

Anticholinesterase Activity of Budmunchiamine Alkaloids Revealed by Comparative Chemical Profiling of Two Albizia spp., Molecular Docking and Dynamic Studies
 ,
,  ,
,  ,
,  , , , ,
, , , ,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Acetylcholinesterase Inhibitory Activities of A. lucidior and A. procera Leaves
2.2. Total Phenolic and Flavonoid Contents of A. lucidior and A. procera Leaves
2.3. Dereplication of Metabolites via UHPLC-ESI-QTOF-MS/MS-Based Molecular Networking
Identification of Budmunchiamine Alkaloids

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Rt (min) | Name | Ion m/z ppm | Molecular Formula | MS/MS Fragmentation Product Ions | Ref. | Al | Ap |
|---|---|---|---|---|---|---|---|---|
| 1 | 0.40 | Anthranilic acid | 138.0551 | C7H8NO2+ | 120.0349; 94.0651; 92.0497 | [40] | √ | - |
| 2 | 1.62 | Herniarin | 177.0538 | C10H9O3+ | 149.0575; 145.0266; 133.0982 117.0317 | [41] | - | √ |
| 3 | 2.08 | Budmunchiamine L6 | 465.4159 | C28H57N4O+ | 380.3221; 278.2137; 266.1944 224.2000; 183.1474; 143.1170 100.0759; 86.0960; 58.0654 | [34] | √ | - |
| 4 | 2.55 | Budmunchiamine L5 | 493.4481 | C30H61N4O+ | 408.3579; 294.2570; 288.2337 255.0398; 252.2334; 183.1498 157.1707; 100.0764;98.0973 84.0818; 72.0818; 58.0661 | [34] | √ | - |
| 5 | 2.60 | 6′-Hydroxy budmunchiamine C | 497.4792 | C29H61N4O2+ | 412.3906; 310.2748; 256.2644 183.1497; 157.1703; 100.0759 98.0969; 86.0967; 72.0812 58.0658 | [32] | √ | - |
| 6 | 2.60 | Budmunchiamine B | 425.4220 | C25H53N4O+ | 340.3301; 295.7907; 269.2570 238.2140; 184.2034; 170.1875 157.1669; 100.0730; 98.0940 86.0938; 72.0785; 58.0633 | [42] | √ | - |
| 7 | 2.63 | Budmunchiamine-H/I | 481.4490 | C28H57N4O2+ | 351.3365; 308.2587; 282.2809 266.2484; 254.2483; 183.1493 143.1547; 112.1121; 100.0760 84.0815; 58.0660 | [31] | √ | - |
| 8 | 2.67 | Budmunchiamine D/E | 495.4641 | C29H59N4O2+ | 410.3745; 308.2592; 298.2726 254.2486; 183.1498; 157.1702 100.0760; 98.0969; 86.0968 72.0812; 58.0660 | [31] | √ | - |
| 9 | 2.94 | 6′-Hydroxy-normethyl budmunchiamine K | 511.4934 | C30H63N4O2+ | 381.3848; 338.3056; 324.3253 284.2954; 283.2834; 183.1490 143.1542; 112.1119; 100.0755 84.0808; 72.0809; 58.0565 | [32] | √ | - |
| 10 | 2.96 | 6′-Hydroxy budmunchiamine K | 525.5092 | C31H65N4O2+ | 440.4201; 338.3040; 297.2970 284.2934; 183.1471;157.1680 100.0738; 98.0947; 86.0945 72.0792; 58.0640 | [33] | √ | - |
| 11 | 2.99 | Budmunchiamine A | 453.4532 | C27H57N4O+ | 368.3624; 297.2891; 266.2470 212.2360 183.1475; 157.1683 100.0742; 98.0950; 86.0949 72.0796 58.06420 | [42] | √ | - |
| 12 | 3.24 | Budmunchiamine F | 439.4381 | C26H55N4O+ | 382.3778; 283.2806; 262.2502 227.2287: 212.2357; 185.1633 143.1524; 112.1106; 100.0740 | [31] | √ | - |
| 13 | 3.28 | Budmunchiamine G | 467.4694 | C28H59N4O+ | 337.3575; 294.2788 283.3109 252.2686; 240.2690; 183.1492 143.1536; 112.1117; 100.0755 84.0809; 72.0808; 58.0655 | [33] | √ | - |
| 14 | 3.31 | Budmunchiamine C | 481.4908 | C29H61N4O+ | 396.3958; 297.2891; 294.2795 240.2703; 183.1492; 157.1700 100.0756; 98.0963 86.0967 72.0809; 58.0655 | [42] | √ | - |
