
Multi-Omics Profiling Identifies Candidate Genes Controlling Seed Size in Peanut

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Phenotypic Variation in Seed Size between Hybs and WT Lines
2.2. Hybs Did Not Show Large Chromosome Structural Variations by Karyotype Analysis and Whole-Genome Resequencing
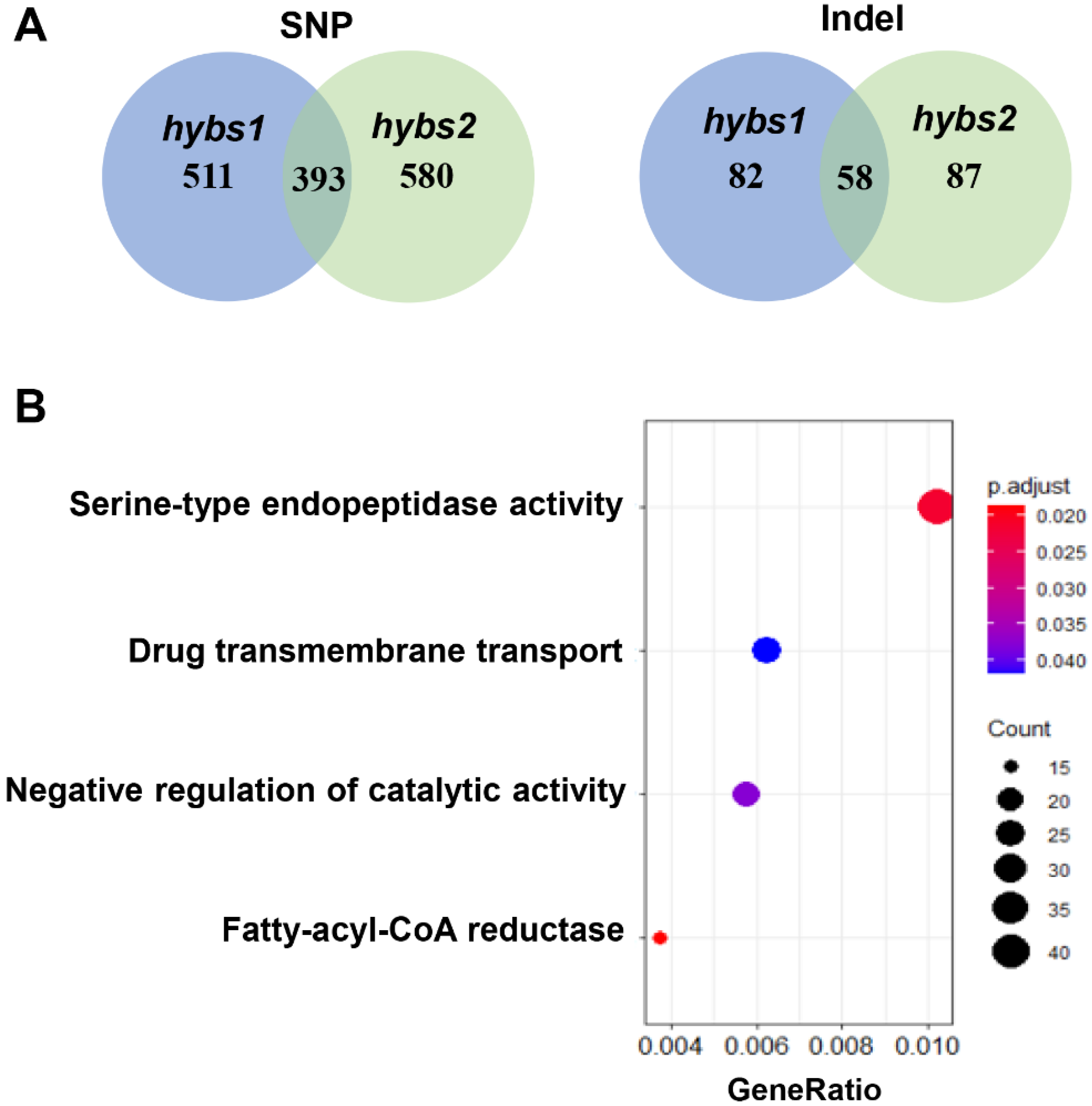
2.3. Single Nucleotide Polymorphism (SNP) and Indel Analysis in WT and hybs Mutants
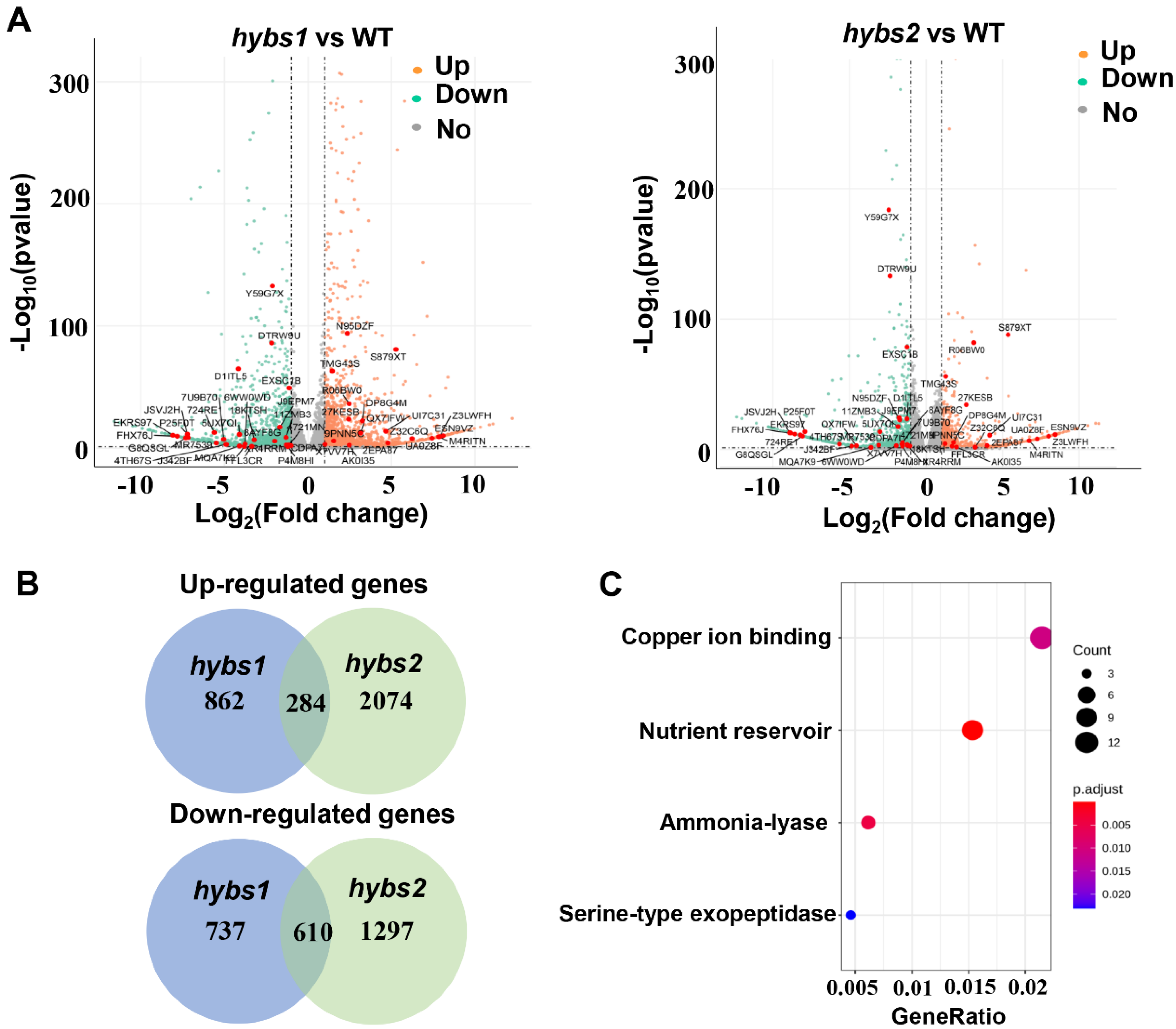
2.4. RNA-Seq Analyses on Seed at the Developmental Stage of WT, Hybs1, and Hybs2
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. DNA Extraction and WGS
4.3. RNA Isolation, RNA-Seq, and Differential Genes Expression Analyses
4.4. qRT-PCR Analysis
4.5. Fluorescence in Situ Hybridization (FISH) and Genomic in Situ Hybridization (GISH)
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kochert, G.; Stalker, H.T.; Gimenes, M.; Galgaro, L.; Lopes, C.R.; Moore, K. RFLP And Cytogenetic Evidence on the Origin and Evolution of Allotetraploid Domesticated Peanut, Arachis hypogaea (Leguminosae). Am. J. Bot. 1996, 83, 1282–1291. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, K.; Luo, L.; Lv, Y.; Li, Y.; Zhu, S.; Luo, B.; Wan, Y.; Zhang, X.; Liu, F. Identification of Peanut Aux/IAA Genes and Functional Prediction during Seed Development and Maturation. Plants 2022, 11, 472. [Google Scholar] [CrossRef]
- Abdurakhmonov, I.Y.; Abdukarimov, A. Application of association mapping to understanding the genetic diversity of plant germplasm resources. Int. J. Plant Genom. 2008, 2008, 574927. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Huang, L.; Ren, X.; Pandey, M.K.; Wu, B.; Chen, Y.; Zhou, X.; Chen, W.; Xia, Y.; Li, Z.; et al. Genetic Variation and Association Mapping of Seed-Related Traits in Cultivated Peanut (Arachis hypogaea L.) Using Single-Locus Simple Sequence Repeat Markers. Front. Plant Sci. 2017, 8, 2105. [Google Scholar] [CrossRef] [Green Version]
- Shirasawa, K.; Koilkonda, P.; Aoki, K.; Hirakawa, H.; Tabata, S.; Watanabe, M.; Hasegawa, M.; Kiyoshima, H.; Suzuki, S.; Kuwata, C.; et al. In silico Polymorphism Analysis for the Development of Simple Sequence Repeat and Transposon Markers and Construction of Linkage Map in Cultivated Peanut. BMC Plant Biol. 2012, 12, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Xu, R.; Li, Y. Molecular Networks of Seed Size Control in Plants. Annu. Rev. Plant Biol. 2019, 70, 435–463. [Google Scholar] [CrossRef]
