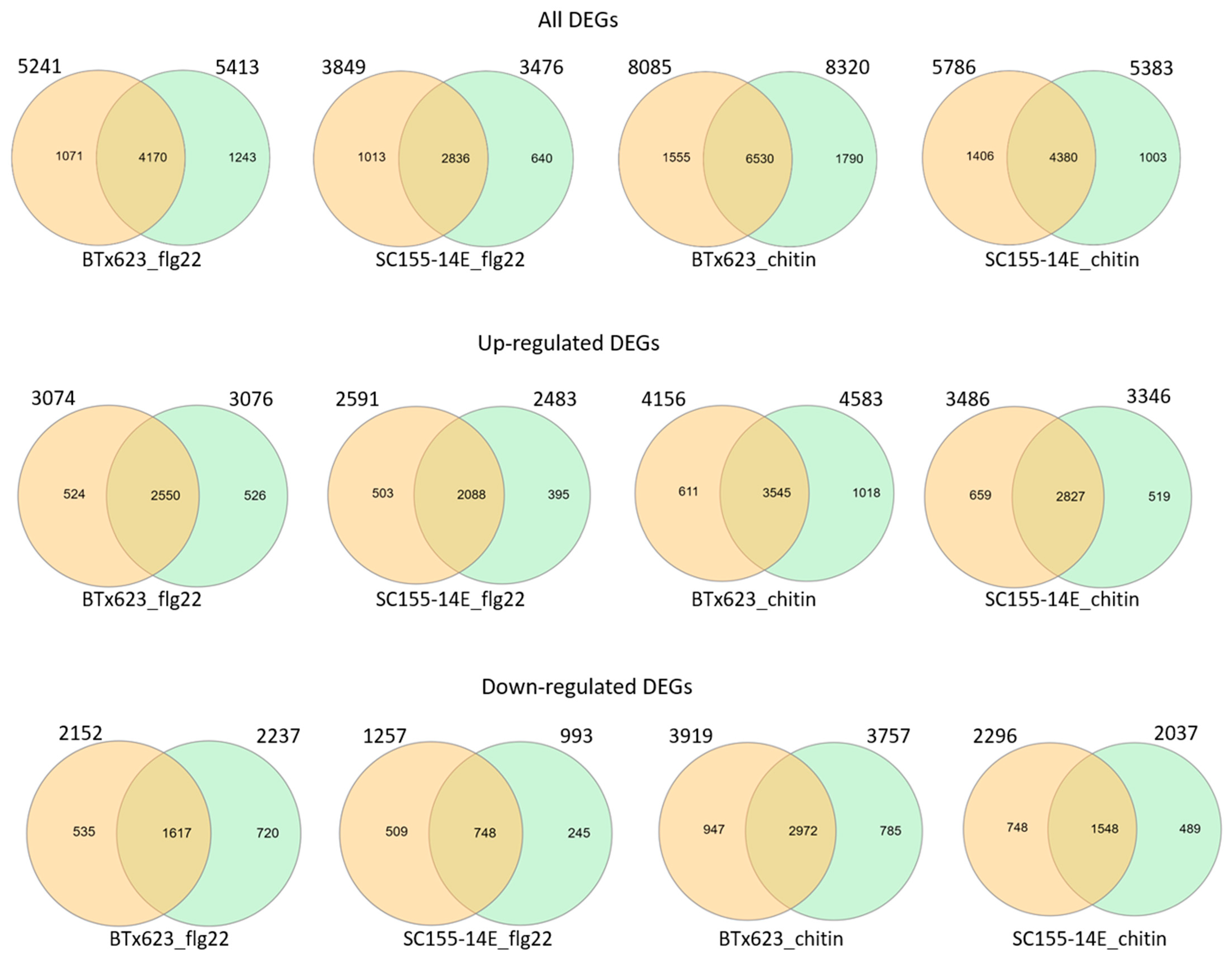
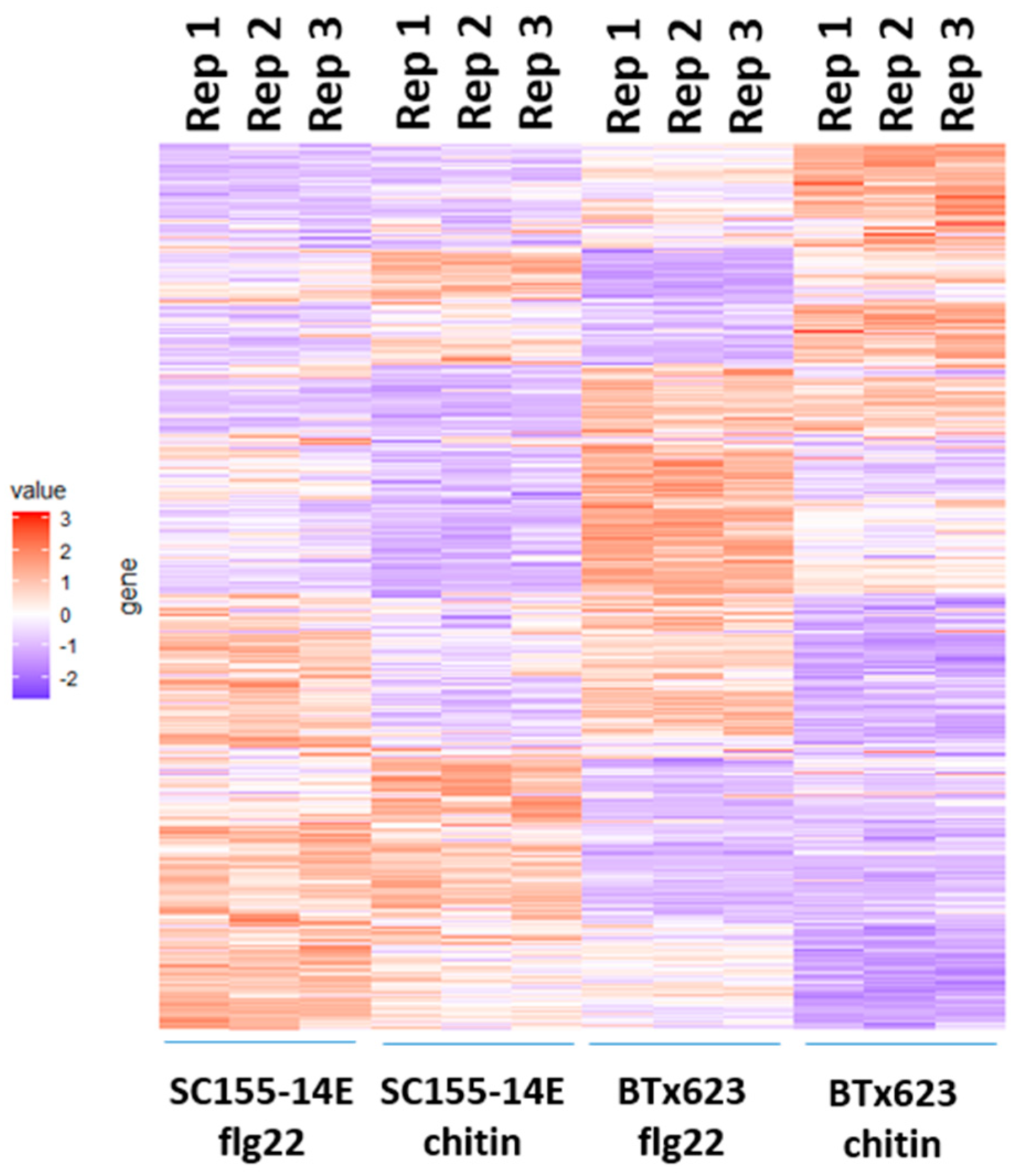
2.2. Differentially Expressed Genes (DEGs) of BTx623 and SC155-14E in Response to MAMP Treatments
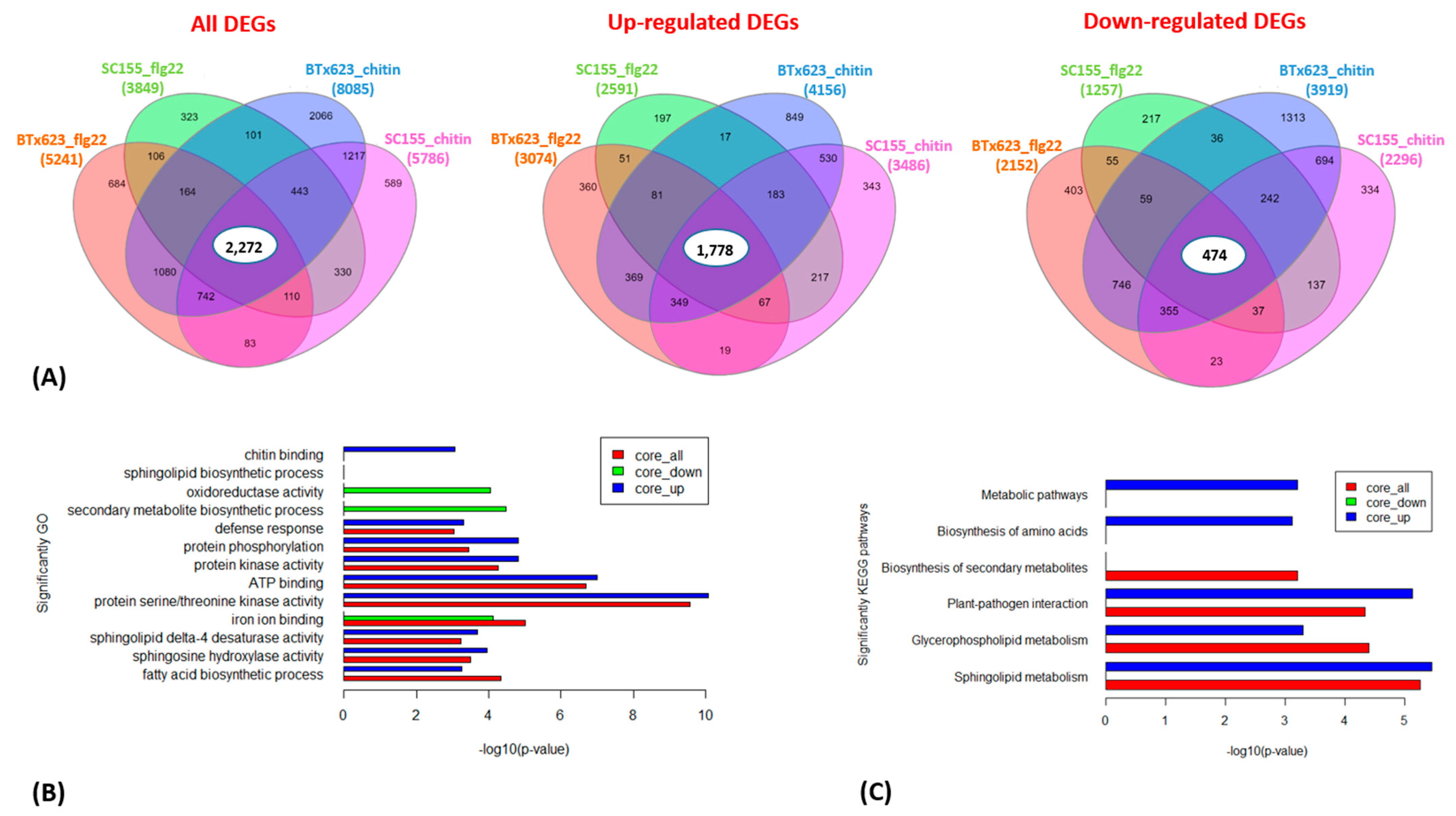
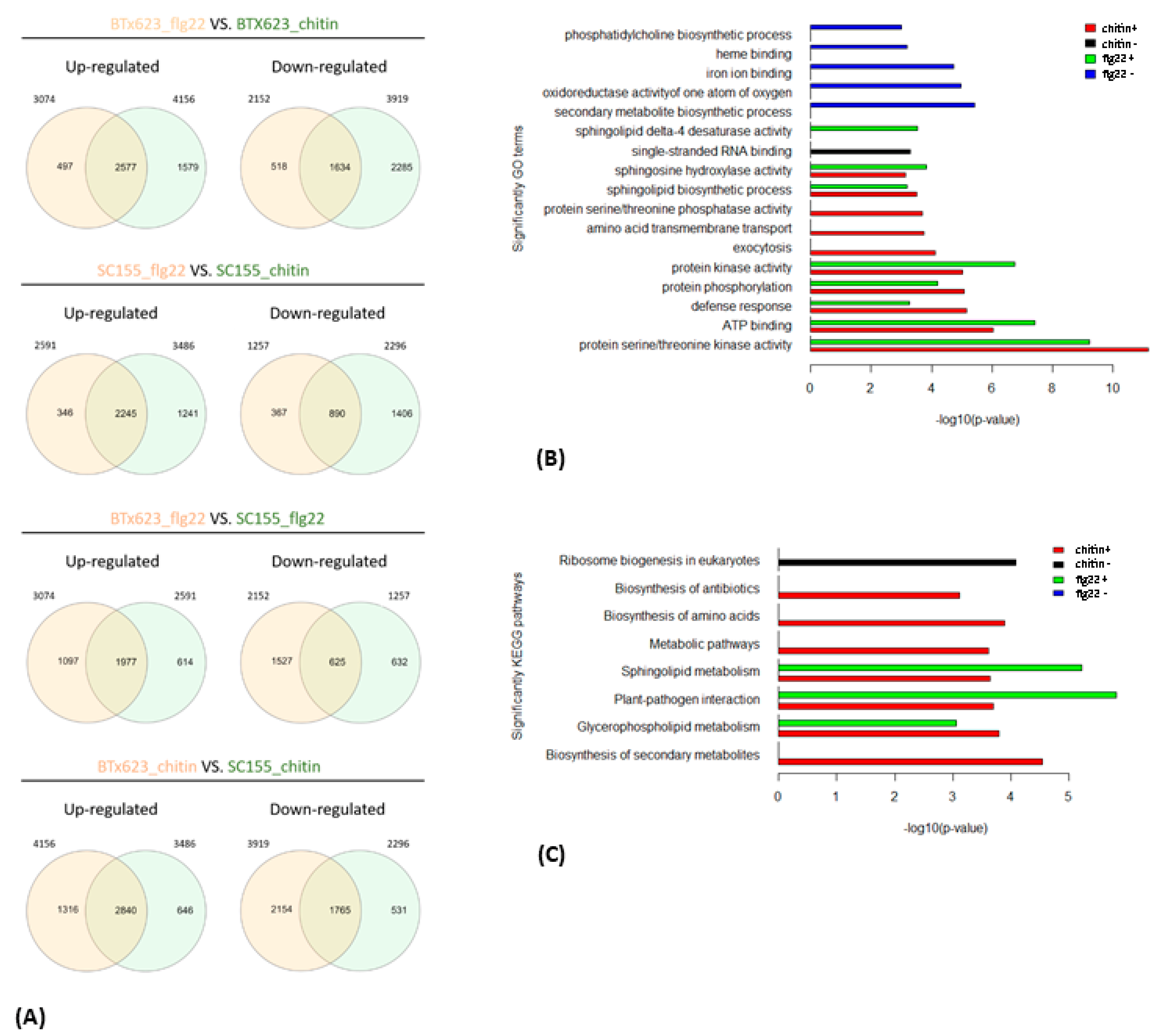
2.3. Gene Functional Enrichment Analysis Comparing the MTI Response of BTx623 and SC155-14E
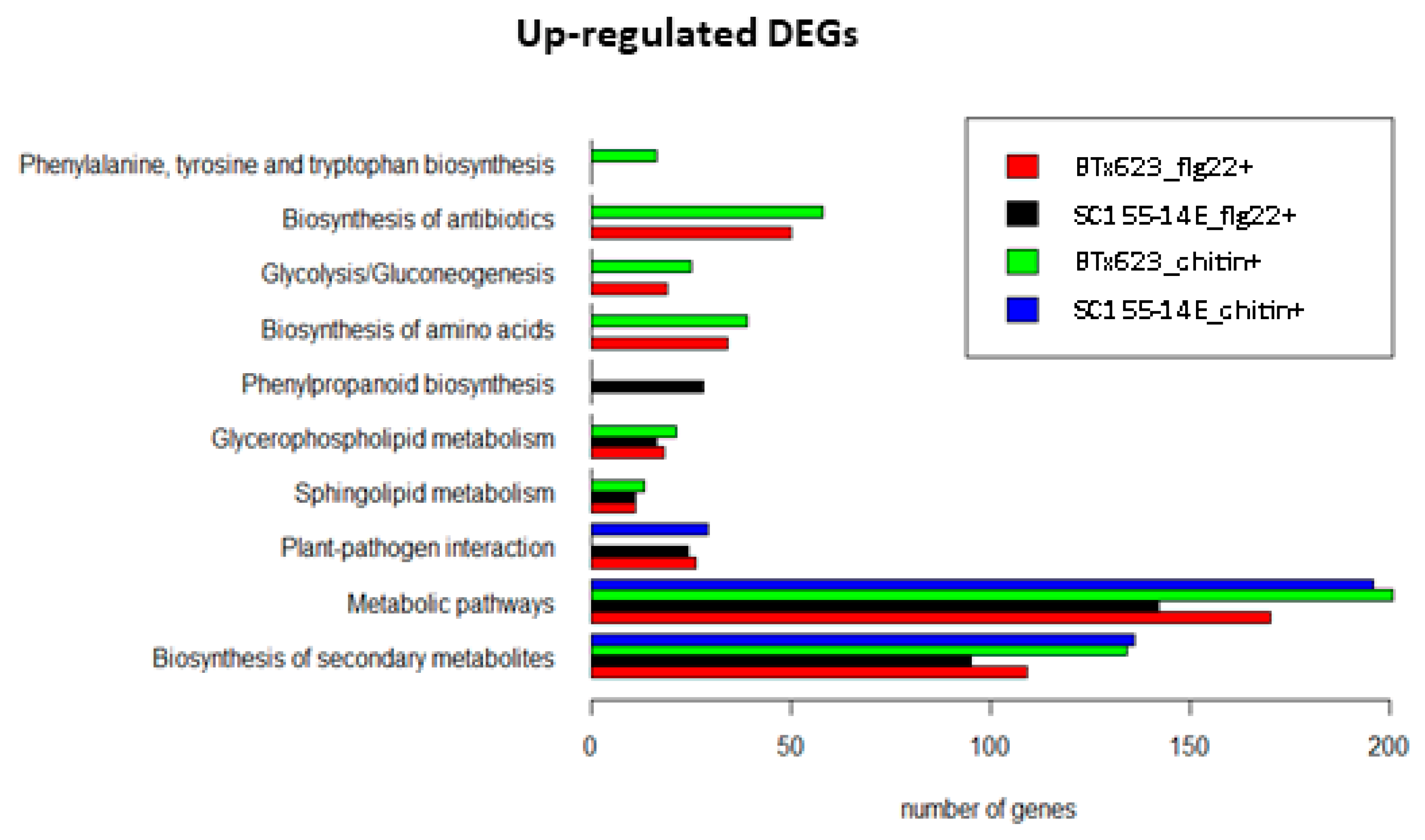
2.4. KEGG Pathway Enrichment Analysis of the MTI Response of BTx623 and SC155-14E
2.5. Validation of the RNA-Seq Data Using Quantitative RT-PCR of Select Genes
2.6. Conclusions
3. Materials and Methods
3.1. Plant Materials
3.2. ROS Assays
3.3. Sample Treatment with MAMPs
3.4. RNA Extraction, Sequencing and Library Construction
3.5. Mapping and Processing of RNA-Seq Reads
3.6. Bioinformatic Analysis of RNA-Seq Data
3.7. Identification of Differentially Expressed Genes
3.8. PCA Plot
3.9. Heat Map of DEGs
3.10. Gene Ontology Enrichment
3.11. KEGG Pathway Enrichment
3.12. GEO Accession Number
3.13. qRT-PCR to Validate RNA Sequence Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Faulkner, C.; Robatzek, S. Plants and pathogens: Putting infection strategies and defence mechanisms on the map. Curr. Opin. Plant Biol. 2012, 15, 699–707. [Google Scholar] [CrossRef]
- Segonzac, C.; Zipfel, C. Activation of plant pattern-recognition receptors by bacteria. Curr. Opin. Microbiol. 2011, 14, 54–61. [Google Scholar] [CrossRef]
- Newman, M.-A.; Sundelin, T.; Nielsen, J.T.; Erbs, G. MAMP (microbe-associated molecular pattern) triggered immunity in plants. Front. Plant Sci. 2013, 4, 139. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Li, L.; Macho, A.P.; Han, Z.; Hu, Z.; Zipfel, C.; Zhou, J.-M.; Chai, J. Structural Basis for flg22-Induced Activation of the Arabidopsis FLS2-BAK1 Immune Complex. Science 2013, 342, 624–628. [Google Scholar] [CrossRef]
- Zipfel, C.; Robatzek, S.; Navarro, L.; Oakeley, E.J.; Jones, J.; Felix, G.; Boller, T. Bacterial disease resistance in Arabidopsis through flagellin perception. Nat. Cell Biol. 2004, 428, 764–767. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, J.; Zipfel, C. Plant pattern recognition receptor complexes at the plasma membrane. Curr. Opin. Plant Biol. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Balint-kurti, P. The plant hypersensitive response: Concepts, control and consequences. Mol. Plant Pathol. 2019, 20, 1163–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomma, B.P.; Nürnberger, T.; Joosten, M.H. Of PAMPs and effectors: The Blurred PTI-ETI dichotomy. Plant Cell 2011, 23, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHale, L.; Tan, X.; Koehl, P.; Michelmore, R.W. Plant NBS-LRR proteins: Adaptable guards. Genome Biol. 2006, 7, 212. [Google Scholar] [CrossRef] [Green Version]
- Conrath, U. Systemic acquired resistance. Plant Signal. Behav. 2006, 1, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, C. Early molecular events in PAMP-triggered immunity. Curr. Opin. Plant Biol. 2009, 12, 414–420. [Google Scholar] [CrossRef]
- Valdes-Lopez, O.; Thibivilliers, S.; Qiu, J.; Xu, W.Z.; Nguyen, T.H.N.; Libault, M.; Le, B.H.; Goldberg, R.B.; Hill, C.B.; Hartman, G.L.; et al. Identification of Quantitative Trait Loci Controlling Gene Expression during the Innate Immunity Response of Soybean. Plant Physiol. 2011, 157, 1975–1986. [Google Scholar] [CrossRef] [Green Version]
- Valdés-López, O.; Khan, S.M.; Schmitz, R.J.; Cui, S.; Qiu, J.; Joshi, T.; Xu, D.; Diers, B.; Ecker, J.R.; Stacey, G. Genotypic variation of gene expression during the soybean innate immunity response. Plant Genet. Resour. Util. 2014, 12, S27–S30. [Google Scholar] [CrossRef]
- Veluchamy, S.; Hind, S.R.; Dunham, D.M.; Martin, G.B.; Panthee, D.R. Natural Variation for Responsiveness to flg22, flgII-28, and csp22 and Pseudomonas syringae pv. tomato in Heirloom Tomatoes. PLoS ONE 2014, 9, e106119. [Google Scholar] [CrossRef]
- Vetter, M.; Karasov, T.L.; Bergelson, J. Differentiation between MAMP Triggered Defenses in Arabidopsis thaliana. PLoS Genet. 2016, 12, e1006068. [Google Scholar] [CrossRef]
- Vetter, M.; Kronholm, I.; He, F.; Häweker, H.; Reymond, M.; Bergelson, J.; Robatzek, S.; de Meaux, J. Flagellin perception varies quantitatively in arabidopsis thaliana and its relatives. Mol. Biol. Evol. 2012, 29, 1655–1667. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Valdés-López, O.; Arellano, C.; Stacey, G.; Balint-Kurti, P. Genetic dissection of the maize (Zea mays L.) MAMP response. Theor. Appl. Genet. 2017, 130, 1155–1168. [Google Scholar] [CrossRef]
