
Identification and Characterization of Verticillium nonalfalfae-Responsive MicroRNAs in the Roots of Resistant and Susceptible Hop Cultivars




Abstract
:1. Introduction
2. Results
2.1. High-Throughput Sequencing of H. lupulus miRNAs
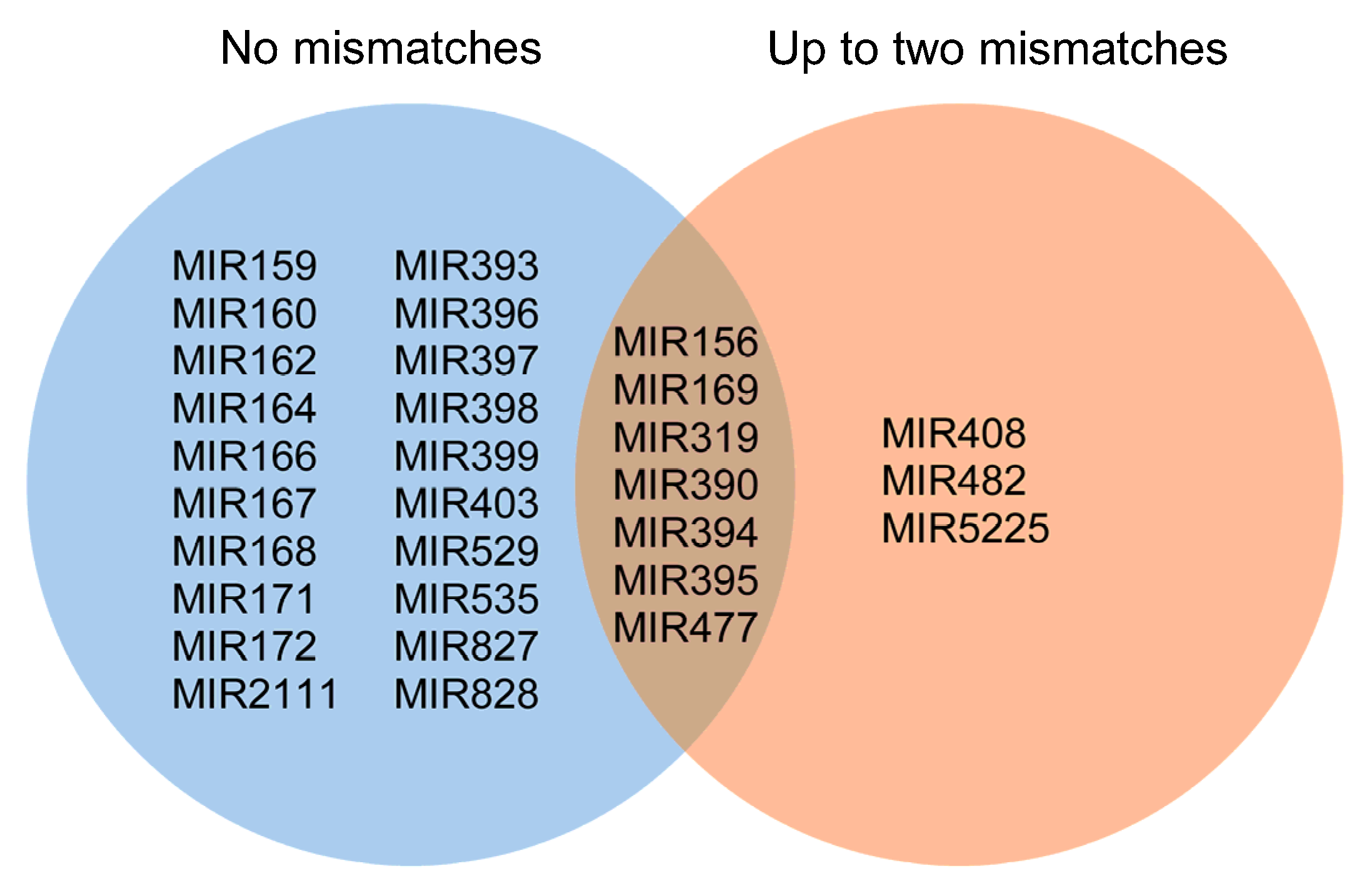
2.2. Known and Novel miRNAs Identified in Hop Root Tissue
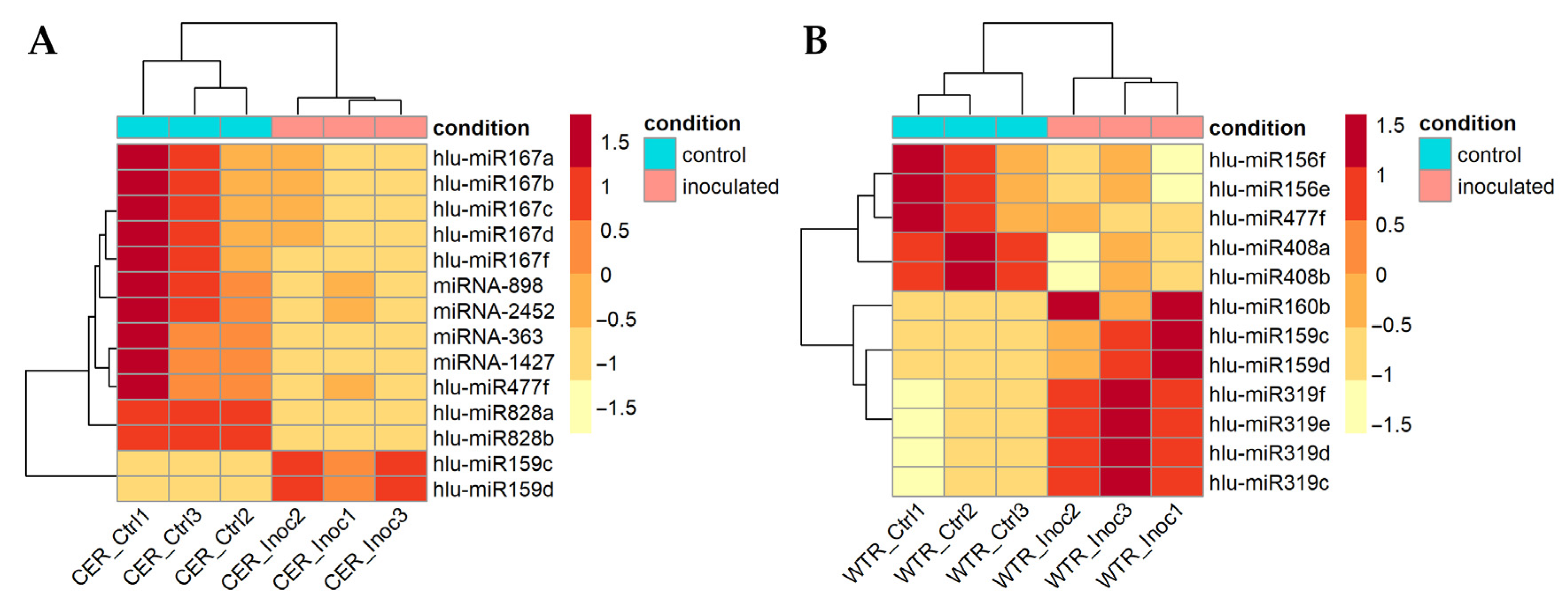
2.3. Differentially Expressed miRNAs between V. nonalfalfae-Inoculated and Control Samples
2.4. Differentially Expressed MiRNAs between Susceptible and Resistant Hop Cultivars
2.5. MiRNA Target Prediction, GO Analysis of miRNA Targets and Protein-Protein Interaction Network Analysis
3. Discussion
4. Materials and Methods
4.1. Inoculation of Hop Plants
4.2. Small RNA Isolation, Library Construction and Sequencing
4.3. Prediction, Identification and Differential Expression Analysis of miRNAs in Hops
4.4. In Silico Prediction of MiRNA Targets of Differentially Expressed MiRNAs, Gene Ontology and Protein–Protein Network Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Steenackers, B.; De Cooman, L.; De Vos, D. Chemical transformations of characteristic hop secondary metabolites in relation to beer properties and the brewing process: A review. Food Chem. 2015, 172, 742–756. [Google Scholar] [CrossRef]
- Hrncic, M.K.; Spaninger, E.; Kosir, I.J.; Knez, Z.; Bren, U. Hop Compounds: Extraction Techniques, Chemical Analyses, Antioxidative, Antimicrobial, and Anticarcinogenic Effects. Nutrients 2019, 11, 257. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Gao, X.; Deeb, D.; Arbab, A.S.; Dulchavsky, S.A.; Gautam, S.C. Anticancer agent xanthohumol inhibits IL-2 induced signaling pathways involved in T cell proliferation. J. Exp. Ther. Oncol. 2012, 10, 1–8. [Google Scholar]
- Radišek, S. Verticillium Wilt. In Compendium of Hop Diseases and Pests; Mahaffee, W., Pethybridge, S., Gent, D.H., Eds.; The American Phytopathological Society: St. Paul, MI, USA, 2009; pp. 33–36. [Google Scholar]
- Radisek, S.; Jakse, J.; Javornik, B. Genetic variability and virulence among Verticillium albo-atrum isolates from hop. Eur. J. Plant Pathol. 2006, 116, 301–314. [Google Scholar] [CrossRef]
- Chisholm, S.T.; Coaker, G.; Day, B.; Staskawicz, B.J. Host-microbe interactions: Shaping the evolution of the plant immune response. Cell 2006, 124, 803–814. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, Q.L.; Zhang, Y.; Xia, R.; Meyers, B.C. Small RNAs Add Zing to the Zig-Zag-Zig Model of Plant Defenses. Mol. Plant Microbe Interact. 2016, 29, 165–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Pignatta, D.; Bendix, C.; Brunkard, J.O.; Cohn, M.M.; Tung, J.; Sun, H.Y.; Kumar, P.; Baker, B. MicroRNA regulation of plant innate immune receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 1790–1795. [Google Scholar] [CrossRef] [Green Version]
