
Comparative Transcriptome Analysis of Two Sugarcane Cultivars in Response to Paclobutrazol Treatment

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Statistics of RNA−seq Data
2.2. Functional Annotation for Unigenes
2.3. Differential Expression Analysis for Sensitive Cultivar LC05−136
2.4. Differential Expression Analysis for Non−Sensitive Cultivar GGZ001
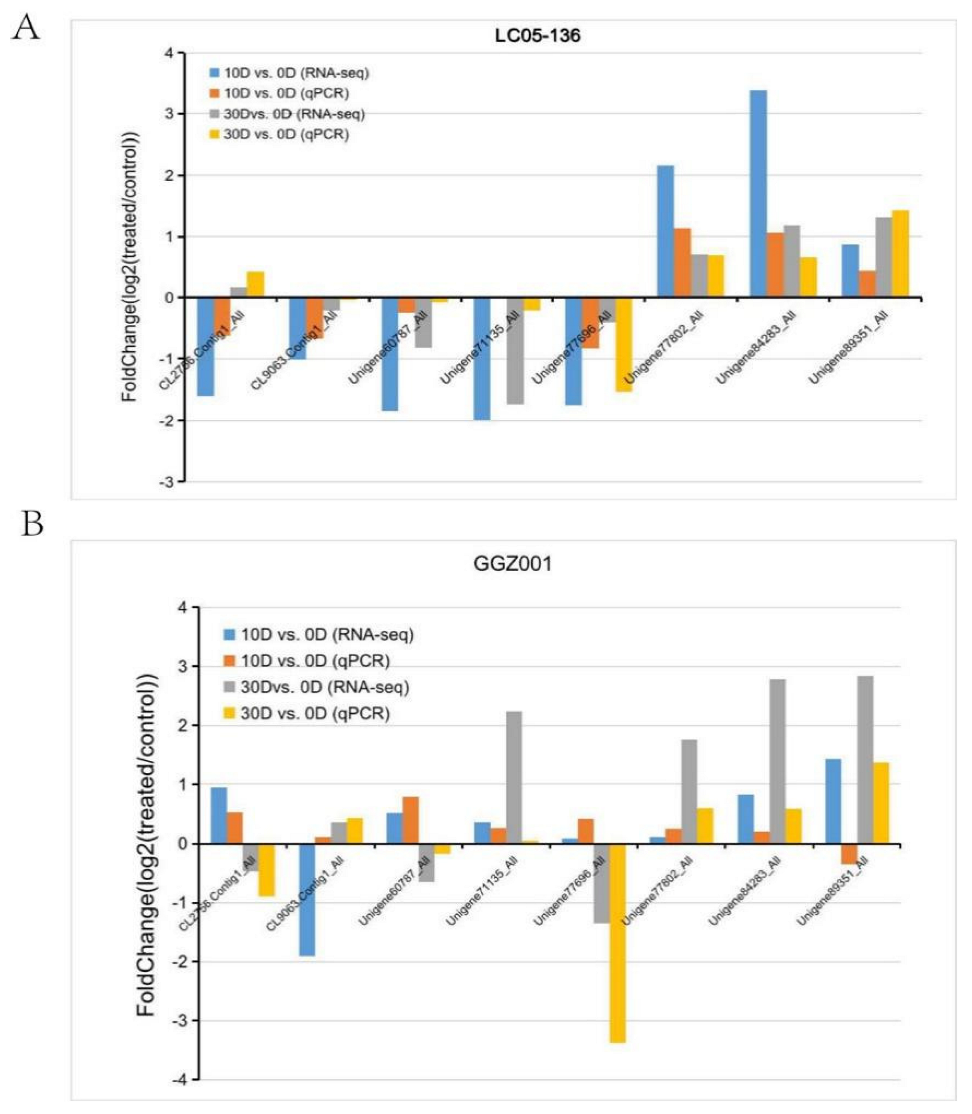
2.5. Validation of DEGs within Sensitive and Non−Sensitive Sugarcane Cultivars
2.6. GO Functional Analysis of DEGs within Sensitive and Non−Sensitive Sugarcane Cultivars
2.7. KEGG Enrichment Analysis of DEGs within Sensitive and Non−Sensitive Sugarcane Cultivars
2.8. DEGs Involved in Metabolism for Sensitive Cultivar LC05−136
2.9. DEGs Associated with Plant–Pathogen Interaction for Non−Sensitive Cultivar GGZ001
3. Discussion
4. Materials and Methods
4.1. Plant Materials and RNA Isolation
4.2. cDNA Library Construction and Sequencing
4.3. De Novo Assembly and Functional Annotation
4.4. Analysis of Differentially Expressed Genes (DEGs)
4.5. RT−qPCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Racedo, J.; Gutiérrez, L.; Perera, M.F.; Ostengo, S.; Pardo, E.M.; Cuenya, M.I.; Welin, B.; Castagnaro, A.P. Genome−wide association mapping of quantitative traits in a breeding population of sugarcane. BMC Plant Biol. 2016, 16, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, T.; Kevers, C.; Penel, C.; Greppin, H.; Reid, D.M.; Thorpe, T.A. Plant hormones and plant growth regulators in plant tissue culture. Vitr. Cell. Dev. Biol. Plant 1996, 32, 272–289. [Google Scholar] [CrossRef]
- Khalil, I.; Rahman, H. Effect of paclobutrazol on growth, chloroplast pigments and sterol biosynthesis of maize (Zea mays L.). Plant Sci. 1995, 105, 15–21. [Google Scholar] [CrossRef]
- Xia, X.; Tang, Y.; Wei, M.; Zhao, D. Effect of paclobutrazol application on plant photosynthetic performance and leaf greenness of herbaceous peony. Horticulturae 2018, 4, 5. [Google Scholar] [CrossRef]
- Kamran, M.; Wennan, S.; Ahmad, I.; Xiangping, M.; Wenwen, C.; Xudong, Z.; Siwei, M.; Khan, A.; Qingfang, H.; Tiening, L. Application of paclobutrazol affect maize grain yield by regulating root morphological and physiological characteristics under a semi−arid region. Sci. Rep. 2018, 8, 1–15. [Google Scholar]
- Tesfahun, W. A review on: Response of crops to paclobutrazol application. Cogent Food Agric. 2018, 4, 1525169. [Google Scholar] [CrossRef]