- Lloyd, S.R.; Ridout, C.; Schoonbeek, H.-J. Methods to Quantify PAMP-triggered oxidative burst, MAP kinase phosphorylation, gene expression, and lignification in Brassicas. Plant Pattern Recognit. Recept. Methods Protoc. 2017, 1578, 325–335. [Google Scholar]
- Lloyd, S.R.; Schoonbeek, H.; Trick, M.; Zipfel, C.; Ridout, C.J. Methods to study PAMP-triggered immunity in Brassica species. Mol. Plant Microbe Interact. 2014, 27, 286–295. [Google Scholar] [CrossRef] [Green Version]
- Zeidler, D.; Zahringer, U.; Gerber, I.; Dubery, I.; Hartung, T.; Bors, W.; Hutzler, P.; Durner, J. From The Cover: Innate immunity in Arabidopsis thaliana: Lipopolysaccharides activate nitric oxide synthase (NOS) and induce defense genes. Proc. Natl. Acad. Sci. USA 2004, 101, 15811–15816. [Google Scholar] [CrossRef] [Green Version]
- Dillon, S.L.; Shapter, F.; Henry, R.J.; Cordeiro, G.A.; Izquierdo, L.; Lee, L.S. Domestication to Crop Improvement: Genetic Resources for Sorghum and Saccharum (Andropogoneae). Ann. Bot. 2007, 100, 975–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A.; et al. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551–556. [Google Scholar] [CrossRef] [Green Version]
- Sharma, I.; Kumari, N.; Sharma, V. Sorghum fungal diseases. In Sustainable Agriculture Reviews. Sustainable Agriculture Reviews; Lichtfouse, E., Goyal, A., Eds.; Springer: Cham, Switzerland, 2015; Volume 16. [Google Scholar] [CrossRef]
- Ali, M.; Warren, H.; Latin, R. Relationship between anthracnose leaf blight and losses in grain yield of sorghum. Plant Dis. 1987, 71, 803–806. [Google Scholar] [CrossRef]
- Tesso, T.; Perumal, R.; Little, C.; Adeyanju, A.; Radwan, G.; Prom, L.; Magill, C. Sorghum pathology and biotechnology—A fungal disease perspective: Part II. Anthracnose, stalk rot, and downy mildew. Eur. J. Plant Sci. Biotechnol. 2011, 6, 31–34. [Google Scholar]