- Ogawa-Ohnishi, M.; Matsubayashi, Y. Identification of Three Potent Hydroxyproline O-galactosyltransferases in Arabidopsis. Plant J. 2015, 81, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yamamoto, T.; Segami, S.; Horikawa, M.; Chaya, G.; Kitano, H.; Iwasaki, Y.; Miura, K. gw2 Mutation Increases Grain Width and Culm Thickness in Rice (Oryza sativa L.). Breed. Sci. 2020, 70, 456–461. [Google Scholar] [CrossRef]
- Jia, H.; Li, M.; Li, W.; Liu, L.; Jian, Y.; Yang, Z.; Shen, X.; Ning, Q.; Du, Y.; Zhao, R.; et al. A Serine/threonine Protein Kinase Encoding gene KERNEL NUMBER PER ROW6 Regulates Maize Grain Yield. Nat. Commun. 2020, 11, 988. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Liu, H.; Hong, Y.; Li, H.; Liu, H.; Li, X.; Wen, S.; Zhou, G.; Li, S.; Chen, X.; et al. Consensus Map Integration and QTL Meta-analysis Narrowed a Locus for Yield Yraits to 0.7 cM and Refined a Region for Late Leaf Spot Resistance Traits to 0.38 cM on Linkage Group A05 in Peanut (Arachis hypogaea L.). BMC Genom. 2018, 19, 887. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hu, X.; Miao, H.; Chu, Y.; Cui, F.; Yang, W.; Wang, C.; Shen, Y.; Xu, T.; Zhao, L.; et al. QTL Identification for Seed Weight and Size Based on a High-density SLAF-seq Genetic Map in Peanut (Arachis hypogaea L.). BMC Plant Biol. 2019, 19, 537. [Google Scholar] [CrossRef] [PubMed]
- Gangurde, S.S.; Wang, H.; Yaduru, S.; Pandey, M.K.; Fountain, J.C.; Chu, Y.; Isleib, T.; Holbrook, C.C.; Xavier, A.; Culbreath, A.K.; et al. Nested-association Mapping (NAM)-based Genetic Dissection Uncovers Candidate Genes for Seed and Pod Weights in Peanut (Arachis hypogaea). Plant Biotechnol. J. 2020, 18, 1457–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhang, X.; Zhao, K.; Zhao, K.; Qu, C.; Gao, G.; Gong, F.; Ma, X.; Yin, D. Comprehensive Transcriptome Analyses Reveal Candidate Genes for Variation in Seed Size/Weight During Peanut (Arachis hypogaea L.) Domestication. Front. Plant Sci. 2021, 12, 666483. [Google Scholar] [CrossRef] [PubMed]