| 15 | 3.69 | Normethyl budmunchiamine K | 495.5003 | C30H63N4O+ | 322.3106; 283.3145; 268.3006 183.1488; 143.1538; 112.1120 100.0756; 84.0809; 72.0806 58.0655 | [33] | √ | - |
| 16 | 3.90 | Budmunchiamine K | 509.5168 | C31H65N4O+ | 424.4266; 322.3114 297.3351 268.3004; 183.1490; 157.1700 100.0756; 98.0965; 86.0964 72.0807; 58.0655 | [33] | √ | - |
| 17 | 4.79 | β-sitosterol | 415.2124 | C29H51O+ | 273.0765;135.0809;119.0864 107.0864 | [43] | √ | √ |
| 18 | 4.85 | Stigmasterol | 395.3678 | C29H47+ [M-H2O + H]+ | 255.2623; 173.1322;159.1180 147.31178; 83.0863 | [44] | √ | - |
| 19 | 4.97 | Stearidonic acid | 277.2166 | C18H29O2+ | 259.2056; 235.1665; 149.1333 135.1173; 121.1012; 107.0858 93.0702 | [45] | √ | √ |
| 20 | 5.79 | Arachidonic acid | 305.2466 | C20H33O2+ | 259.02036; 135.1159; 121.1000 107.0845; 93.0691; 55.0539 | [45] | √ | √ |
| 21 | 5.85 | Linolenic acid ethyl ester | 307.2260 | C20H35O2+ | 261.2217; 243.2109;135.1185 123.1165; 109.1006; 95.0854 81.0701; 67.0544 | [46] | √ | √ |
| 22 | 6.18 | 3-O-[glucosyl (1⟶3)-glucoside]-28-O-[rhamnosyl (1⟶2) arabinoside] zanhic acid | 1121.6241 | C53H85O25+ | 959.5738; 843.3981; 519.2929 | [47] | √ | √ |
| 23 | 6.21 | Pheophorbide B | 607.2561 | C35H35N4O6+ | 579.2601; 547.2351; 519.2391 505.2262; 475.2124; 447.2188 433.2024; 419.2213 | [41] | √ | √ |
| 24 | 6.48 | Pheophorbide A | 593.2781 | C35H37N4O5+ | 533.2570; 505.2267; 461.2358 447.2196; 433.2369; 307.2628 177.1113; 133.0853 | [41] | √ | √ |
| 25 | 7.04 | 3-O-[arabinosyl(1→6)] -2-acetamido-2-deoxy-glucosyl oleanolic acid | 792.5636 | C43H70NO12+ | 457.3664; 336.2624; 335.2592 | [48] | √ | √ |
| 26 | 7.12 | 3-O-glucoside-28-O-[rhamnosyl-(1→2)-arabinoside] medicagenic acid | 965.6190 | C47H74O19Na | 803.5684; 687.3933; 525.3411 | [49] | √ | √ |
| 27 | 7.78 | Sapindoside B | 905.5798 | C46H74O16Na | 627.2820; 495.2353 | [50] | √ | - |
| 28 | 8.31 | Julibroside JA2 | 911.6747 | C47H75O17+ | 633.4474; 471.2672 | [51] | √ | √ |
| 29 | 8.37 | Julibroside JA3 | 952.7286 | C49H78NO17+ | 822.7053; 674.5065 | [51] | √ | - |
| 30 | 8.48 | 3-O-[rhamnosyl (1→2)-arabinosyl(1→2)glucoside]-2-hydroxy oleanolic acid | 913.6884 | C47H77O17+ | 635.4620 | [49] | √ | - |
| 31 | 8.93 | Sapinoside A | 773.5747 | C41H66O12Na | 495.2767 | [50] | - | √ |
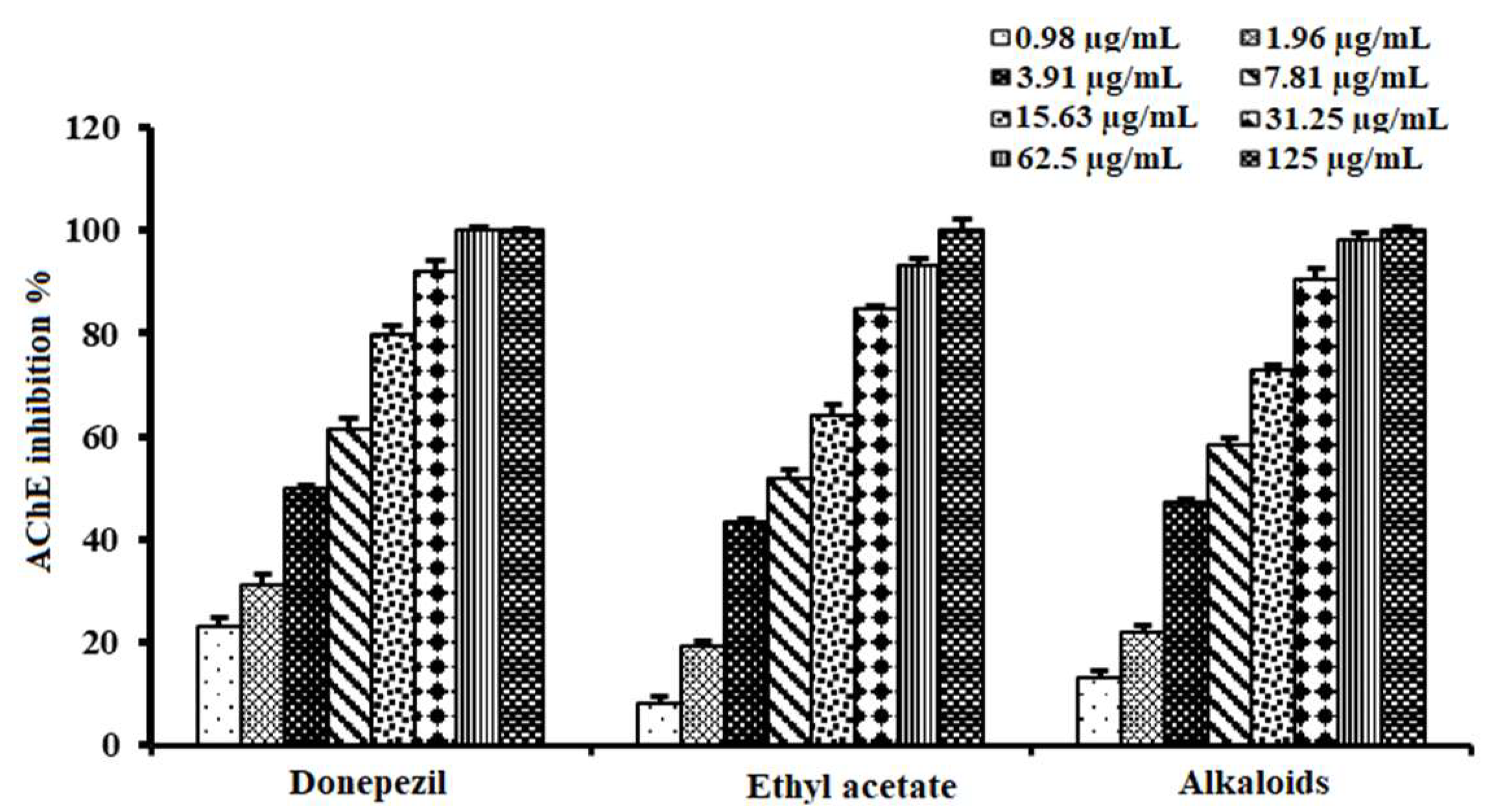
2.4. Acetylcholinesterase Inhibitory Activity of Alkaloid-Rich Fraction of A. lucidior Ethyl Acetate Fraction
2.5. Molecular Docking Simulation
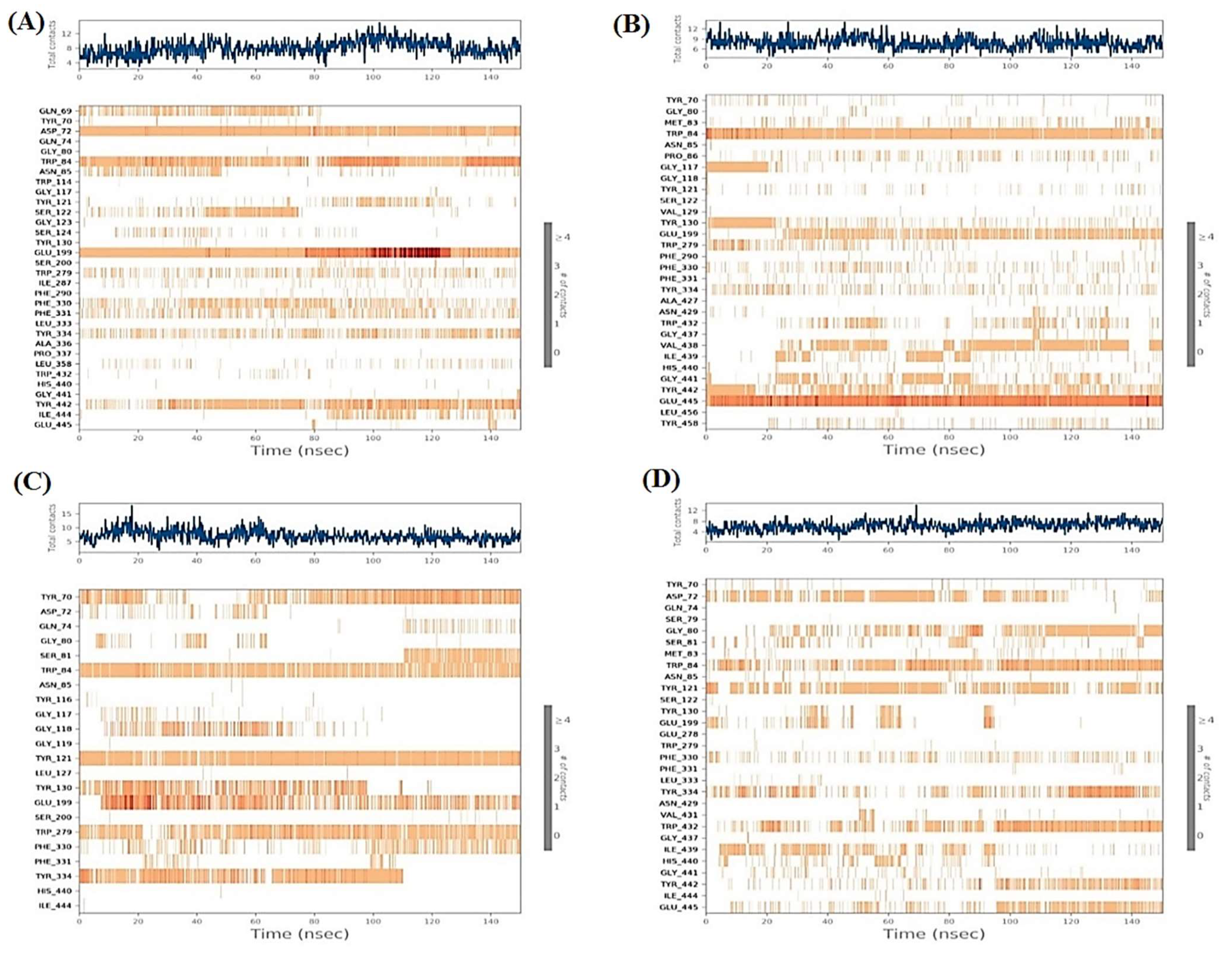
2.6. Molecular Dynamics (MD) Simulations
2.6.1. Histograms Analysis
2.6.2. Heat Maps Analysis
2.7. MD Trajectory Analysis and Prime MM-GBSA Calculations
3. Materials and Methods
3.1. Plant Material, Extraction and Fractionation of Plants
3.2. In Vitro Acetylcholinesterase Assay
3.3. Determination of Total Phenolic and Flavonoid Contents
3.4. UHPLC-QTOF-MS/MS Profiling of the Crude Extracts and Fractions
3.5. GNPS Feature-Based Molecular MS/MS Network
3.6. Preparation and Purification of Alkaloid-Rich Fraction
3.7. Docking Studies and Validation of the MOE Software
3.8. Molecular Dynamics Simulations