- Lo, S.C.; Hipskind, J.D.; Nicholson, R.L. cDNA Cloning of a Sorghum Pathogenesis-Related Protein (PR-10) and Differential Expression of Defense-Related Genes Following Inoculation with Cochliobolus heterostrophus or Colletotrichum sublineolum. Mol. Plant Microbe Interact. 1999, 12, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Shetty, N.P.; Jorgensen, H.J.L.; Jensen, J.D.; Collinge, D.B.; Shetty, H.S. Roles of reactive oxygen species in interactions between plants and pathogens. Eur. J. Plant Pathol. 2008, 121, 267–280. [Google Scholar] [CrossRef]
- Lo, S.C.; de Verdier, K.; Nicholson, R.L. Accumulation of 3-deoxyanthocyanidin phytoalexins and resistance to Colletotrichum sublineolum in sorghum. Physiol. Mol. Plant Pathol. 1999, 55, 263–273. [Google Scholar] [CrossRef]
- Abreha, K.B.; Ortiz, R.; Carlsson, A.S.; Geleta, M. Understanding the Sorghum–Colletotrichum sublineola Interactions for Enhanced Host Resistance. Front. Plant Sci. 2021, 12, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Stutts, L.R.; Vermerris, W. Elucidating anthracnose resistance mechanisms in sorghum—A review. Phytopathology 2020, 110, 1863–1876. [Google Scholar] [CrossRef]
- Erayman, M.; Turktas, M.; Akdogan, G.; Gurkok, T.; Inal, B.; Ishakoglu, E.; Ilhan, E.; Unver, T. Transcriptome analysis of wheat inoculated with Fusarium graminearum. Front. Plant Sci. 2015, 6, 867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kebede, A.Z.; Johnston, A.; Schneiderman, D.; Bosnich, W.; Harris, L.J. Transcriptome profiling of two maize inbreds with distinct responses to Gibberella ear rot disease to identify candidate resistance genes. BMC Genom. 2018, 19, 131. [Google Scholar] [CrossRef] [Green Version]
- Matić, S.; Bagnaresi, P.; Biselli Orru’, L.; Carneiro, G.; Siciliano, I.; Valé, G.; Gullino, M.L.; Spadaro, D. Comparative transcriptome profiling of resistant and susceptible rice genotypes in response to the seedborne pathogen Fusarium fujikuroi. BMC Genom. 2006, 17, 608. [Google Scholar] [CrossRef] [Green Version]
- Shu, X.; Livingston, D.P.; Woloshuk, C.P.; Payne, G.A. Comparative Histological and Transcriptional Analysis of Maize Kernels Infected with Aspergillus flavus and Fusarium verticillioides. Front. Plant Sci. 2017, 8, 2075. [Google Scholar] [CrossRef]