- Kumar, S.; Ghatty, S.; Satyanarayana, J.; Guha, A.; Chaitanya, B.S.K.; Reddy, A.R. Paclobutrazol treatment as a potential strategy for higher seed and oil yield in field−grown Camelina sativa L. Crantz. BMC Res. Notes 2012, 5, 137. [Google Scholar] [CrossRef]
- Yancheva, S.; Kondakova, V. Plant Tissue Culture Technology: Present and Future Development. In Bioprocessing of Plant In Vitro Systems; Pavlov, A., Bley, T., Eds.; Reference Series in Phytochemistry; Springer: Cham, Switzerland, 2018; pp. 39–63. [Google Scholar]
- Daniels, D.; Panti, N.; Guerra, D.; Williams, S. Effects of Different Paclobutrazol−Cultar Concentrations on the Micropropagation of Sugarcane (Saccharum officinarum) Variety CPCL99−4455. J. Adv. Biotechnol. 2018, 7, 1011–1018. [Google Scholar] [CrossRef]
- Liu, J.; Li, S.; Tan, F.; Liu, X.; He, Y.; Wu, K. Effects of Seed Soaking with Paclobutrazol on Tillering and Physiological Characteristics of Sugarcane Seedlings. Asian Agric. Res. 2017, 9, 65–69. [Google Scholar]
- Owens, N.D.L.; De Domenico, E.; Gilchrist, M.J. An RNA−seq protocol for differential expression analysis. Cold Spring Harb. Protoc. 2019, 2019, pdb-prot098368. [Google Scholar] [CrossRef]
- Gomez−Casati, D.F.; Pagani, M.A.; Busi, M.V.; Bhadauria, V. Omics Approaches for the Engineering of Pathogen Resistant Plants. Curr. Issues Mol. Biol. 2016, 19, 89–98. [Google Scholar] [PubMed]
- Naidoo, S.; Visser, E.A.; Zwart, L.; du Toit, Y.; Bhadauria, V.; Shuey, L.S. Dual rna−seq to elucidate the plant–pathogen duel. Curr. Issues Mol. Biol. 2018, 27, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Ntambo, M.S.; Meng, J.Y.; Rott, P.C.; Henry, R.J.; Zhang, H.L.; Gao, S.J. Comparative transcriptome profiling of resistant and susceptible sugarcane cultivars in response to infection by Xanthomonas albilineans. Int. J. Mol. Sci. 2019, 20, 6138. [Google Scholar] [CrossRef] [PubMed]
- Belesini, A.A.; Carvalho, F.M.S.; Telles, B.R.; de Castro, G.M.; Giachetto, P.F.; Vantini, J.S.; Carlin, S.D.; Cazetta, J.O.; Pinheiro, D.G.; Ferro, M.I.T. De novo transcriptome assembly of sugarcane leaves submitted to prolonged water−deficit stress. Genet. Mol. Res. 2017, 16. [Google Scholar] [CrossRef]
- Que, Y.X.; Su, Y.C.; Guo, J.L.; Wu, Q.B.; Xu, L.P. A global view of transcriptome dynamics during Sporisorium scitamineum challenge in sugarcane by RNA−seq. PLoS ONE 2014, 9, e106476. [Google Scholar] [CrossRef]