3.9. MD Trajectory Analysis and Prime MM-GBSA Calculations
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gauthier, S.; Rosa-Neto, P.; Morais, J.A.; Webster, C. World Alzheimer Report 2021: Journey through the diagnosis of dementia. Alzheimer’s Dis. Int. 2021. [Google Scholar]
- Schachter, A.S.; Davis, K.L. Alzheimer’s disease. Dialogues Clin. Neurosci. 2000, 2, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [Green Version]
- Akram, M.; Nawaz, A. Effects of medicinal plants on Alzheimer’s disease and memory deficits. Neural Regen. Res. 2017, 12, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Roy, A. Role of medicinal plants against Alzheimer’s disease. Int. J. Complement. Alt. Med. 2018, 11, 205–208. [Google Scholar] [CrossRef] [Green Version]
- Essono Mintsa, M.; Otogo N’nang, E.; Choque, É.; Siah, A.; Jacquin, J.; Muchembled, J.; Molinié, R.; Roulard, R.; Cailleu, D.; Beniddir, M.A.; et al. Combined LC-MS/MS and Molecular Networking Approach Reveals Antioxidant and Antimicrobial Compounds from Erismadelphus exsul Bark. Plants 2022, 11, 1505. [Google Scholar] [CrossRef]
- Arora, N.; Banerjee, A.K. Dereplication in natural product discovery. Curr. Top. Med. Chem. 2019, 19, 101–102. [Google Scholar] [CrossRef] [PubMed]
- Jarmusch, A.K.; Aron, A.T.; Petras, D.; Phelan, V.V.; Bittremieux, W.; Acharya, D.D.; Ahmed, M.M.; Bauermeister, A.; Bertin, M.J.; Boudreau, P.D.; et al. A Universal Language for Finding Mass Spectrometry Data Patterns. bioRxiv 2022. [Google Scholar] [CrossRef]
- Hegazi, N.M.; Khattab, A.R.; Frolov, A.; Wessjohann, L.A.; Farag, M.A. Authentication of saffron spice accessions from its common substitutes via a multiplex approach of UV/VIS fingerprints and UPLC/MS using molecular networking and chemometrics. Food Chem. 2022, 367, 130739. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinzi, L.; Rastelli, G. Molecular docking: Shifting paradigms in drug discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wang, Q.; Ye, Y.; Liu, Z.; Sun, H. The ethnopharmacology, phytochemistry, pharmacology and toxicology of genus Albizia: A review. J. Ethnopharmacol. 2020, 257, 112677. [Google Scholar] [CrossRef] [PubMed]
- Risa, A.; Risa, J.; Adsersen, A.; Stafford, G.I.; Van Staden, J.; Jäger, A.K. Acetylcholinesterase inhibitory activity of plants used as memory-enhancers in traditional South African medicine. S. Afr. J. Bot. 2004, 70, 664–666. [Google Scholar] [CrossRef] [Green Version]
- Stafford, G.I.; Pedersen, M.E.; van Staden, J.; Jäger, A.K. Review on plants with CNS-effects used in traditional South African medicine against mental diseases. J. Ethnopharmacol. 2008, 119, 513–537. [Google Scholar] [CrossRef] [PubMed]
- Ingkaninan, K.; Temkitthawon, P.; Chuenchom, K.; Yuyaem, T.; Thongnoi, W. Screening for acetylcholinesterase inhibitory activity in plants used in Thai traditional rejuvenating and neurotonic remedies. J. Ethnopharmacol. 2003, 89, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Eldeen, I.M.S.; Elgorashi, E.E.; Van Staden, J. Antibacterial, anti-inflammatory, anti-cholinesterase and mutagenic effects of extracts obtained from some trees used in South African traditional medicine. J. Ethnopharmacol. 2005, 102, 457–464. [Google Scholar] [CrossRef]