- Mizuno, H.; Kawahigashi, H.; Kawahara, Y.; Kanamori, H.; Ogata, J.; Minami, H.; Itoh, T.; Matsumoto, T. Global transcriptome analysis reveals distinct expression among duplicated genes during sorghum-Bipolaris sorghicola interaction. BMC Plant Biol. 2012, 12, 121. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.; Liu, J.; Din, G.M.U.; Zhang, H.; Du, Z.; Chen, W.; Liu, T.; Zhang, J.; Zhao, S.; Gao, L. Transcriptome analysis of wheat spikes in response to Tilletia controversa Kühn which cause wheat dwarf bunt. Sci. Rep. 2020, 10, 21567–21579. [Google Scholar] [CrossRef]
- Wang, L.; Chen, M.; Zhu, F.; Fan, T.; Zhang, J.; Lo, C. Alternative splicing is a Sorghum bicolor defense response to fungal infection. Planta 2020, 251, 14–27. [Google Scholar] [CrossRef]
- Kimball, J.; Cui, Y.; Chen, D.; Brown, P.; Rooney, W.L.; Stacey, G.; Balint-Kurti, P.J. Identification of QTL for Target Leaf Spot resistance in Sorghum bicolor and investigation of relationships between disease resistance and variation in the MAMP response. Sci. Rep. 2019, 9, 18285. [Google Scholar] [CrossRef] [Green Version]
- Patil, N.Y.; Klein, R.R.; Williams, C.L.; Collins, S.D.; Knoll, J.E.; Burrell, A.M.; Anderson, W.F.; Rooney, W.L.; Klein, P.E. Quantitative trait loci associated with anthracnose resistance in sorghum. Crop Sci. 2017, 57, 877–890. [Google Scholar] [CrossRef] [Green Version]
- D’Ambrosio, J.M.; Couto, D.; Fabro, G.; Scuffi, D.; Lamattina, L.; Munnik, T.; Andersson, M.X.; Alvarez, M.E.; Zipfel, C.; Laxalt, A.M. Phospholipase C2 Affects MAMP-Triggered Immunity by Modulating ROS Production. Plant Physiol. 2017, 175, 970–981. [Google Scholar] [CrossRef] [Green Version]
- Wojtaszek, P. Oxidative burst: An early plant response to pathogen infection. Biochem. J. 1997, 322, 681–692. [Google Scholar] [CrossRef] [Green Version]
- Herrera-Vã¡squez, A.; Salinas, P.; Holuigue, L. Salicylic acid and reactive oxygen species interplay in the transcriptional control of defense genes expression. Front. Plant Sci. 2015, 6, 171. [Google Scholar] [CrossRef] [Green Version]
- Segonzac, C.; Feike, D.; Gimenez-Ibanez, S.; Hann, D.R.; Zipfel, C.; Rathjen, J.P. Hierarchy and roles of pathogen-associated molecular patter-induced responses in Nicotiana benthamiana. Plant Physiol. 2011, 156, 687–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinoza, C.; Liang, Y.; Stacey, G. Chitin receptor CERK 1 links salt stress and chitin-triggered innate immunity in Arabidopsis. Plant J. 2017, 89, 984–995. [Google Scholar] [CrossRef] [Green Version]