- D’Ario, M.; Griffiths-Jones, S.; Kim, M. Small RNAs: Big Impact on Plant Development. Trends Plant Sci. 2017, 22, 1056–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto-Suarez, M.; Baldrich, P.; Weigel, D.; Rubio-Somoza, I.; San Segundo, B. The Arabidopsis miR396 mediates pathogen-associated molecular pattern-triggered immune responses against fungal pathogens. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Navarro, L.; Dunoyer, P.; Jay, F.; Arnold, B.; Dharmasiri, N.; Estelle, M.; Voinnet, O.; Jones, J.D.G. A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science 2006, 312, 436–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, J.; Gao, L.; Yang, Y.; Zhai, J.X.; Arikit, S.; Yu, Y.; Duan, S.Y.; Chan, V.; Xiong, Q.; Yan, J.; et al. Roles of small RNAs in soybean defense against Phytophthora sojae infection. Plant J. 2014, 79, 928–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, B.; Kalsi, H.S.; Godbole, P.; Malankar, N.; Thiagarayaselvam, A.; Siddappa, S.; Thulasiram, H.V.; Chakrabarti, S.K.; Banerjee, A.K. MiRNA160 is associated with local defense and systemic acquired resistance against Phytophthora infestans infection in potato. J. Exp. Bot. 2018, 69, 2023–2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagadeeswaran, G.; Zheng, Y.; Li, Y.F.; Shukla, L.I.; Matts, J.; Hoyt, P.; Macmil, S.L.; Wiley, G.B.; Roe, B.A.; Zhang, W.X.; et al. Cloning and characterization of small RNAs from Medicago truncatula reveals four novel legume-specific microRNA families. New Phytol. 2009, 184, 85–98. [Google Scholar] [CrossRef]
- Yin, Z.J.; Li, Y.; Han, X.L.; Shen, F.F. Genome-Wide Profiling of miRNAs and Other Small Non-Coding RNAs in the Verticillium dahliae-Inoculated Cotton Roots. PLoS ONE 2012, 7, e35765. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.X.; Yan, Q.; Gan, S.P.; Xue, D.; Dou, D.L.; Guo, N.; Xing, H. Overexpression of gma-miR1510a/b suppresses the expression of a NB-LRR domain gene and reduces resistance to Phytophthora sojae. Gene 2017, 621, 32–39. [Google Scholar] [CrossRef]
- Lu, S.F.; Sun, Y.H.; Chiang, V.L. Stress-responsive microRNAs in Populus. Plant J. 2008, 55, 131–151. [Google Scholar] [CrossRef]
- Chen, L.; Ren, Y.Y.; Zhang, Y.Y.; Xu, J.C.; Zhang, Z.Y.; Wang, Y.W. Genome-wide profiling of novel and conserved Populus microRNAs involved in pathogen stress response by deep sequencing. Planta 2012, 235, 873–883. [Google Scholar] [CrossRef]
- Jakse, J.; Cerenak, A.; Radisek, S.; Satovic, Z.; Luthar, Z.; Javornik, B. Identification of quantitative trait loci for resistance to Verticillium wilt and yield parameters in hop (Humulus lupulus L.). Theor. Appl. Genet. 2013, 126, 1431–1443. [Google Scholar] [CrossRef]
- Majer, A.; Javornik, B.; Cerenak, A.; Jakse, J. Development of novel EST-derived resistance gene markers in hop (Humulus lupulus L.). Mol. Breed. 2014, 33, 61–74. [Google Scholar] [CrossRef]
- Diwan, N.; Fluhr, R.; Eshed, Y.; Zamir, D.; Tanksley, S.D. Mapping of Ve in tomato: A gene conferring resistance to the broad-spectrum pathogen, Verticillium dahliae race 1. Theor. Appl. Genet. 1999, 98, 315–319. [Google Scholar] [CrossRef]