- Dhanya, K.; Satish, S.; Hegde, K.; Shabaraya, A.R. Investigation on Learning and Memory Enhancing activity of Essential Oil in Albizia julibrissin Flowers in Experimental Mice. Asian J. Biomed. Pharm. Sci. 2016, 6, 11–15. [Google Scholar]
- Sonibare, M.A.; Ayoola, I.O.; Elufioye, T.O. Antioxidant and acetylcholinesterase inhibitory activities of leaf extract and fractions of Albizia adianthifolia (Schumach) WF Wright. J. Basic Clin. Physiol. Pharmacol. 2017, 28, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Rasool, M.; Malik, A.; Waquar, S.; Tul-Ain, Q.; Jafar, T.H.; Rasool, R.; Kalsoom, A.; Ghafoor, M.A.; Sehgal, S.A.; Gauthaman, K.; et al. In-silico characterization and in-vivo validation of Albiziasaponin-A, Iso-Orientin, and Salvadorin using a rat model of alzheimer’s disease. Front. Pharmacol. 2018, 9, 730. [Google Scholar] [CrossRef]
- Saleem, U.; Raza, Z.; Anwar, F.; Ahmad, B.; Hira, S.; Ali, T. Experimental and computational studies to characterize and evaluate the therapeutic effect of Albizia lebbeck (L.) seeds in Alzheimer’s disease. Medicina 2019, 55, 184. [Google Scholar] [CrossRef] [Green Version]
- Yadav, S.; Bhadoria, B.K. Novel biflavonoids from the leaves of Leucaena diversifolia and Albizia procera and their protein binding efficiency. J. Indian Chern. Soc. 2004, 81, 392–394. [Google Scholar] [CrossRef]
- Melek, F.; Ghaly, N.S.; El-Kady, M.; Nabil, M. Flavonoids from Albizia procera. Egy. J. Pure Appl. Sci. 2011, 49, 79–82. [Google Scholar]
- Sivakrishnan, S.; Muthu, A.K.; Kavitha, J. GC-MS analysis of ethanolic extract of aerial parts of Albizia procera (Roxb.) Benth. Int. J. Pharm. Pharm. Sci. 2013, 5, 702–704. [Google Scholar]
- Wankhade, M.; Mulani, R. Chromatographic screening and phytochemical investigationon leaf and Bark methanolic extracts of Albizia procera (Roxb.) Benth. Int. J. Innov. Pharm. Sci. Res. 2015, 3, 1662–1674. [Google Scholar]
- Sivakrishnan, S.; Muthu, A.K.; Veeramani, G. Isolation of active compounds from aerial parts of Albizia procera. J. Glob. Pharma Technol. 2019, 11, 1–6. [Google Scholar]
- Srivastava, V.; Verma, S.K.; Panwar, S.; Deep, P.; Verma, S. A brief review on phytopharmacological reports on Albizia procera. Asian J. Pharm. Sci. 2020, 6, 144–149. [Google Scholar] [CrossRef]
- Pradhan, A.; Bhuyan, S.; Chhetri, K.; Mandal, S.; Bhattacharyya, A. Saponins from Albizia procera extract: Surfactant activity and preliminary analysis. Colloids Surf. A Physicochem. Eng. Asp. 2022, 643, 128778. [Google Scholar] [CrossRef]
- Hussein, M.E.; Mohamed, O.G.; El-Fishawy, A.M.; El-Askary, H.I.; El-Senousy, A.S.; El-Beih, A.A.; Nossier, E.S.; Naglah, A.M.; Almehizia, A.A.; Tripathi, A.; et al. Identification of Antibacterial Metabolites from Endophytic Fungus Aspergillus fumigatus, Isolated from Albizia lucidior Leaves (Fabaceae), Utilizing Metabolomic and Molecular Docking Techniques. Molecules 2022, 27, 1117. [Google Scholar] [CrossRef]