- Ramonell, K.M.; Zhang, B.; Ewing, R.M.; Chen, Y.; Xu, D.; Stacey, G.; Somerville, S. Microarray analysis of chitin elicitation in Arabidopsis thaliana. Mol. Plant Pathol. 2002, 3, 301–311. [Google Scholar] [CrossRef]
- Wan, J.; Zhang, X.-C.; Neece, D.; Ramonell, K.M.; Clough, S.; Kim, S.Y.; Stacey, M.G.; Stacey, G. A LysM receptor-like kinase plays a critical role in chitin signaling and fungal resistance in Arabidopsis. Plant Cell 2008, 20, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Jingchu, L.; He, K.; Tang, X.; Li, Z.; Lv, L.; Zhao, Y.; Luo, J.; Gao, G. An Arabidopsis Transcriptional Regulatory Map Reveals Distinct Functional and Evolutionary Features of Novel Transcription Factors. Mol. Biol. Evol. 2015, 32, 1767–1773. [Google Scholar]
- Jin, J.; Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Kong, L.; Luo, J.; Gao, G. Plant TFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stringlis, I.; Proietti, S.; Hickman, R.; Van Verk, M.C.; Zamioudis, C.; Pieterse, C.M.J. Root transcriptional dynamics induced by beneficial rhizobacteria and microbial immune elicitors reveal signatures of adaptation to mutualists. Plant J. 2017, 93, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Stintzi, A.; Heitz, T.; Prasad, V.; Wiedemann-Merdinoglu, S.; Kauffmann, S.; Geoffroy, P.; Legrand, M.; Fritig, B. Plant ‘pathogenesis-related’ proteins and their role in defense against pathogens. Biochimie 1993, 75, 687–706. [Google Scholar] [CrossRef]
- Agrios, G.N. How plants defend themselves against pathogens, Chapter Six. In Plant Pathology, 5th ed.; Academic Press: Cambridge, MA, USA, 2005. [Google Scholar] [CrossRef]
- Fernandes, H.; Michalska, K.; Sikorski, M.; Jaskolski, M. Structural and functional aspects of PR-10 proteins. FEBS J. 2013, 280, 1169–1199. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-J.; Ekramoddoullah, A.K.M.; Yu, X. Differential expression of multiple PR10 proteins in western white pine following wounding, fungal infection and cold-hardening. Physiol. Plant. 2003, 119, 544–553. [Google Scholar] [CrossRef] [Green Version]