- Kawchuk, L.M.; Hachey, J.; Lynch, D.R.; Kulcsar, F.; van Rooijen, G.; Waterer, D.R.; Robertson, A.; Kokko, E.; Byers, R.; Howard, R.J.; et al. Tomato Ve disease resistance genes encode cell surface-like receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 6511–6515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fradin, E.F.; Abd-El-Haliem, A.; Masini, L.; van den Berg, G.C.M.; Joosten, M.H.A.J.; Thomma, B.P.H.J. Interfamily Transfer of Tomato Ve1 Mediates Verticillium Resistance in Arabidopsis. Plant Physiol. 2011, 156, 2255–2265. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Fradin, E.; de Jonge, R.; van Esse, H.P.; Smit, P.; Liu, C.M.; Thomma, B.P.H.J. Optimized Agroinfiltration and Virus-Induced Gene Silencing to Study Ve1-Mediated Verticillium Resistance in Tobacco. Mol. Plant Microbe Interact. 2013, 26, 182–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Zhang, Z.; Seidl, M.F.; Majer, A.; Jakse, J.; Javornik, B.; Thomma, B.P.H.J. Broad taxonomic characterization of Verticillium wilt resistance genes reveals an ancient origin of the tomato Ve1 immune receptor. Mol. Plant Pathol. 2017, 18, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Mandelc, S.; Timperman, I.; Radisek, S.; Devreese, B.; Samyn, B.; Javornik, B. Comparative proteomic profiling in compatible and incompatible interactions between hop roots and Verticillium albo-atrum. Plant Physiol.Biochem. 2013, 68, 23–31. [Google Scholar] [CrossRef]
- Progar, V.; Jakse, J.; Stajner, N.; Radisek, S.; Javornik, B.; Berne, S. Comparative transcriptional analysis of hop responses to infection with Verticillium nonalfalfae. Plant Cell Rep. 2017, 36, 1599–1613. [Google Scholar] [CrossRef] [Green Version]
- Cregeen, S.; Radisek, S.; Mandelc, S.; Turk, B.; Stajner, N.; Jakse, J.; Javornik, B. Different Gene Expressions of Resistant and Susceptible Hop Cultivars in Response to Infection with a Highly Aggressive Strain of Verticillium albo-atrum. Plant Mol. Biol. Rep. 2015, 33, 689–704. [Google Scholar] [CrossRef] [Green Version]
- Hill, S.T.; Sudarsanam, R.; Henning, J.; Hendrix, D. HopBase: A unified resource for Humulus genomics. Database-Oxf. 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Padgitt-Cobb, L.K.; Kingan, S.B.; Wells, J.; Elser, J.; Kronmiller, B.; Moore, D.; Concepcion, G.; Peluso, P.; Rank, D.; Jaiswal, P.; et al. A phased, diploid assembly of the Cascade hop (Humulus lupulus) genome reveals patterns of selection and haplotype variation. bioRxiv 2019, 786145. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.K.; Sun, Y. miR-PREFeR: An accurate, fast and easy-to-use plant miRNA prediction tool using small RNA-Seq data. Bioinformatics 2014, 30, 2837–2839. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39, D152–D157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, B.A.; Petrov, A.I.; Burkov, B.; Finn, R.D.; Bateman, A.; Szymanski, M.; Karlowski, W.M.; Gorodkin, J.; Seemann, S.E.; Cannone, J.J.; et al. RNAcentral: A hub of information for non-coding RNA sequences. Nucleic Acids Res. 2019, 47, D221–D229. [Google Scholar] [CrossRef] [Green Version]