- Hussein, M.E.; El-Senousy, A.S.; El-Askary, H.I.; El Fishawy, A.M.; Hamed, A.A.; Youness, R.A. Pseurotin A Halts Hepatocellular Carcinoma Oncogenic Potential Through Tuning miR-30a and let-7i Tumor Suppressor miRNAs. Egypt. J. Chem. 2022, 65, 6186. [Google Scholar] [CrossRef]
- Roseiro, L.B.; Rauter, A.P.; Serralheiro, M.L.M. Polyphenols as acetylcholinesterase inhibitors: Structural specificity and impact on human disease. Nutr. Aging 2012, 1, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Pezzuto, J.M.; Mar, W.; Lin, L.-Z.; Cordell, G.A.; Neszmélyi, A.; Wagner, H. Budmunchiamines D–I from Albizia amara. Phytochemistry 1992, 31, 1795–1800. [Google Scholar] [CrossRef]
- Rukunga, G.M.; Waterman, P.G. Spermine alkaloids from Albizia schimperana. Phytochemistry 1996, 42, 1211–1215. [Google Scholar] [CrossRef]
- Rukunga, G.M.; Waterman, P.G. New macrocyclic spermine (Budmunchiamine) Alkaloids from Albizia gummifera: With some observations on the structure−Activity relationships of the Budmunchiamines. J. Nat. Prod. 1996, 59, 850–853. [Google Scholar] [CrossRef] [PubMed]
- Dixit, A.K.; Misra, L.N. Macrocyclic budmunchiamine alkaloids from Albizia lebbek. J. Nat. Prod. 1997, 60, 1036–1037. [Google Scholar] [CrossRef]
- Samoylenko, V.; Jacob, M.R.; Khan, S.I.; Zhao, J.; Tekwani, B.L.; Midiwo, J.O.; Walker, L.A.; Muhammad, I. Antimicrobial, antiparasitic and cytotoxic spermine alkaloids from Albizia schimperiana. Nat. Prod. Commun. 2009, 4, 791–796. [Google Scholar] [CrossRef] [Green Version]
- Thippeswamy, S.; Mohana, D.C.; Abhishek, R.U.; Manjunath, K. Evaluation of some pharmacological activities of Budmunchiamine-A isolated from Albizia amara. Braz. J. Microbiol. 2015, 46, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Thippeswamy, S.; Mohana, D.C.; Abhishek, R.U.; Manjunath, K. Efficacy of bioactive compounds isolated from Albizia amara and Albizia saman as source of antifungal and antiaflatoxigenic agents. J. Verbr. Lebensm. 2013, 8, 297–305. [Google Scholar] [CrossRef]
- Jarrell, J.T.; Gao, L.; Cohen, D.S.; Huang, X. Network medicine for Alzheimer’s disease and traditional Chinese medicine. Molecules 2018, 23, 1143. [Google Scholar] [CrossRef] [Green Version]
- Bhadra, S.; Dalai, M.K.; Chanda, J.; Mukherjee, P.K. Evaluation of bioactive compounds as acetylcholinesterase inhibitors from medicinal plants. In Evidence-Based Validation of Herbal Medicine; Mukherjee, P.K., Ed.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 273–306. [Google Scholar]
- Chobot, V.; Hadacek, F.; Weckwerth, W.; Kubicova, L. Iron chelation and redox chemistry of anthranilic acid and 3-hydroxyanthranilic acid: A comparison of two structurally related kynurenine pathway metabolites to obtain improved insights into their potential role in neurological disease development. J. Organomet. Chem. 2015, 782, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Rosa, W.; da Silva Domingos, O.; de Oliveira Salem, P.P.; Caldas, I.S.; Murgu, M.; Lago, J.H.G.; Sartorelli, P.; Dias, D.F.; Chagas-Paula, D.A.; Soares, M.G. In vivo anti-inflammatory activity of Fabaceae species extracts screened by a new ex vivo assay using human whole blood. Phytochem. Anal. 2021, 32, 859–883. [Google Scholar] [CrossRef] [PubMed]
