
Genome Survey Sequencing and Genetic Background Characterization of Ilex chinensis Sims (Aquifoliaceae) Based on Next-Generation Sequencing



Abstract
:1. Introduction
2. Results
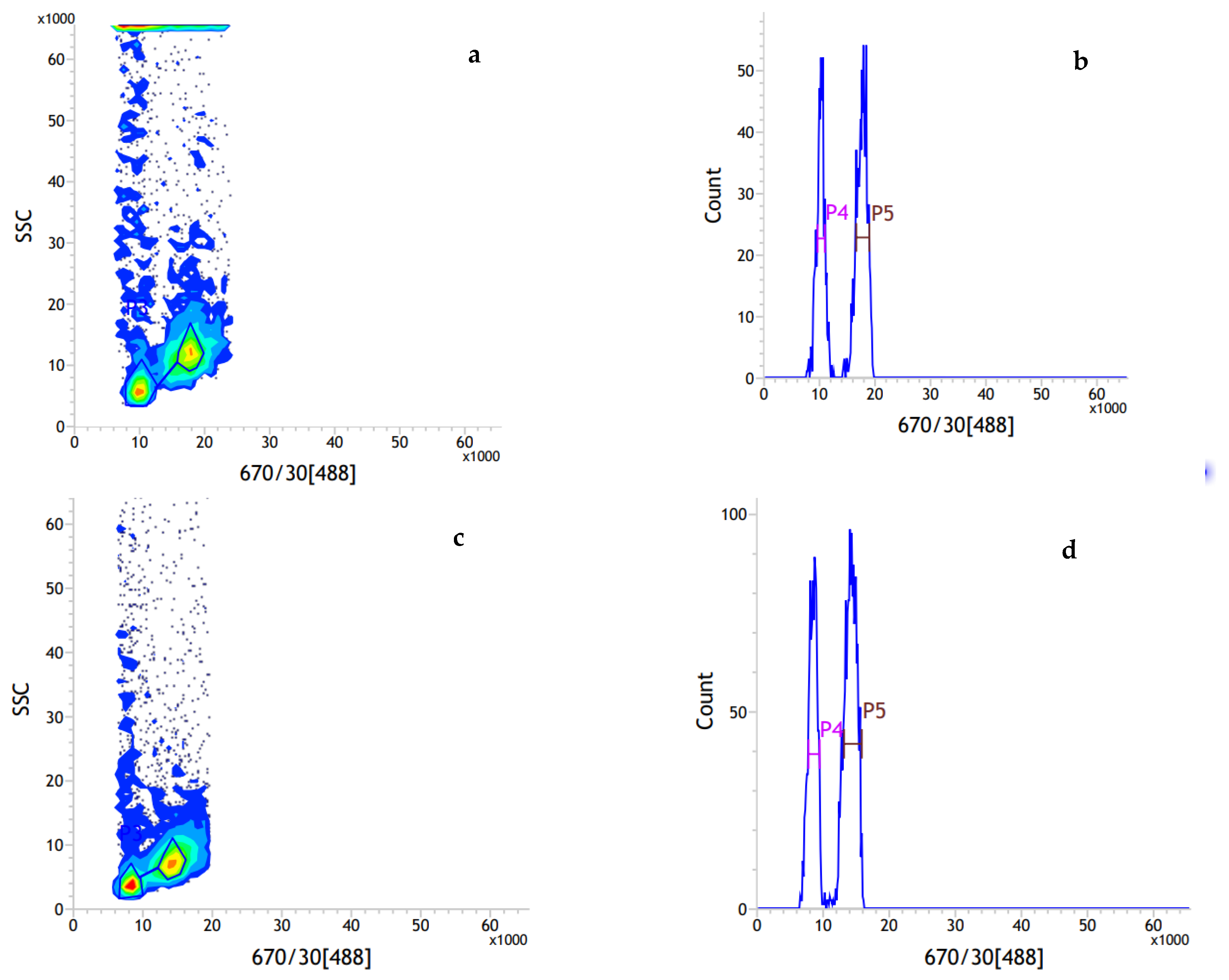
2.1. Genome Size Estimation by Flow Cytometry
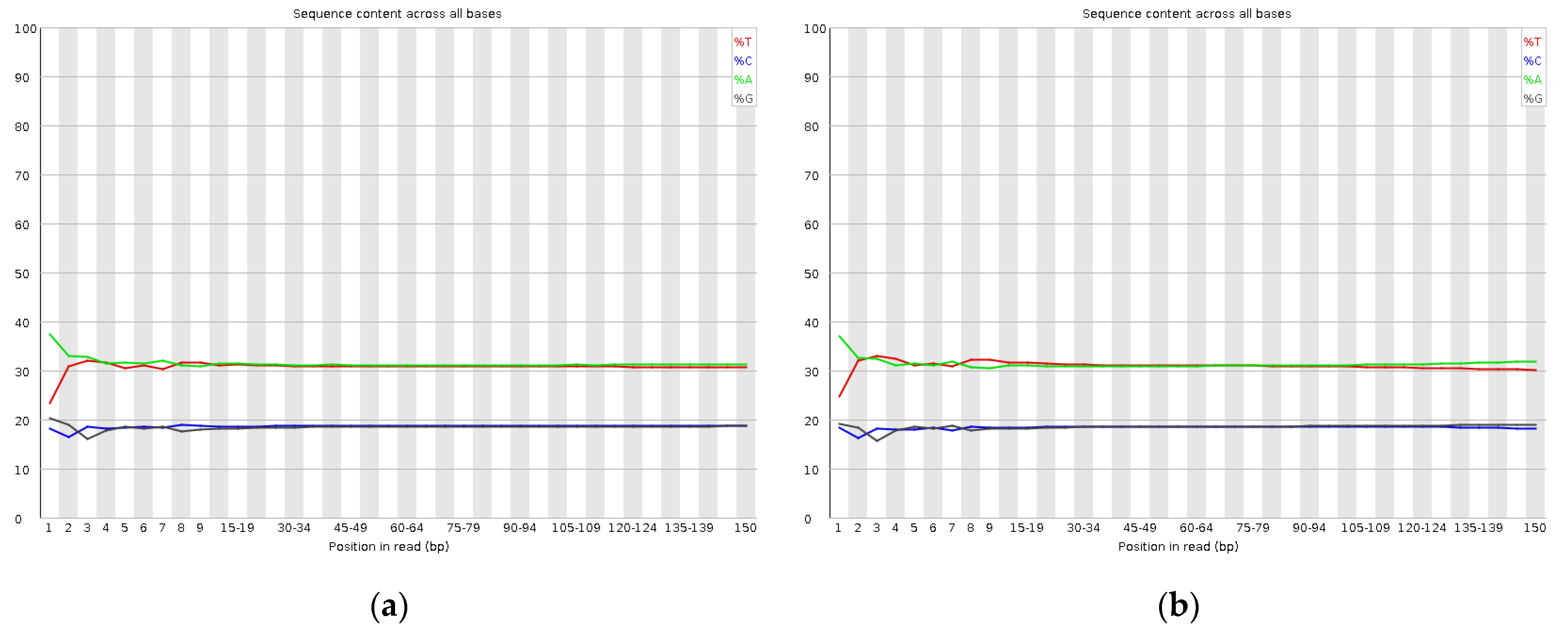
2.2. Sequencing and Quality Evaluation of Ilex chinensis
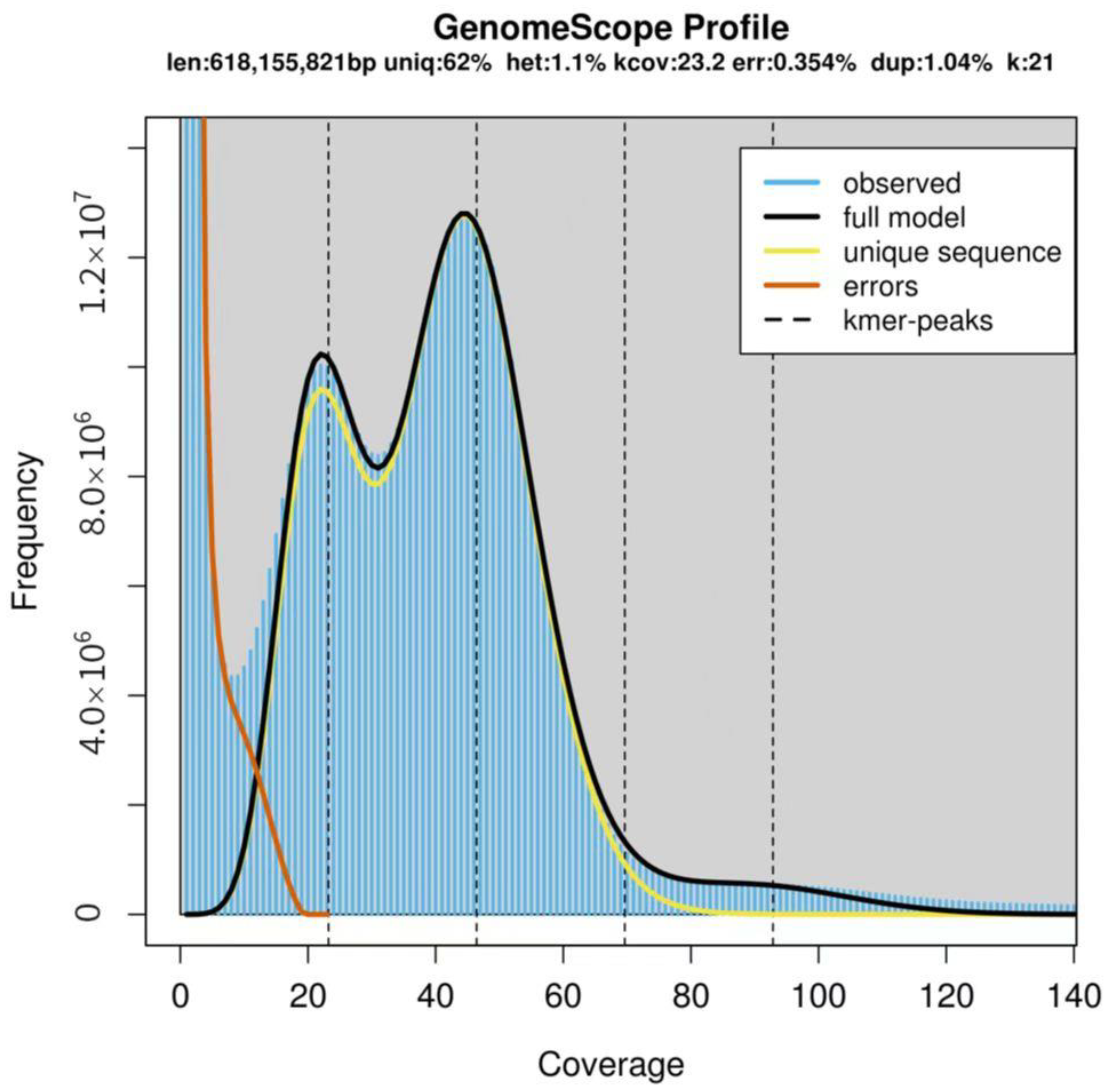
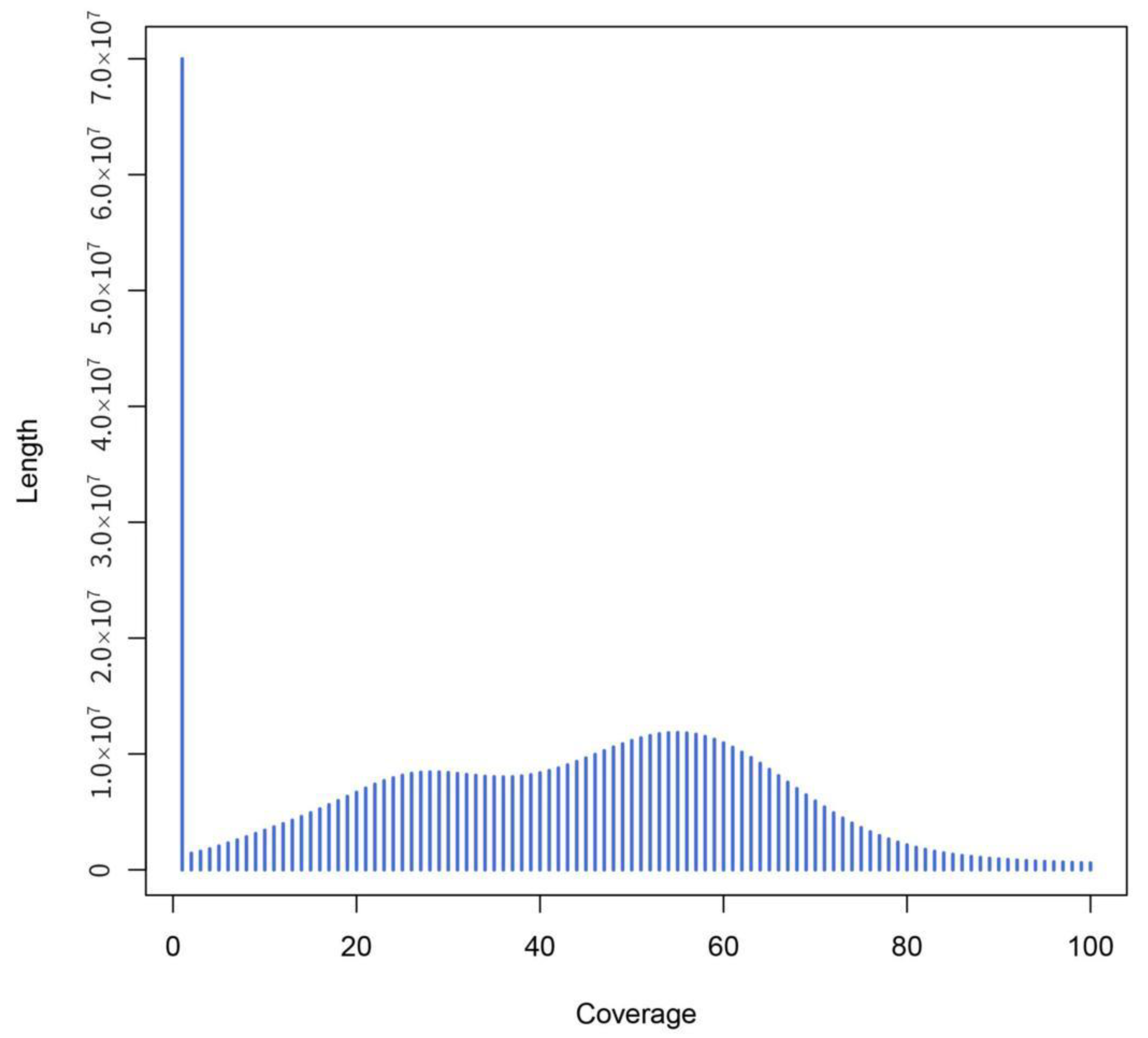
2.3. Genome Size Estimation by K-Mer Analysis
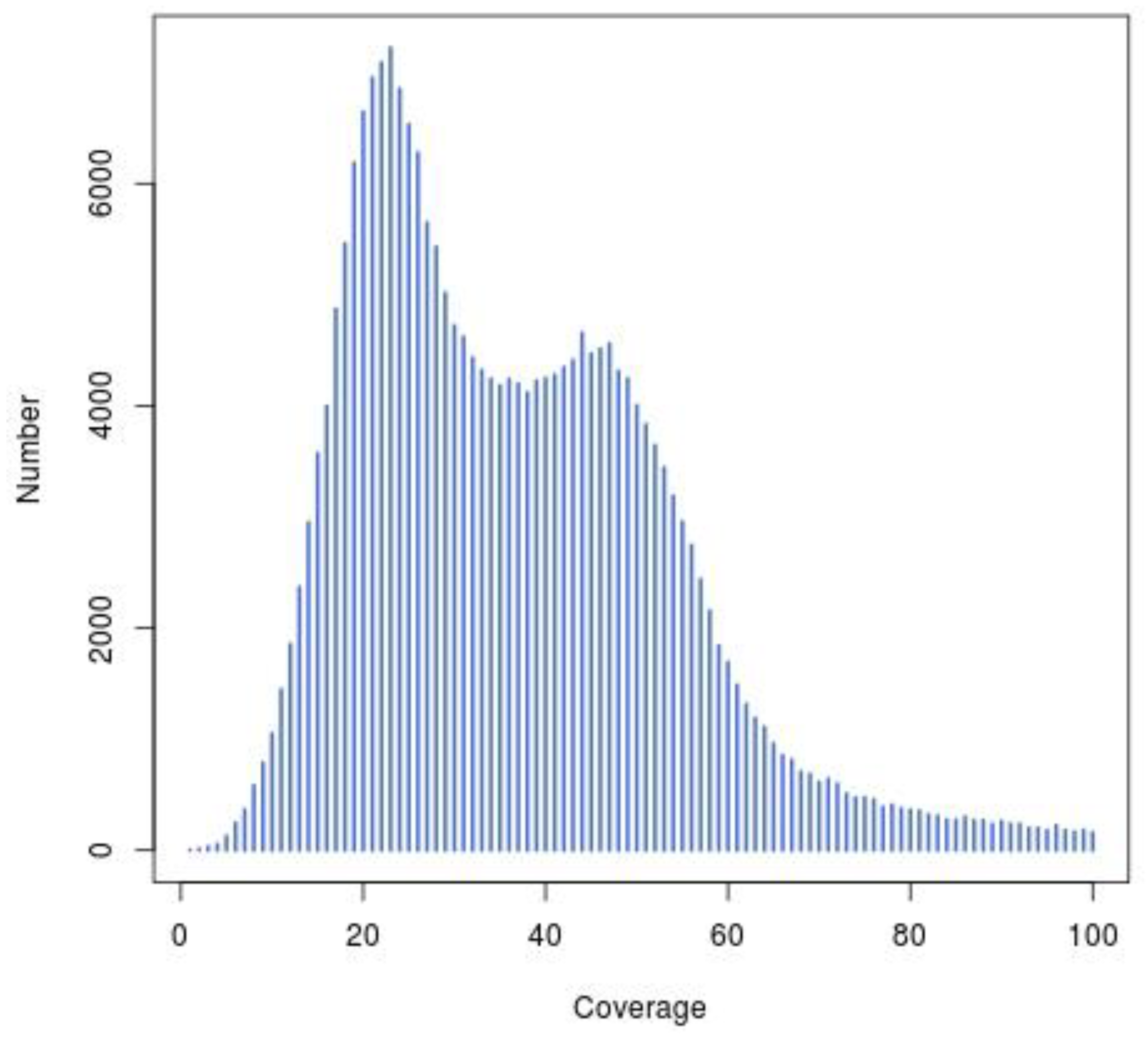
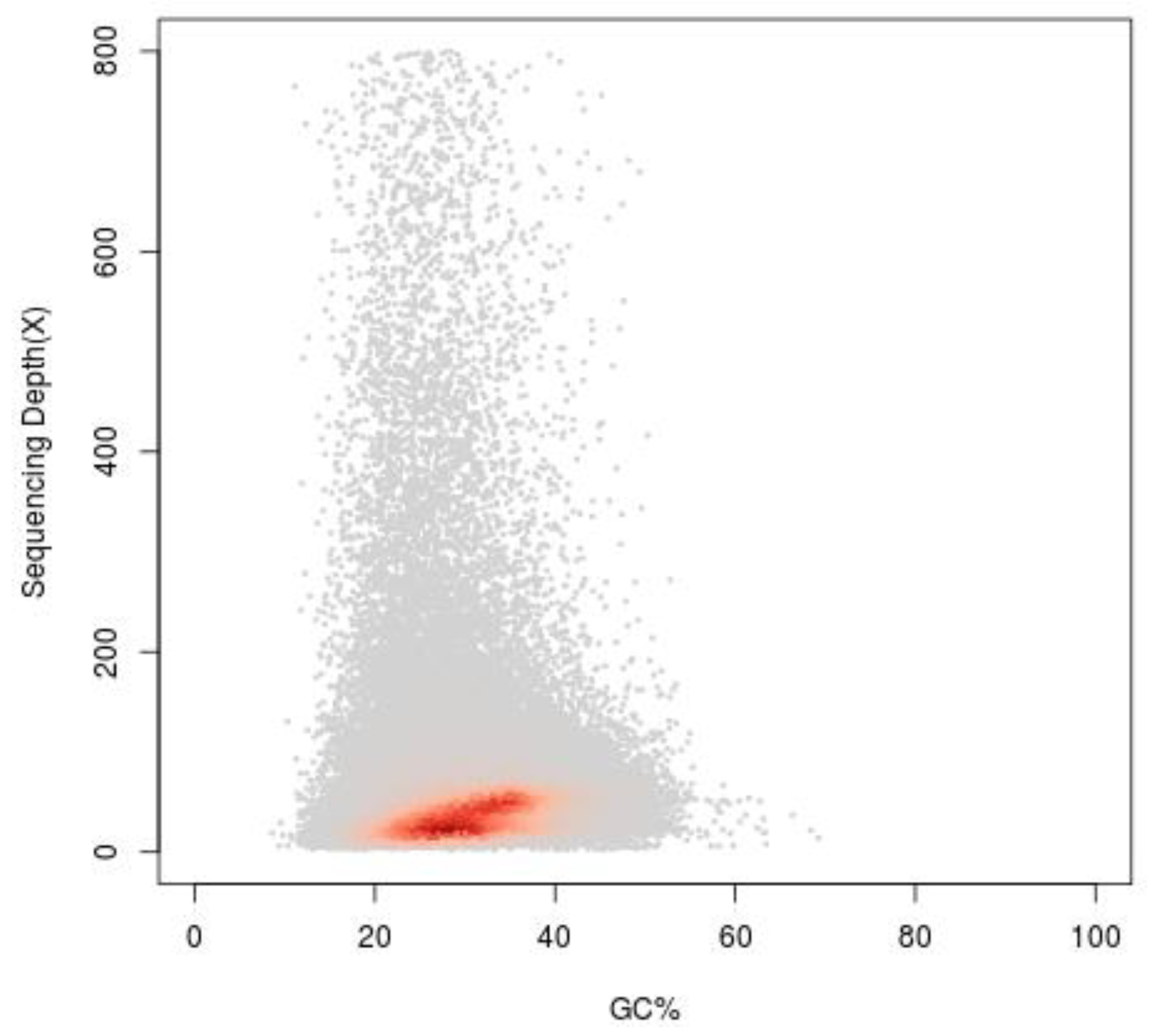
2.4. Genome Assembly and Guanine plus Cytosine (GC) Content Analysis
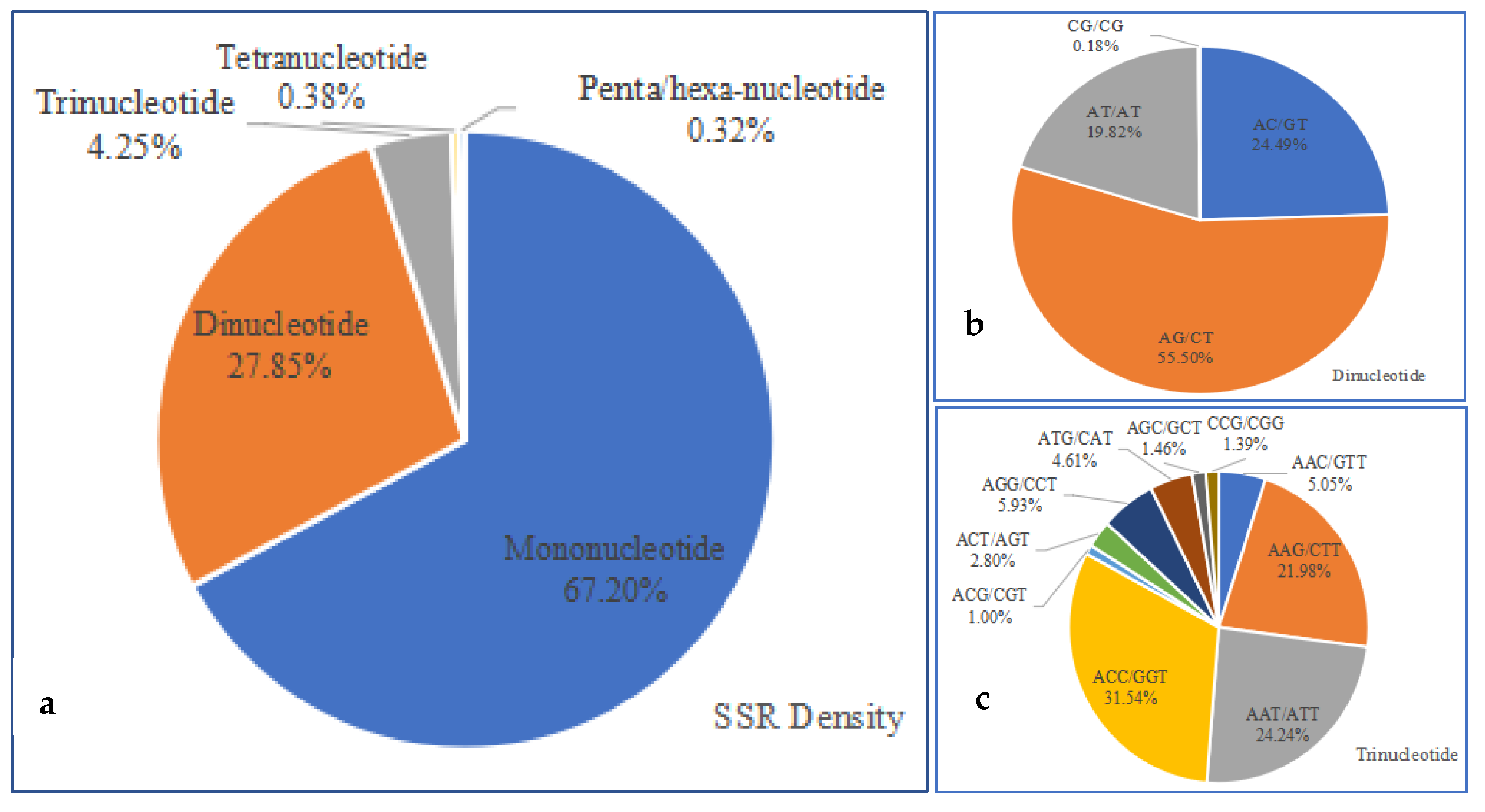