- Ambros, V.; Bartel, B.; Bartel, D.P.; Burge, C.B.; Carrington, J.C.; Chen, X.M.; Dreyfuss, G.; Eddy, S.R.; Griffiths-Jones, S.; Marshall, M.; et al. A uniform system for microRNA annotation. RNA 2003, 9, 277–279. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.B.; Zhuang, Z.H.; Zhao, P.X.C. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Qi, L.; Zhang, Z. Isolation and characterization of a novel wall-associated kinase gene TaWAK5 in wheat (Triticum aestivum). Crop. J. 2014, 2, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Li, C.L.; Li, D.Q.; Li, J.; Shao, F.J.; Lu, S.F. Characterization of the polyphenol oxidase gene family reveals a novel microRNA involved in posttranscriptional regulation of PPOs in Salvia miltiorrhiza. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.P.; Jiang, X.L.; Zhang, B.Y.; Su, X.H. Involvement of microRNA-Mediated Gene Expression Regulation in the Pathological Development of Stem Canker Disease in Populus trichocarpa. PLoS ONE 2012, 7, e44968. [Google Scholar] [CrossRef]
- Gupta, O.P.; Permar, V.; Koundal, V.; Singh, U.D.; Praveen, S. MicroRNA regulated defense responses in Triticum aestivum L. during Puccinia graminis f.sp. tritici infection. Mol. Biol. Rep. 2012, 39, 817–824. [Google Scholar] [CrossRef]
- Millar, A.A.; Lohe, A.; Wong, G. Biology and Function of miR159 in Plants. Plants-Basel 2019, 8, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, T.; Liu, Z.H.; Dai, X.H.; Xiang, F.N. Primary root growth in Arabidopsis thaliana is inhibited by the miR159 mediated repression of MYB33, MYB65 and MYB101. Plant Sci. 2017, 262, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Palatnik, J.F.; Wollmann, H.; Schommer, C.; Schwab, R.; Boisbouvier, J.; Rodriguez, R.; Warthmann, N.; Allen, E.; Dezulian, T.; Huson, D.; et al. Sequence and expression differences underlie functional specialization of Arabidopsis MicroRNAs miR159 and miR319. Dev. Cell 2007, 13, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, R.S.; Li, J.Y.; Stahle, M.I.; Dubroue, A.; Gubler, F.; Millar, A.A. Genetic analysis reveals functional redundancy and the major target genes of the Arabidopsis miR159 family. Proc. Natl. Acad. Sci. USA 2007, 104, 16371–16376. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.S.; Woo, M.O.; Koh, E.B.; Lee, J.; Ham, T.H.; Seo, H.S.; Koh, H.J. Teosinte Branched 1 modulates tillering in rice plants. Plant Cell Rep. 2012, 31, 57–65. [Google Scholar] [CrossRef]
- Gaudin, A.C.M.; McClymont, S.A.; Soliman, S.S.M.; Raizada, M.N. The effect of altered dosage of a mutant allele of Teosinte branched 1 (tb1-ref) on the root system of modern maize. Bmc Genet. 2014, 15, 23. [Google Scholar] [CrossRef] [Green Version]
- Mishra, A.K.; Duraisamy, G.S.; Matousek, J.; Radisek, S.; Javornik, B.; Jakse, J. Identification and characterization of microRNAs in Humulus lupulus using high-throughput sequencing and their response to Citrus bark cracking viroid (CBCVd) infection. BMC Genom. 2016, 17, 919. [Google Scholar] [CrossRef] [Green Version]
- Bhogale, S.; Mahajan, A.S.; Natarajan, B.; Rajabhoj, M.; Thulasiram, H.V.; Banerjee, A.K. MicroRNA156: A Potential Graft-Transmissible MicroRNA That Modulates Plant Architecture and Tuberization in Solanum tuberosum ssp. andigena. Plant Physiol. 2014, 164, 1011–1027. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhang, Z.; Liu, D.; Zhang, K.; Li, A.; Mao, L. SQUAMOSA promoter-binding protein-like transcription factors: Star players for plant growth and development. J. Integr. Plant Biol. 2010, 52, 946–951. [Google Scholar] [CrossRef]