- Bertioli, D.J.; Cannon, S.B.; Froenicke, L.; Huang, G.; Farmer, A.D.; Cannon, E.K.S.; Liu, X.; Gao, D.; Clevenger, J.; Dash, S.; et al. The Genome Sequences of Arachis duranensis and Arachis ipaensis, the Diploid Ancestors of Cultivated Peanut. Nat. Genet. 2016, 48, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Yin, D.; Ji, C.; Ma, X.; Li, H.; Zhang, W.; Li, S.; Liu, F.; Zhao, K.; Li, F.; Li, K.; et al. Genome of an Allotetraploid Wild Peanut Arachis monticola: A de novo assembly. Gigascience 2018, 7, giy066. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.; Chen, H.; Yang, M.; Wang, J.; Pandey, M.K.; Zhang, C.; Chang, W.-C.; Zhang, L.; Zhang, X.; Tang, R.; et al. The Genome of Cultivated Peanut Provides Insight into Legume Karyotypes, Polyploid Evolution and Crop Domestication. Nat. Genet. 2019, 51, 865–876. [Google Scholar] [CrossRef] [Green Version]
- Bertioli, D.J.; Jenkins, J.; Clevenger, J.; Dudchenko, O.; Gao, D.; Seijo, G.; Leal-Bertioli, S.C.M.; Ren, L.; Farmer, A.D.; Pandey, M.K.; et al. The Genome Sequence of Segmental Allotetraploid Peanut Arachis hypogaea. Nat. Genet. 2019, 51, 877–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Yang, Q.; Li, H.; Li, H.; Hong, Y.; Pan, L.; Chen, N.; Zhu, F.; Chi, X.; Zhu, W.; et al. Transcriptome-wide Sequencing Provides Insights into Geocarpy in Peanut (Arachis hypogaea L.). Plant Biotechnol. J. 2016, 14, 1215–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevenger, J.; Chu, Y.; Scheffler, B.; Ozias-Akins, P. A Developmental Transcriptome Map for Allotetraploid Arachis hypogaea. Front. Plant Sci. 2016, 7, 1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, P.; Bajaj, P.; Pazhamala, L.T.; Nayak, S.N.; Pandey, M.K.; Chitikineni, A.; Huai, D.; Khan, A.W.; Desai, A.; Jiang, H.; et al. Arachis hypogaea Gene Expression Atlas for Fastigiata Subspecies of Cultivated Groundnut to Accelerate Functional and Translational Genomics applications. Plant Biotechnol. J. 2020, 18, 2187–2200. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Wang, Q.; Li, L.; Lang, T.; Guo, J.; Wang, S.; Sun, Z.; Han, S.; Huang, B.; Dong, W.; et al. Physical Mapping of Repetitive Oligonucleotides Facilitates the Establishment of a Genome Map-based Karyotype to Identify Chromosomal Variations in Peanut. BMC Plant Biol. 2021, 21, 107. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Wang, Q.; Li, Z.; Cheng, H.; Li, Z.; Liu, X.; Song, W.; Appels, R.; Zhao, H. Expression of TaCYP78A3, a Gene Encoding Cytochrome P450 CYP78A3 Protein in Wheat (Triticum aestivum L.), Affects Seed Size. Plant J. 2015, 83, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Luo, G.; Song, Y.; Xu, J.; Ji, J.; Zhang, C.; Gregová, E.; Yang, W.; Li, X.; Sun, J.; et al. A Novel NAC Family Transcription Factor SPR Suppresses Seed Storage Protein Synthesis in Wheat. Plant Biotechnol. J. 2021, 19, 992–1007. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, N.; Maji, S.; Waseem, M.; Thakur, P.; Kumar, V.; Parida, S.K.; Thakur, J.K. The Mediator Subunit OsMED15a is a Transcriptional Co-regulator of Seed Size/weight–modulating Genes in Rice. Biochim. Et Biophys. Acta (BBA)-Gene Regul. Mech. 2019, 1862, 194432. [Google Scholar] [CrossRef]
- Huang, L.; He, H.; Chen, W.; Ren, X.; Chen, Y.; Zhou, X.; Xia, Y.; Wang, X.; Jiang, X.; Liao, B.; et al. Quantitative Trait Locus Analysis of Agronomic and Quality-related Traits in Cultivated Peanut (Arachis hypogaea L.). Theor. Appl. Genet. 2015, 128, 1103–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Jiao, Y.; Cheng, L.; Huang, L.; Liao, B.; Tang, M.; Ren, X.; Zhou, X.; Chen, Y.; Jiang, H. Quantitative Trait Locus Analysis for Pod- and Kernel-related Traits in the Cultivated Peanut (Arachis hypogaea L.). BMC Genet. 2016, 17, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.; Ren, X.; Li, Z.; Xu, Z.; Li, X.; Huang, L.; Zhou, X.; Chen, Y.; Chen, W.; Lei, Y.; et al. Co-localization of major quantitative trait loci for pod size and weight to a 3.7 cM interval on chromosome A05 in cultivated peanut (Arachis hypogaea L.). BMC Genom. 2017, 18, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.; Guo, J.; Ren, X.; Chen, W.; Huang, L.; Zhou, X.; Chen, Y.; Liu, N.; Xiong, F.; Lei, Y.; et al. Chromosomes A07 and A05 associated with stable and major QTLs for pod weight and size in cultivated peanut (Arachis hypogaea L.). Theor. Appl. Genet. 2018, 131, 267–282. [Google Scholar] [CrossRef]
- Alyr, M.H.; Pallu, J.; Sambou, A.; Nguepjop, J.R.; Seye, M.; Tossim, H.-A.; Djiboune, Y.R.; Sane, D.; Rami, J.-F.; Fonceka, D. Fine-Mapping of a Wild Genomic Region Involved in Pod and Seed Size Reduction on Chromosome A07 in Peanut (Arachis hypogaea L.). Genes 2020, 11, 1402. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Chee, P.; Isleib, T.G.; Holbrook, C.C.; Ozias-Akins, P. Major seed size QTL on chromosome A05 of peanut (Arachis hypogaea) is conserved in the US mini core germplasm collection. Mol. Breed. 2019, 40, 6. [Google Scholar] [CrossRef]