- Popaj, K.; Hesse, M. Syntheses of the Macrocyclic Spermine Alkaloids (±)-Budmunchiamine A–C. Helv. Chim. Acta 2001, 84, 180–186. [Google Scholar] [CrossRef]
- Wang, Y.; Jiao, J.; Yang, Y.; Yang, M.; Zheng, Q. Screening and identification for immunological active components from andrographis herba using macrophage biospecific extraction coupled with UPLC/Q-TOF-MS. Molecules 2018, 23, 1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Wang, S.; Yang, R.; Mao, J.; Jiang, J.; Wang, X.; Zhang, W.; Zhang, Q.; Li, P. Simultaneous determination of tocopherols, carotenoids and phytosterols in edible vegetable oil by ultrasound-assisted saponification, LLE and LC-MS/MS. Food Chem. 2019, 289, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Ristivojević, P.; Jovanović, V.; Opsenica, D.M.; Park, J.; Rollinger, J.M.; Velicković, T.Ć. Rapid analytical approach for bioprofiling compounds with radical scavenging and antimicrobial activities from seaweeds. Food Chem. 2021, 334, 127562. [Google Scholar] [CrossRef]
- Ma, X.; Shao, S.; Xiao, F.; Zhang, H.; Zhang, R.; Wang, M.; Li, G.; Yan, M. Platycodon grandiflorum extract: Chemical composition and whitening, antioxidant, and anti-inflammatory effects. RSC Adv. 2021, 11, 10814–10826. [Google Scholar] [CrossRef]
- Wang, G.; Wang, J.; Liu, W.; Nisar, M.F.; El-Esawi, M.A.; Wan, C. Biological activities and chemistry of triterpene saponins from Medicago species: An update review. Evid. Based Complement. Altern. Med. 2021, 2021, 6617916. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Samadi, A.K.; Rao, K.V.; Cohen, M.S.; Timmermann, B.N. Cytotoxic oleanane-type saponins from Albizia inundata. J. Nat. Prod. 2011, 74, 477–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tava, A.; Mella, M.; Avato, P.; Argentieri, M.P.; Bialy, Z.; Jurzysta, M. Triterpenoid glycosides from leaves of Medicago arborea L. J. Agric. Food. Chem. 2005, 53, 9954–9965. [Google Scholar] [CrossRef]
- Ling, Y.; Zhang, Q.; Zhong, W.; Chen, M.; Gong, H.; He, S.; Liang, R.; Lv, J.; Song, L. Rapid identification and analysis of the major chemical constituents from the fruits of Sapindus mukorossi by HPLC-ESI-QTOF-MS/MS. Nat. Prod.Res. 2020, 34, 2144–2150. [Google Scholar] [CrossRef]
- Kinjo, J.; Araki, K.; Fukui, K.; Higuchi, H.; Ikeda, T.; Nohara, T.; Ida, Y.; Takenoto, N.; Miyakoshi, M.; Shoji, J. Six new triterpenoidal glycosides including two new sapogenols from Albizziac Cortex. V. Chem. Pharm. Bull. 1992, 40, 3269–3273. [Google Scholar] [CrossRef] [Green Version]
- El-Masry, R.M.; Al-Karmalawy, A.A.; Alnajjar, R.; Mahmoud, S.H.; Mostafa, A.; Kadry, H.H.; Abou-Seri, S.M.; Taher, A.T. Newly synthesized series of oxoindole–oxadiazole conjugates as potential anti-SARS-CoV-2 agents: In silico and in vitro studies. New J. Chem. 2022, 46, 5078–5090. [Google Scholar] [CrossRef]