- Stone, J.M.; Liang, X.; Nekl, E.R.; Stiers, J.J. Arabidopsis AtSPL14, a plant-specific SBP-domain transcription factor, participates in plant development and sensitivity to fumonisin B1. Plant J. 2005, 41, 744–754. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Shao, F.J.; Macmillan, C.; Wilson, I.W.; van der Merwe, K.; Hussey, S.G.; Myburg, A.A.; Dong, X.M.; Qiu, D.Y. Genomewide analysis of the lateral organ boundaries domain gene family in Eucalyptus grandis reveals members that differentially impact secondary growth. Plant Biotechnol. J. 2018, 16, 124–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi-Ito, K.; Iwamoto, K.; Fukuda, H. LOB DOMAIN-CONTAINING PROTEIN 15 Positively Regulates Expression of VND7, a Master Regulator of Tracheary Elements. Plant Cell Physiol. 2018, 59, 989–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soyano, T.; Thitamadee, S.; Machida, Y.; Chua, N.H. ASYMMETRIC LEAVES2-LIKE19/LATERAL ORGAN BOUNDARIES DOMAIN30 and ASL20/LBD18 Regulate Tracheary Element Differentiation in Arabidopsis. Plant Cell 2008, 20, 3359–3373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reusche, M.; Thole, K.; Janz, D.; Truskina, J.; Rindfleisch, S.; Drubert, C.; Polle, A.; Lipka, V.; Teichmann, T. Verticillium Infection Triggers VASCULAR-RELATED NAC DOMAIN7-Dependent de Novo Xylem Formation and Enhances Drought Tolerance in Arabidopsis. Plant Cell 2012, 24, 3823–3837. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; Lei, Y.; Wang, L.; Liu, J.; Tang, Y.; Zhang, Z.; Chen, A.; Peng, Q.; Yang, Z.; Wu, J. The ghr-miR164 and GhNAC100 module participates in cotton plant defence against Verticillium dahliae. bioRxiv 2018, 440826. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Xia, Y.; Lin, S.; Wang, Y.; Guo, B.; Song, X.; Ding, S.; Zheng, L.; Feng, R.; Chen, S.; et al. Osa-miR164a targets OsNAC60 and negatively regulates rice immunity against the blast fungus Magnaporthe oryzae. Plant J. 2018, 95, 584–597. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.S.; Xie, Q.; Fei, J.F.; Chua, N.H. MicroRNA directs mRNA cleavage of the transcription factor NAC1 to downregulate auxin signals for Arabidopsis lateral root development. Plant Cell 2005, 17, 1376–1386. [Google Scholar] [CrossRef] [Green Version]
- Bari, R.; Jones, J. Role of plant hormones in plant defence responses. Plant Mol. Biol. 2009, 69, 473–488. [Google Scholar] [CrossRef]
- Tiwari, S.B.; Hagen, G.; Guilfoyle, T. The roles of auxin response factor domains in auxin-responsive transcription. Plant Cell 2003, 15, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.W.; Wang, L.J.; Mao, Y.B.; Cai, W.J.; Xue, H.W.; Chen, X.Y. Control of root cap formation by microRNA-targeted auxin response factors in Arabidopsis. Plant Cell 2005, 17, 2204–2216. [Google Scholar] [CrossRef] [Green Version]