- Mondal, S.; Badigannavar, A.M. Identification of major consensus QTLs for seed size and minor QTLs for pod traits in cultivated groundnut (Arachis hypogaea L.). 3 Biotech 2019, 9, 347. [Google Scholar] [CrossRef]
- Wang, Z.; Tao, S.; Liu, S.; Jia, M.; Cui, D.; Sun, G.; Deng, Z.; Wang, F.; Kong, X.; Fu, M.; et al. A Multi-Omics Approach for Rapid Identification of Large Genomic Lesions at the Wheat Dense Spike (wds) Locus. Front. Plant Sci. 2022, 13, 850302. [Google Scholar] [CrossRef] [PubMed]
- Komura, S.; Jinno, H.; Sonoda, T.; Oono, Y.; Handa, H.; Takumi, S.; Yoshida, K.; Kobayashi, F. Genome sequencing-based coverage analyses facilitate high-resolution detection of deletions linked to phenotypes of gamma-irradiated wheat mutants. BMC Genom. 2022, 23, 111. [Google Scholar] [CrossRef]
- Liu, G.-S.; Li, H.-L.; Grierson, D.; Fu, D.-Q. NAC Transcription Factor Family Regulation of Fruit Ripening and Quality: A Review. Cells 2022, 11, 525. [Google Scholar] [CrossRef]
- Singh, S.; Koyama, H.; Bhati, K.K.; Alok, A. The biotechnological importance of the plant-specific NAC transcription factor family in crop improvement. J. Plant Res. 2021, 134, 475–495. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.S.; Kim, Y.S.; Redillas, M.C.F.R.; Jang, G.; Jung, H.; Bang, S.W.; Choi, Y.D.; Ha, S.-H.; Reuzeau, C.; Kim, J.-K. OsNAC5 overexpression enlarges root diameter in rice plants leading to enhanced drought tolerance and increased grain yield in the field. Plant Biotechnol. J. 2013, 11, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Mathew, I.E.; Das, S.; Mahto, A.; Agarwal, P. Three Rice NAC Transcription Factors Heteromerize and Are Associated with Seed Size. Front. Plant Sci. 2016, 7, 1638. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Chen, W.; Dong, J.; Li, J.; Yang, F.; Wu, Z.; Zhou, H.; Wang, W.; Zhuang, C. Overexpression of miR164b-resistant OsNAC2 improves plant architecture and grain yield in rice. J. Exp. Bot. 2018, 69, 1533–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, D.R.; Schuler, M.A.; Paquette, S.M.; Werck-Reichhart, D.; Bak, S. Comparative Genomics of Rice and Arabidopsis. Analysis of 727 Cytochrome P450 Genes and Pseudogenes from a Monocot and a Dicot. Plant Physiol. 2004, 135, 756–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, D.R. Plant Cytochrome P450s from Moss to Poplar. Phytochem. Rev. 2006, 5, 193–204. [Google Scholar] [CrossRef]