- Ling, H.; Huang, N.; Wu, Q.B.; Su, Y.C.; Peng, Q.; Ahmad, W.; Gao, S.W.; Su, W.H.; Que, Y.X.; Xu, L.P. Transcriptional insights into the sugarcane−Sorghum mosaic virus interaction. Trop. Plant Biol. 2018, 11, 163–176. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Gao, S.W.; Su, Y.C.; Lin, Z.L.; Guo, J.L.; Li, M.J.; Wang, Z.T.; Que, Y.X.; Xu, L.P. Transcripts and low nitrogen tolerance: Regulatory and metabolic pathways in sugarcane under low nitrogen stress. Environ. Exp. Bot. 2019, 163, 97–111. [Google Scholar] [CrossRef]
- Almeida, C.M.A.; Donato, V.M.T.S.; Amaral, D.O.J.; Lima, G.S.A.; Brito, G.G.D.; Lima, M.M.D.A.; Correia, M.T.S.; Silva, M.V. Differential gene expression in sugarcane induced by salicylic acid and under water deficit conditions. Agric. Sci. Res. J. 2013, 3, 38–44. [Google Scholar]
- Ibrahim, H.A.; Abdellatif, Y.M.R. Effect of maltose and trehalose on growth, yield and some biochemical components of wheat plant under water stress. Ann. Agric. Sci. 2016, 61, 267–274. [Google Scholar] [CrossRef]
- Huang, B.H.; Xu, X.Q.; Chen, X.H.; Xu, X.P.; Zhang, C.Y.; Lin, Y.L.; Lai, Z.X. Transcriptome Analysis of the Effect of Paclobutrazol on the Growth of Hippeastrum. China J. Trop. Crops 2022, 43, 185–195. [Google Scholar]
- Wit, M.; Galvão, V.; Fankhauser, C. Light−Mediated Hormonal Regulation of Plant Growth and Development. Annu. Rev. Plant Biol. 2016, 67, 513–537. [Google Scholar] [CrossRef] [PubMed]
- Rossato Jr, J.A.S.; Madaleno, L.L.; Mutton, M.J.R.; Higley, L.G.; Fernandes, O.A. Photosynthesis, yield and raw material quality of sugarcane injured by multiple pests. PeerJ 2019, 7, e6166. [Google Scholar] [CrossRef]
- Nair, V.D.; Jaleel, C.A.; Gopi, R.; Panneerselvam, R. Changes in growth and photosynthetic characteristics of Ocimum sanctum under growth regulator treatments. Front. Biol. China 2009, 4, 192–199. [Google Scholar] [CrossRef]
- Cohen, Y.; Aloni, D.D.; Adur, U.; Hazon, H. Characterization of growth−retardant effects on vegetative growth of date palm seedlings. J. Plant Growth Regul. 2013, 32, 533–541. [Google Scholar] [CrossRef]
- Ozmen, A.D.; Tukan, F.O. Effects of paclobutrazol on response of two barley cultivars to salt stress. Biol. Plant. 2003, 46, 263–268. [Google Scholar] [CrossRef]
- Lin, K.H.; Pai, F.H.; Hwang, S.Y.; Lo, H.F. Pre−treating with paclobutrazol enhanced chilling tolerance of sweetpotato. Plant Growth Regul. 2006, 49, 249–262. [Google Scholar] [CrossRef]