- Mallory, A.C.; Bartel, D.P.; Bartel, B. MicroRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response genes. Plant Cell 2005, 17, 1360–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhao, S.L.; Li, J.L.; Hu, X.H.; Wang, H.; Cao, X.L.; Xu, Y.J.; Zhao, Z.X.; Xiao, Z.Y.; Yang, N.; et al. Osa-miR169 Negatively Regulates Rice Immunity against the Blast Fungus Magnaporthe oryzae. Front. Plant Sci. 2017, 8, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorin, C.; Declerck, M.; Christ, A.; Blein, T.; Ma, L.; Lelandais-Briere, C.; Njo, M.F.; Beeckman, T.; Crespi, M.; Hartmann, C. A miR169 isoform regulates specific NF-YA targets and root architecture in Arabidopsis. New Phytol. 2014, 202, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Dodds, P.N.; Rathjen, J.P. Plant immunity: Towards an integrated view of plant-pathogen interactions. Nat. Rev. Genet. 2010, 11, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.T.; Ramachandran, S.R.; Zhai, Y.; Bu, C.Y.; Pappu, H.R.; Hulbert, S.H. A novel fungal effector from Puccinia graminis suppressing RNA silencing and plant defense responses. New Phytol. 2019, 222, 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Pulsifer, I.P.; Kluge, S.; Rowland, O. Arabidopsis LONG-CHAIN ACYL-COA SYNTHETASE 1 (LACS1), LACS2, and LACS3 facilitate fatty acid uptake in yeast. Plant Physiol. Biochem. 2012, 51, 31–39. [Google Scholar] [CrossRef]
- Deng, Y.X.; Lu, S.F. Biosynthesis and Regulation of Phenylpropanoids in Plants. CRC Crit. Rev. Plant Sci. 2017, 36, 257–290. [Google Scholar] [CrossRef]
- Bonar, N.; Liney, M.; Zhang, R.X.; Austin, C.; Dessoly, J.; Davidson, D.; Stephens, J.; McDougall, G.; Taylor, M.; Bryan, G.J.; et al. Potato miR828 Is Associated With Purple Tuber Skin and Flesh Color. Front. Plant Sci. 2018, 9, 1742. [Google Scholar] [CrossRef] [Green Version]
- Aasland, R.; Stewart, A.F.; Gibson, T. The SANT domain: A putative DNA-binding domain in the SWI-SNF and ADA complexes, the transcriptional co-repressor N-CoR and TFIIIB. Trends Biochem. Sci. 1996, 21, 87–88. [Google Scholar] [CrossRef]
- Chi, M.; Liu, C.M.; Su, Y.Q.; Tong, Y.W.; Liu, H.Y. Bioinformatic prediction of upstream microRNAs of PPO and novel microRNAs in potato. Can. J. Plant Sci. 2015, 95, 871–877. [Google Scholar] [CrossRef] [Green Version]
- Ren, G.H.; Wang, B.J.; Zhu, X.D.; Mu, Q.; Wang, C.; Tao, R.; Fang, J.G. Cloning, expression, and characterization of miR058 and its target PPO during the development of grapevine berry stone. Gene 2014, 548, 166–173. [Google Scholar] [CrossRef]
- Schindler, C.; Chen, Y.; Pu, J.; Guo, X.L.; Bonifacino, J.S. EARP is a multisubunit tethering complex involved in endocytic recycling. Nat. Cell Biol. 2015, 17, 639–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flajsman, M.; Radisek, S.; Javornik, B. Pathogenicity Assay of Verticillium nonalfalfae on Hop Plants. Bio-Protoc. 2017, 7, e2171. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Axtell, M.J.; Meyers, B.C. Revisiting Criteria for Plant MicroRNA Annotation in the Era of Big Data. Plant Cell 2018, 30, 272–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, R.; Bernhart, S.H.; Siederdissen, C.H.Z.