2.5. Identification and Characteristics of Microsatellite Motifs
3. Discussion
3.1. Genome Size of Ilex chinensis
3.2. The Genome Characteristics of Ilex chinensis
3.3. Characteristics of Microsatellites in Ilex chinensis
4. Materials and Methods
4.1. Plant Materials
4.2. Genome Size Estimation by Flow Cytometry
4.3. Genome Sequencing
4.4. Estimation of Genome Size, Heterozygosity, and Repeat Rate
4.5. Genome Assembly and Guanine plus Cytosine (GC) Content Analysis
4.6. Identification and Verification of Microsatellite Motifs
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hu, S.Y. The genus Ilex in China. J. Arnold Arbor. 1949, 30, 233–344. [Google Scholar] [CrossRef]
- Chen, W.W.; Xiao, Z.Z.; Tong, X.; Liu, Y.P.; Li, Y.Y. Development and characterization of 25 microsatellite primers for Ilex chinensis (Aquifoliaceae). Appl. Plant Sci. 2015, 3, 1500057. [Google Scholar] [CrossRef] [PubMed]
- Chinese Pharmacopoeia Commission. Pharmacopoeia of the People’s Republic of China; China Medical Science Press: Beijing, China, 2015; Volume 1, p. 100. [Google Scholar]
- Shao, S.Y.; Li, R.F.; Sun, H.; Li, S. New triterpenoid saponins from the leaves of Ilex chinensis and their hepatoprotective activity. Chin. J. Nat. Med. 2021, 19, 376–384. [Google Scholar] [CrossRef]
- Li, R.F.; Xia, W.Q.; Liu, J.B.; Cui, B.S.; Hou, Q.; Sun, H.; Li, S. New triterpenoid saponins from the leaves of Ilex chinensis. Fitoterapia 2018, 131, 134–140. [Google Scholar] [CrossRef]