- Srivastav, M.; Kishor, A.; Dahuja, A.; Sharma, R.R. Effect of paclobutrazol and salinity on ion leakage, proline content and activities of antioxidant enzymes in mango (Mangifera indica L.). Sci. Hortic. 2010, 125, 785–788. [Google Scholar] [CrossRef]
- Asad, S.; Arshad, M.; Mansoor, S.; Zafar, Y. Effect of various amino acids on shoot regeneration of sugarcane (Sacchrum officinarum L.). Afr. J. Biotechnol. 2009, 8, 1214–1218. [Google Scholar]
- Chen, L.; Song, Y.; Li, S.; Zhang, L.; Zou, C.; Yu, D. The role of WRKY transcription factors in plant abiotic stresses. Biochim. Biophys. Acta−Gene Regul. Mech. 2012, 1819, 120–128. [Google Scholar] [CrossRef]
- Jiang, J.; Ma, S.; Ye, N.; Jiang, M.; Cao, J.; Zhang, J. WRKY transcription factors in plant responses to stresses. J. Integr. Plant Biol. 2017, 59, 86–101. [Google Scholar] [CrossRef]
- Rushton, P.J.; Somssich, I.E.; Ringler, P.; Shen, Q.J. WRKY transcription factors. Trends Plant Sci. 2010, 15, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Yang, Y.; Zhou, Y.; Zhou, J.; Fan, B.; Yu, J.Q.; Chen, Z. Protein−protein interactions in the regulation of WRKY transcription factors. Mol. Plant 2013, 6, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Chu, N.; Ma, P.; Javed, T.; Zaheer, U.; Huang, M.T.; Fu, H.Y.; Gao, S.J. Genome−wide analysis of mitogen−activated protein (MAP) kinase gene family expression in response to biotic and abiotic stresses in sugarcane. Physiol. Plant. 2021, 171, 86–107. [Google Scholar] [CrossRef] [PubMed]
- Virdi, A.S.; Singh, S.; Singh, P. Abiotic stress responses in plants: Roles of calmodulin−regulated proteins. Front. Plant Sci. 2015, 6, 809. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full−length transcriptome assembly from RNA−Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Martin, G.B.; Bogdanove, A.J.; Sessa, G. Understanding the Functions of Plant Disease Resistance Proteins. Annu. Rev. Plant Biol. 2003, 54, 23–61. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA−Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA−seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Raw Reads (M) | Total Clean Reads (M) | Total Mapping (%) | Clean Reads Q20 (%) | Clean Reads Q30 (%) | Clean Reads Ratio (%) |
|---|---|---|---|---|---|---|
| LC05−136−10D−1 | 77.13 | 70.7 | 82.92 | 95.87 | 87.09 | 91.67 |
| LC05−136−10D−2 | 77.13 | 70.85 | 84.43 | 95.72 | 86.67 | 91.86 |
| LC05−136−10D−3 | 77.13 | 70.85 | 84.11 | 95.64 | 86.48 | 91.87 |
| LC05−136−30D−1 | 77.13 | 71.13 | 84.29 | 96.28 | 87.76 | 92.22 |
| LC05−136−30D−2 | 77.13 | 70.91 | 84.04 | 96.36 | 87.91 | 91.94 |