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.H.; Pan, X.P.; Cox, S.B.; Cobb, G.P.; Anderson, T.A. Evidence that miRNAs are different from other RNAs. Cell Mol. Life Sci. 2006, 63, 246–254. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing, 3.5.1; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Pokorn, T.; Radisek, S.; Javornik, B.; Stajner, N.; Jakse, J. Development of hop transcriptome to support research into host-viroid interactions. PLoS ONE 2017, 12, e0184528. [Google Scholar] [CrossRef] [Green Version]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology, 2.40.0; Bioconductor: Buffalo, NY, USA, 2020. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Num. of Raw Reads | Mean Length of Raw Reads (bp) | Num. of Reads after Processing | Mean Length of Processed Reads (bp) | Reads Mapped to Hop Genome |
|---|---|---|---|---|---|
| CE-Ctrl1 | 10,041,349 | 18 | 4,636,681 | 20 | 2,634,528 (56.82%) |
| CE-Ctrl2 | 6,109,522 | 16 | 2,232,764 | 20 | 1,341,656 (60.09%) |
| CE-Ctrl3 | 8,146,318 | 17 | 3,232,808 | 20 | 2,044,935 (63.26%) |
| CE-Inoc1 | 8,631,607 | 16 | 2,672,954 | 19 | 1,575,284 (58.93%) |
| CE- Inoc2 | 6,991,916 | 18 | 3,466,372 | 20 | 2,000,154 (57.70%) |
| CE- Inoc3 | 6,723,592 | 14 | 1,771,295 | 18 | 1,230,008 (69.44%) |
| WT-Ctrl1 | 6,223,982 | 20 | 3,745,411 | 22 | 2,094,410 (55.92%) |
| WT-Ctrl2 | 5,222,013 | 19 | 2,933,213 | 21 | 1,729,720 (58.97%) |
| WT-Ctrl3 | 7,737,001 | 20 | 4,083,377 | 22 | 2,393,417 (58.61%) |
| WT-Inoc1 | 10,059,037 | 18 | 4,435,862 | 21 | 2,559,244 (57.69%) |
| WT-Inoc2 | 7,537,943 | 18 | 3,698,547 | 20 | 2,125,103 (57.46%) |
| WT-Inoc3 | 6,930,753 | 20 | 3,202,208 | 22 | 1,811,863 (56.58%) |
| miRNA | Log2FC; adj. p ≤ 0.1 | Sig. Interaction; p ≤ 0.05 | Target Transcript (Orthologue ID *) | |
|---|---|---|---|---|
| DE in CE | DE in WT | |||
| hlu-miR156e–f | NS | −0.65 | NS | Squamosa promoter-binding-like protein 15 (W9RJ15; hops transcript: GAAW01048142.1) 4, Squamosa promoter-binding-like protein 6 (W9QNN5) 4, Squamosa promoter-binding-like protein 12 (W9RS10) 4, Squamosa promoter-binding-like protein 7 (W9R3S1) 4, Squamosa promoter-binding-like protein 13 (W9QLM6) 4, Squamosa promoter-binding protein 1 (W9SN75) 4, gag-polypeptide of LTR copia-type (3694.POPTR_0008s08210.1), Protein SCO1 homolog 2 (225117.XP_009378785.1), LOB domain-containing protein (W9SE87) |
| hlu-miR159c–d | 1.28 | 0.95 | NS | Putative anion transporter 3 (W9RGS2) 1,3, Transcription factor GAMYB (W9QVM8) 2,4, Integrase catalytic domain-containing protein (A0A087HSL5) 1,2,3,4, Acetyltransferase At3g50280-like (M5VT21) 2,4, Transcription factor MYB51-like (102107.XP_008226955.1), Cytochrome p450 (57918.XP_004291627.1) |
| hlu-miR160b | NS | 1.21 | NS | Auxin response factor (A0A061FPV2) 3,4, Auxin response factor (W9QUH2) 3,4, Auxin response factor (W9S7Q7) 3,4 |
| hlu-miR164b | NS | NS | −1.4 | NAC domain-containing protein 100 (W9QTW9), NAC domain-containing protein (W9QCM5; hops transcript: GAAW01060518.1) |
| hlu-miR167a–d | −1.5 | NS | 1.35 | Mediator of RNA polymerase II transcription subunit (102107.XP_008227322.1) |