- Xia, W.Q.; Li, R.F.; Liu, J.B.; Cui, B.S.; Hou, Q.; Sun, H.; Li, S. Triterpenoids from the leaves of Ilex chinensis. Phytochemistry 2018, 148, 113–121. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhou, P.; Liu, C.L.; Yu, Y.F.; Zhang, M.; Yang, J.D. Comparison of transcriptomic activity of Ilex integra and I. purpurea roots with Nacl treatments. J. Nanjing For. Univ. 2022, 46, 99–108. [Google Scholar]
- Zhou, Y.W.; Li, N.W.; Chen, H.; Chong, X.R.; Li, Y.L.; Lu, X.P.; Zhou, T.; Zhang, F. The complete chloroplast genome sequence of Ilex chinensis Sims. (Aquifoliaceae), a folk herbal medicine plant in China. Mitochondrial DNA 2021, 6, 1241–1242. [Google Scholar] [CrossRef]
- Xu, K.W.; Wei, X.F.; Lin, C.X.; Zhang, M.; Zhang, Q.; Zhou, P.; Fang, Y.M.; Xue, J.Y.; Duan, Y.F. The chromosome-level holly (Ilex latifolia) genome reveals key enzymes in triterpenoid saponin biosynthesis and fruit color change. Front. Plant. Sci. 2022, 13, 982323. [Google Scholar] [CrossRef]
- Kong, B.L.H.; Nong, W.; Wong, K.H.; Law, S.T.S.; So, W.L.; Chan, J.J.S.; Zhang, J.; Lau, T.W.D.; Hui, J.H.L.; Shaw, P.C. Chromosomal level genome of Ilex asprella and insight into antiviral triterpenoid pathway. Genomics 2022, 114, 110366. [Google Scholar] [CrossRef]