- Ito, T.; Meyerowitz, E.M. Overexpression of a Gene Encoding a Cytochrome P450, CYP78A9, Induces Large and Seedless Fruit in Arabidopsis. Plant Cell 2000, 12, 1541–1550. [Google Scholar] [CrossRef] [Green Version]
- Adamski Nikolai, M.; Anastasiou, E.; Eriksson, S.; O’Neill Carmel, M.; Lenhard, M. Local Maternal Control of Seed Size by KLUH/CYP78A5-dependent Growth Signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 20115–20120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, M.; Zhang, N.; Sauvage, C.; Muños, S.; Blanca, J.; Cañizares, J.; Diez Maria, J.; Schneider, R.; Mazourek, M.; McClead, J.; et al. A Cytochrome P450 Regulates a Domestication Trait in Cultivated Tomato. Proc. Natl. Acad. Sci. USA 2013, 110, 17125–17130. [Google Scholar] [CrossRef] [Green Version]
- van der Knaap, E.; Chakrabarti, M.; Chu, Y.H.; Clevenger, J.P.; Illa-Berenguer, E.; Huang, Z.; Keyhaninejad, N.; Mu, Q.; Sun, L.; Wang, Y.; et al. What Lies Beyond the Eye: The Molecular Mechanisms Regulating Tomato Fruit Weight and Shape. Front. Plant Sci. 2014, 5, 227. [Google Scholar] [CrossRef] [Green Version]
- Monforte, A.J.; Diaz, A.; Caño-Delgado, A.; van der Knaap, E. The Genetic Basis of Fruit Morphology in Horticultural Crops: Lessons from Tomato and Melon. J. Exp. Bot. 2014, 65, 4625–4637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, M.; Du, P.; Liu, H.; Zhang, Z.; Xu, J.; Qin, L.; Huang, B.; Zheng, Z.; Dong, W.; et al. Transcriptome Analysis of Pod Mutant Reveals Plant Hormones are Important Regulators in Controlling Pod Size in Peanut (Arachis hypogaea L.). PeerJ 2022, 10, e12965. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Q.; Su, H.; Liu, K.; Xiao, X.; Li, W.; Sun, Q.; Birchler, J.A.; Han, F. Genome-wide Mapping Reveals R-loops Associated with Centromeric Repeats in Maize. Genome Res. 2021, 31, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Long-read Alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Wang, C.; Su, H.; Birchler, J.A.; Han, F. Phosphorylation of Histone H3 by Haspin Regulates Chromosome Alignment and Segregation during Mitosis in Maize. J. Exp. Bot. 2021, 72, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python Framework to Work with High-throughput Sequencing Data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Liu, Y.; Su, H.; Liu, Y.; Zhang, J.; Dong, Q.; Birchler, J.A.; Han, F. Cohesion and Centromere Activity are Required for Phosphorylation of Histone H3 in Maize. Plant J. 2017, 92, 1121–1131. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | WT vs. Ref | Hybs1 vs. Ref | Hybs2 vs. Ref | Hybs1 Private Variations | Hybs2 Private Variations |