| hlu-miR167f | −2.1 | NS | 1.34 | Mediator of RNA polymerase II transcription subunit (102107.XP_008227322.1) |
| hlu-miR169a–d | NS | NS | −2.2 | Nuclear transcription factor Y subunit A-8 (W9QJW4), ATPase (W9WDM0), Nuclear transcription factor Y subunit A-1 (W9RR19), Nuclear transcription factor Y subunit A-10 (W9SK30) |
| hlu-miR169m–n | NS | NS | −1.95 | Nuclear transcription factor Y subunit A-10 (W9SK30), Nuclear transcription factor Y subunit A-1 (W9RR19), TATA-binding protein-associated factor (W9R0N8), Exo84_C domain-containing protein (W9SE15) |
| hlu-miR171g–h | NS | NS | −1.3 | Scarecrow-like protein 22 (102107.XP_008238556.1), Scarecrow-like protein 6-like (57918.XP_004306953.1), GRAS domain-containing protein (A0A251QFM0; hop transcript: GAAW01082848.1), NAD(P)-bd_dom domain-containing protein (M5VYK9) |
| hlu-miR319c–f | NS | 0.85 | NS | Transcription factor GAMYB (W9QVM8) 2,4, Teosinte branched 1, putative isoform 1 (A0A061GDP3) 4 |
| hlu-miR390a | NS | NS | −2.2 | Regulation of response to stimulus (218851.Aquca_1504_00001.1) ARM repeat superfamily protein isoform 1 (A0A061G6D5) Mitochondrial protein (3827.XP_004514187.1) Cation-transporting ATPase (W9RQ63) |
| hlu-miR408a–b | NS | −1.41 | −3.9 | Long chain acyl-CoA synthetase 8 (W9QFE0) 4 |
| hlu-miR828a–b | −3.50 | NS | 2.5 | Serine/threonine-protein phosphatase (W9QNS5) 2, Nuclear pore membrane glycoprotein (W9S8T7), RNA pol II transcription regulator recruiting activity-ATMYB5 (3649.evm.model.supercontig_96.57), 3-hydroxyisobutyryl-CoA hydrolase (A0A087GEK8) 1 |
| miRNA-363 miRNA-1427 | −2.61 | NS | 2.4 | Polyphenol oxidase (W9S222) 1,2, ER lumen retaining receptor family (F6HCQ4) 1,2, Dynamin-related protein 4C (W9QQY9) 2 |
| miRNA-898 miRNA-2452 | −2.49 | NS | NS | L-threonine ammonia-lyase activity (161934.XP_010694863.1), gag-polypeptide of LTR copia-type (3750.XP_008361163.1), Vacuolar protein sorting-associated protein 54 (W9SA63) 1 |
| miRNA-617 | NS | NS | −6.35 | Wall-associated receptor kinase (F6GSN9), DIS3-like exonuclease 2 (W9QVR2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunej, U.; Jakše, J.; Radišek, S.; Štajner, N. Identification and Characterization of Verticillium nonalfalfae-Responsive MicroRNAs in the Roots of Resistant and Susceptible Hop Cultivars. Plants 2021, 10, 1883. https://doi.org/10.3390/plants10091883
Kunej U, Jakše J, Radišek S, Štajner N. Identification and Characterization of Verticillium nonalfalfae-Responsive MicroRNAs in the Roots of Resistant and Susceptible Hop Cultivars. Plants. 2021; 10(9):1883. https://doi.org/10.3390/plants10091883
Chicago/Turabian StyleKunej, Urban, Jernej Jakše, Sebastjan Radišek, and Nataša Štajner. 2021. "Identification and Characterization of Verticillium nonalfalfae-Responsive MicroRNAs in the Roots of Resistant and Susceptible Hop Cultivars" Plants 10, no. 9: 1883. https://doi.org/10.3390/plants10091883
APA StyleKunej, U., Jakše, J., Radišek, S., & Štajner, N. (2021). Identification and Characterization of Verticillium nonalfalfae-Responsive MicroRNAs in the Roots of Resistant and Susceptible Hop Cultivars. Plants, 10(9), 1883. https://doi.org/10.3390/plants10091883