| LC05−136−30D−3 | 77.13 | 71.34 | 84.13 | 96.53 | 88.37 | 92.5 |
| LC05−136−0D−1 | 75.37 | 69.59 | 84.17 | 96.18 | 87.55 | 92.33 |
| LC05−136−0D−2 | 75.37 | 69.16 | 83.97 | 96.12 | 87.33 | 91.75 |
| LC05−136−0D−3 | 77.13 | 71.19 | 84.25 | 96.25 | 87.76 | 92.31 |
| GGZ001−0D−1 | 73.62 | 68.55 | 86.25 | 96.42 | 88.31 | 93.12 |
| GGZ001−0D−2 | 77.13 | 70.82 | 85.36 | 96.11 | 87.42 | 91.83 |
| GGZ001−0D−3 | 75.37 | 69.71 | 86.72 | 96.11 | 87.39 | 92.49 |
| GGZ001−10D−1 | 77.13 | 71.04 | 85.72 | 95.72 | 86.56 | 92.11 |
| GGZ001−10D−2 | 75.37 | 68.5 | 85.95 | 95.56 | 86.21 | 90.89 |
| GGZ001−10D−3 | 75.37 | 68.78 | 85.31 | 95.62 | 86.34 | 91.26 |
| GGZ001−30D−1 | 77.13 | 71.27 | 86.21 | 95.97 | 86.89 | 92.4 |
| GGZ001−30D−2 | 77.13 | 71.22 | 86.47 | 96.11 | 87.3 | 92.35 |
| GGZ001−30D−3 | 77.13 | 70.82 | 85.88 | 96.07 | 87.22 | 91.82 |
| Unigenes | Number | Percentage (%) |
|---|---|---|
| 200–500 bp length | 43,548 | 18.20 |
| 500–1000 bp length | 39,173 | 16.38 |
| 1000–2000 bp length | 71,079 | 29.71 |
| >2000 bp length | 85,412 | 35.71 |
| Total | 239,212 | 100% |
| Minimum length (bp) | 297 | / |
| Mean length (bp) | 1790 | / |
| Maximum length (bp) | 15,177 | / |
| N50 | 2541 | / |
| N90 | 999 | |
| GC% | 47.64 | / |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, R.; Li, H.; Gui, Y.; Wei, J.; Zhu, K.; Zhou, H.; Lakshmanan, P.; Mao, L.; Lu, M.; Liu, J.; et al. Comparative Transcriptome Analysis of Two Sugarcane Cultivars in Response to Paclobutrazol Treatment. Plants 2022, 11, 2417. https://doi.org/10.3390/plants11182417
Zhang R, Li H, Gui Y, Wei J, Zhu K, Zhou H, Lakshmanan P, Mao L, Lu M, Liu J, et al. Comparative Transcriptome Analysis of Two Sugarcane Cultivars in Response to Paclobutrazol Treatment. Plants. 2022; 11(18):2417. https://doi.org/10.3390/plants11182417
Chicago/Turabian StyleZhang, Ronghua, Haibi Li, Yiyun Gui, Jinju Wei, Kai Zhu, Hui Zhou, Prakash Lakshmanan, Lianying Mao, Manman Lu, Junxian Liu, and et al. 2022. "Comparative Transcriptome Analysis of Two Sugarcane Cultivars in Response to Paclobutrazol Treatment" Plants 11, no. 18: 2417. https://doi.org/10.3390/plants11182417
APA StyleZhang, R., Li, H., Gui, Y., Wei, J., Zhu, K., Zhou, H., Lakshmanan, P., Mao, L., Lu, M., Liu, J., Que, Y., Li, S., & Liu, X. (2022). Comparative Transcriptome Analysis of Two Sugarcane Cultivars in Response to Paclobutrazol Treatment. Plants, 11(18), 2417. https://doi.org/10.3390/plants11182417