- Katilé, S.O.; Perumal, R.; Rooney, W.L.; Prom, L.K.; Magill, C.W.; Katilã, S.O. Expression of pathogenesis-related protein PR-10 in sorghum floral tissues in response to inoculation with Fusarium thapsinum and Curvularia lunata. Mol. Plant Pathol. 2010, 11, 93–103. [Google Scholar] [CrossRef]
- An, C.; Mou, Z. Salicylic acid and defense responses in plants. In Phytohormones: A Window to Metabolism, Signaling and Biotechnological Applications; Tran, L.S., Pal, S., Eds.; Springer: New York, NY, USA, 2014. [Google Scholar] [CrossRef]
- Tugizimana, F.; Djami-Tchatchou, A.T.; Steenkamp, P.A.; Piater, L.A.; Dubery, I.A. metabolomic analysis of defense-related reprogramming in Sorghum bicolor in response to Colletotrichum sublineolum infection reveals a functional metabolic web of phenylpropanoid and flavonoid pathways. Front. Plant Sci. 2019, 9, 1840. [Google Scholar] [CrossRef] [Green Version]
- Jiao, X.; Sherman, B.T.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. DAVID-WS: A stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012, 28, 1805–1806. [Google Scholar] [CrossRef] [Green Version]
- Vogt, T. Phenylpropanoid Biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef] [Green Version]
- Treutter, D. Significance of flavonoids in plant resistance and enhancement of their biosynthesis. Plant Biol. 2005, 7, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wei, W.; Zhang, J.; Li, G.; Gao, K. Structures and antipathogenic fungi activities of flavonoids from pathogen-infected Astragalus adsurgens. Nat. Prod. Res. 2017, 33, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Ibraheem, F.; Gaffoor, I.; Chopra, S. Flavonoid phytoalexin-dependent resistance to anthracnose leaf blight requires a functional yellow seed1 in Sorghum bicolor. Genetics 2010, 184, 915–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felix, G.; Duran, J.D.; Volko, S.; Boller, T. Plants have a sensitive perception system for the most conserved domain of bacterial flagellin. Plant J. 1999, 18, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A quality Control Tool for High Throughput Sequence Data [Online]. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 30 September 2020).