- Elufioye, T.O.; Chinaka, C.G.; Oyedeji, A.O. Antioxidant and anticholinesterase activities of Macrosphyra longistyla (DC) Hiern relevant in the management of Alzheimer’s Disease. Antioxidants 2019, 8, 400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attard, E. A rapid microtitre plate Folin-Ciocalteu method for the assessment of polyphenols. Open Life Sci. 2013, 8, 48–53. [Google Scholar] [CrossRef]
- Kiranmai, M.; Kumar, C.B.M.; Ibrahim, M. Comparison of total flavanoid content of Azadirachta indica root bark extracts prepared by different methods of extraction. Res. J. Pharm. Biol.Chem.Sci. 2011, 2, 254–261. [Google Scholar]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nothias, L.-F.; Petras, D.; Schmid, R.; Dührkop, K.; Rainer, J.; Sarvepalli, A.; Protsyuk, I.; Ernst, M.; Tsugawa, H.; Fleischauer, M.; et al. Feature-based molecular networking in the GNPS analysis environment. Nat. Methods 2020, 17, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Li, W.; Wang, H.; Dong, A. Preparative separation of alkaloids from stem of Euchresta tubulosa dunn. by high-speed counter-current chromatography using stepwise elution. Molecules 2019, 24, 4602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- C.C.G. Inc. Molecular Operating Environment (MOE); C.C.G. Group Inc.: Bengaluru, India, 2016. [Google Scholar]
- Hamed, M.I.A.; Darwish, K.M.; Soltane, R.; Chrouda, A.; Mostafa, A.; Abo Shama, N.M.; Elhady, S.S.; Abulkhair, H.S.; Khodir, A.E.; Abo Elmaaty, A.A.; et al. β-Blockers bearing hydroxyethylamine and hydroxyethylene as potential SARS-CoV-2 Mpro inhibitors: Rational based design, in silico, in vitro, and SAR studies for lead optimization. RSC Adv. 2021, 11, 35536–35558. [Google Scholar] [CrossRef]
- Salem, M.A.; El-Shiekh, R.A.; Aborehab, N.M.; Al-Karmalawy, A.A.; Ezzat, S.M.; Alseekh, S.; Fernie, A.R. Metabolomics driven analysis of Nigella sativa seeds identifies the impact of roasting on the chemical composition and immunomodulatory activity. Food Chem. 2022, 398, 133906. [Google Scholar] [CrossRef] [PubMed]
- Al-Karmalawy, A.A.; Farid, M.M.; Mostafa, A.; Ragheb, A.Y.; Mahmoud, S.H.; Shehata, M.; Abo Shama, N.M.; GabAllah, M.; Mostafa-Hedeab, G.; Marzouk, M.M. Naturally available flavonoid aglycones as potential antiviral drug candidates against SARS-CoV-2. Molecules 2021, 26, 6559. [Google Scholar] [CrossRef] [PubMed]
- Diab, R.T.; Abdel-Sami, Z.K.; Abdel-Aal, E.H.; Al-Karmalawy, A.A.; Abo-Dya, N.E. Design and synthesis of a new series of 3, 5-disubstituted-1, 2, 4-oxadiazoles as potential colchicine binding site inhibitors: Antiproliferative activity, molecular docking, and SAR studies. New J. Chem. 2021, 45, 21657–21669. [Google Scholar] [CrossRef]
- Release, S. 3: Desmond molecular dynamics system. In Maest.-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2017. [Google Scholar]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Neria, E.; Fischer, S.; Karplus, M. Simulation of activation free energies in molecular systems. J. Chem. Phys. 1996, 105, 1902–1921. [Google Scholar] [CrossRef]
- Manual, D.U. Desmond2. 2. 2009. Available online: https://www.cines.fr/wp-content/uploads/2014/01/des22_user_manual.pdf (accessed on 5 August 2022).