|---|---|---|---|---|---|
| SNP | 370,912 | 334,753 | 368,220 | 94,719 | 111,194 |
| Indel | 98,445 | 84,850 | 97,956 | 32,012 | 38,600 |
| Hybs1 | Hybs2 | |||
|---|---|---|---|---|
| SNP | Indel | SNP | Indel | |
| Promoter | 6114 | 3375 | 6787 | 3995 |
| Terminator | 5321 | 2826 | 5929 | 3299 |
| 5′ UTR | 336 | 298 | 359 | 361 |
| 3′ UTR | 520 | 308 | 553 | 383 |
| Exon | 2919 | 863 | 3291 | 1037 |
| Intron | 4220 | 2295 | 5053 | 2745 |
| Intergenic | 87580 | 28870 | 102850 | 34838 |
| Splice site acceptor | 41 | 48 | 26 | 44 |
| Splice site donor | 843 | 27 | 1012 | 20 |
| Start lost | 8 | 13 | 6 | 16 |
| Stop gained | 4 | 21 | 4 | 19 |
| Stop lost | 6 | 3 | 7 | 5 |
| Mutant | Gene_ID | Chromosome | Physical Position (bp) | Reference | Variation | SNP/Indel Effect | Predicated Gene Function |
|---|---|---|---|---|---|---|---|
| Hybs1 | Tifrunner.gnm1.ann1.TMG43S | B04 | 21,074,785 | T | C | Amino acid change | Cytochrome P450 superfamily protein |
| Hybs2 | Tifrunner.gnm1.ann1.TMG43S | B04 | 21,074,785 | T | C | Amino acid change | Cytochrome P450 superfamily protein |
| Hybs1 | Tifrunner.gnm1.ann1.CDPA7L | A08 | 30,827,924 | C | CA | Frameshift | NAC transcription factor |
| Hybs2 | Tifrunner.gnm1.ann1.CDPA7L | A08 | 30,827,924 | C | CA | Frameshift | NAC transcription factor |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Yi, C.; Liu, Q.; Wang, C.; Wang, W.; Han, F.; Hu, X. Multi-Omics Profiling Identifies Candidate Genes Controlling Seed Size in Peanut. Plants 2022, 11, 3276. https://doi.org/10.3390/plants11233276
Liu Y, Yi C, Liu Q, Wang C, Wang W, Han F, Hu X. Multi-Omics Profiling Identifies Candidate Genes Controlling Seed Size in Peanut. Plants. 2022; 11(23):3276. https://doi.org/10.3390/plants11233276
Chicago/Turabian StyleLiu, Yang, Congyang Yi, Qian Liu, Chunhui Wang, Wenpeng Wang, Fangpu Han, and Xiaojun Hu. 2022. "Multi-Omics Profiling Identifies Candidate Genes Controlling Seed Size in Peanut" Plants 11, no. 23: 3276. https://doi.org/10.3390/plants11233276
APA StyleLiu, Y., Yi, C., Liu, Q., Wang, C., Wang, W., Han, F., & Hu, X. (2022). Multi-Omics Profiling Identifies Candidate Genes Controlling Seed Size in Peanut. Plants, 11(23), 3276. https://doi.org/10.3390/plants11233276