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Libault, M.; Farmer, A.; Brechenmacher, L.; Drnevich, J.; Langley, R.J.; Bilgin, D.D.; Radwan, Q.; Neece, D.J.; Clough, S.J.; May, G.D.; et al. Complete transcriptome of the soybean root hairs cell, a single-cell model, and its alteration in response to Bradyrhizobium japonicum infection. Plant Physiol. 2010, 152, 541–552. [Google Scholar] [CrossRef] [Green Version]
- Libault, M.; Thibivilliers, S.; Bilgin, D.D.; Radwan, O.; Benitez, M.; Clough, S.J.; Stacey, G. Identification of four soybean reference genes for gene expression normalization. Plant Genome 2008, 1, 44–54. [Google Scholar] [CrossRef]
- Reddy, P.S.; Dumbala, D.S.; Sivasakthi, K.; Bhatnagar-Mathur, P.; Vadez, V.; Sharma, K.K. Evaluation of sorghum [Sorghum bicolor (L.)] reference genes in various tissues and under abiotic stress conditions for quantitative Real-Time PCR data normalization. Front. Plant Sci. 2016, 7, 529. [Google Scholar]
- Ramakers, C.; Ruijter, M.J.; Deprez, H.R.; Moorman, A.F. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neuro Sci. Lett. 2003, 339, 62–66. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Annotation | Expression Ratio of flg22 Treated (RNA-seq) | Expression Ratio of flg22 Treated (qRT-PCR) | Expression Ratio of Chitin Treated (RNA-seq) | Expression Ratio of Chitin Treated (qRT-PCR) | Category |
|---|---|---|---|---|---|---|
| SC155-14E/BTx623 | SC155-14E/BTx623 | SC155-14E/BTx623 | SC155-14E/BTx623 | |||
| SORBI_3006G217900 | integral component of membrane [GO:0016021]; ATP binding [GO:0005524]; protein kinase activity [GO:0004672] | 0.925 | primer set 1: 0.87 primer set 2: 0.79 | 1.13 | primer set 1: 1.24 primer set 2: 1.36 | a |
| SORBI_3002G260900 | integral component of membrane [GO:0016021]; ATP binding [GO:0005524]; protein kinase activity [GO:0004672] | 0.9 | primer set 1:1.39 primer set 2: 1.22 | 1.04 | primer set 1: 0.99 primer set 2: 0.91 | a |
| SORBI_3004G052500 | hypothetical protein | 29,666 | primer set 1: 50,000 primer set 2:60,000 | 30,000 | primer set 1: 40,000 primer set 2: 40,000 | b |
| SORBI_3006G261500 | oxidoreductase activity [GO:0016491] | 15.3 | primer set 1: 20.8 Primer set 2: 19.6 | 16.6 | primer set 1: 13.5 Primer set 2: 13.6 | c |
| SORBI_3007G120401 | hypothetical protein | 3.64 | primer set 1: 2.3 Primer set 2: 2.8 | 2.72 | primer set 1: 2.4 primer set 2: 2.1 | c |
| SORBI_3007G074200 | hypothetical protein | 1.44 | Primer set 1: 1.7 primer set 2: 1.65 | 1.96 | primer set 1: 1.36 Primer set 2: 1.43 | a |
| SORBI_3008G191300 | ATP binding [GO:0005524] | 2.4 | primer set 1: 2.8 primer set 2: 3.0 | 4.15 | primer set 1: 2.8 primer set 2: 2.7 | c |
| SORBI_3010G117800 | hypothetical protein | 2.51 | primer set 1: 1.8 Primer set 2: 1.5 | 2.79 | primer set 1: 1.54 Primer set 2: 2.2 | c |
| SORBI_3003G036200 | chloroplast [GO:0009507]; malate dehydrogenase (decarboxylating) (NAD+) activity [GO:0004471]; malate dehydrogenase (decarboxylating) (NADP+) activity [GO:0004473]; metal ion binding [GO:0046872]; NAD binding [GO:0051287]; malate metabolic process [GO:0006108]; pyruvate metabolic process [GO:0006090] | 1.06 | primer set 1: 1.38 primer set 2: 1.2 | 1.02 | primer set 1:1.2 primer set 2: 1.1 | a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, Y.; Chen, D.; Jiang, Y.; Xu, D.; Balint-Kurti, P.; Stacey, G. Variation in Gene Expression between Two Sorghum bicolor Lines Differing in Innate Immunity Response. Plants 2021, 10, 1536. https://doi.org/10.3390/plants10081536
Cui Y, Chen D, Jiang Y, Xu D, Balint-Kurti P, Stacey G. Variation in Gene Expression between Two Sorghum bicolor Lines Differing in Innate Immunity Response. Plants. 2021; 10(8):1536. https://doi.org/10.3390/plants10081536
Chicago/Turabian StyleCui, Yaya, Dongqin Chen, Yuexu Jiang, Dong Xu, Peter Balint-Kurti, and Gary Stacey. 2021. "Variation in Gene Expression between Two Sorghum bicolor Lines Differing in Innate Immunity Response" Plants 10, no. 8: 1536. https://doi.org/10.3390/plants10081536
APA StyleCui, Y., Chen, D., Jiang, Y., Xu, D., Balint-Kurti, P., & Stacey, G. (2021). Variation in Gene Expression between Two Sorghum bicolor Lines Differing in Innate Immunity Response. Plants, 10(8), 1536. https://doi.org/10.3390/plants10081536