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]








| Extract/Fraction | IC50 (µg/mL) |
|---|---|
| Ethanolic extract of A. procera | 43.50 ± 2.10 e |
| Ethanolic extract of A. lucidior | 24.89 ± 1.60 d |
| Petroleum ether fraction of A. lucidior | ND |
| Dichloromethane fraction of A. lucidior | 17.40 ± 1.30 c |
| Ethyl acetate fraction of A. lucidior | 6.90 ± 0.96 b |
| n-butanol fraction of A. lucidior | >125 f |
| Donepezil | 3.90 ± 0.72 a |
| Tested Samples | IC50 (µg/mL) |
|---|---|
| Ethyl acetate fraction | 6.90 ± 0.96 b |
| Alkaloid-rich fraction | 5.26 ± 0.62 a |
| Donepezil | 3.90 ± 0.72 a |
| Compounds | a S RMSD | 2D Interaction | 3D Interaction | 3D Positioning |
|---|---|---|---|---|
| Normethyl budmunchiamine K | −10.24 1.97 |  |  |  |
| Budmunchiamine L5 | −9.81 1.82 |  |  |  |
| Donepezil | −8.03 1.64 |  |  |  |
| MF2 700 | −7.88 1.29 |  |  |  |
| Complexes | Normethylbudmunchiamine K-1OCE | Budmunchiamine L5-1OCE | Donepezil -1OCE | MF2 700 -1OCE |
|---|---|---|---|---|
| ΔG Binding | −90.69 | −76.83 | −68.99 | −64.55 |
| Coulomb | −32.56 | −150.52 | −44.38 | −51.57 |
| Covalent | 5.14 | 3.72 | 1.36 | 1.37 |
| H-bond | −2.26 | −1.81 | −0.63 | −1.28 |
| Lipo | −32.61 | −35.25 | −30.90 | −23.44 |
| Bind Packing | −6.94 | 0 | −7.66 | 0 |
| Solv_GB | 48.55 | 180.17 | 65.41 | 54.60 |
| VdW | −70.01 | −73.12 | −52.18 | −44.22 |
| St. Dev. | 7.02 | 4.88 | 4.51 | 5.62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussein, M.E.; Mohamed, O.G.; El-Fishawy, A.M.; El-Askary, H.I.; Hamed, A.A.; Abdel-Aziz, M.M.; Alnajjar, R.; Belal, A.; Naglah, A.M.; Almehizia, A.A.; et al. Anticholinesterase Activity of Budmunchiamine Alkaloids Revealed by Comparative Chemical Profiling of Two Albizia spp., Molecular Docking and Dynamic Studies. Plants 2022, 11, 3286. https://doi.org/10.3390/plants11233286
Hussein ME, Mohamed OG, El-Fishawy AM, El-Askary HI, Hamed AA, Abdel-Aziz MM, Alnajjar R, Belal A, Naglah AM, Almehizia AA, et al. Anticholinesterase Activity of Budmunchiamine Alkaloids Revealed by Comparative Chemical Profiling of Two Albizia spp., Molecular Docking and Dynamic Studies. Plants. 2022; 11(23):3286. https://doi.org/10.3390/plants11233286
Chicago/Turabian StyleHussein, Mai E., Osama G. Mohamed, Ahlam M. El-Fishawy, Hesham I. El-Askary, Ahmed A. Hamed, Marwa M. Abdel-Aziz, Radwan Alnajjar, Amany Belal, Ahmed M. Naglah, Abdulrahman A. Almehizia, and et al. 2022. "Anticholinesterase Activity of Budmunchiamine Alkaloids Revealed by Comparative Chemical Profiling of Two Albizia spp., Molecular Docking and Dynamic Studies" Plants 11, no. 23: 3286. https://doi.org/10.3390/plants11233286
APA StyleHussein, M. E., Mohamed, O. G., El-Fishawy, A. M., El-Askary, H. I., Hamed, A. A., Abdel-Aziz, M. M., Alnajjar, R., Belal, A., Naglah, A. M., Almehizia, A. A., Al-Karmalawy, A. A., Tripathi, A., & El Senousy, A. S. (2022). Anticholinesterase Activity of Budmunchiamine Alkaloids Revealed by Comparative Chemical Profiling of Two Albizia spp., Molecular Docking and Dynamic Studies. Plants, 11(23), 3286. https://doi.org/10.3390/plants11233286