- Yao, X.; Lu, Z.Q.; Song, Y.; Hu, X.D.; Corlett, R.T. A chromosome-scale genome assembly for the holly (Ilex polyneura) provides insights into genomic adaptations to elevation in Southwest China. Hortic. Res. 2022, 9, uhab049. [Google Scholar] [CrossRef]
- Liang, X.Y.; Bai, T.D.; Wang, J.Z.; Jiang, W.X. Genome survey and development of 13 SSR markers in Eucalyptus cloeziana by NGS. J. Genet. 2022, 101, 39. [Google Scholar] [CrossRef] [PubMed]
- Morozova, O.; Marra, M.A. Applications of next-generation sequencing technologies in functional genomics. Genomics 2008, 92, 255–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.J.; Liu, M.X.; Lu, X.Y.; Sun, S.S.; Cheng, Y.W.; Ya, H.Y. Genome survey sequencing and identification of genomic SSR markers for Rhododendron micranthum. Biosci. Rep. 2020, 40, BSR20200988. [Google Scholar] [CrossRef] [PubMed]
- Ziya Motalebipour, E.; Kafkas, S.; Khodaeiaminjan, M.; Çoban, N.; Gözel, H. Genome survey of pistachio (Pistacia vera L.) by next generation sequencing: Development of novel SSR markers and genetic diversity in Pistacia species. BMC Genom. 2016, 17, 998. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Zhang, X.J.; Yuan, J.B.; Li, F.H.; Chen, X.H.; Zhao, Y.Z.; Huang, L.; Zheng, H.K.; Xiang, J.H. Genome survey and high-density genetic map construction provide genomic and genetic resources for the Pacific White Shrimp Litopenaeus vannamei. Sci. Rep. 2015, 5, 15612. [Google Scholar] [CrossRef] [Green Version]
- De la Rosa, L.; Zambrana, E.; Ramirez-Parra, E. Molecular bases for drought tolerance in common vetch: Designing new molecular breeding tools. BMC Plant Biol. 2020, 20, 71. [Google Scholar] [CrossRef]
- Li, J.M.; Li, S.Q.; Kong, L.J.; Wang, L.H.; Wei, A.Z.; Liu, Y.L. Genome survey of Zanthoxylum bungeanum and development of genomic-SSR markers in congeneric species. Biosci. Rep. 2020, 40, BSR20201101. [Google Scholar] [CrossRef]
- Lim, L.W.K.; Chung, H.H.; Hussain, H.; Gan, H.M. Genome survey of sago palm (Metroxylon sagu Rottboll). Plant Gene 2021, 28, 100341. [Google Scholar] [CrossRef]
- Fawad, A.; Nadeem, M.A.; Habyarimana, E.; Altaf, M.T.; Barut, M.; Kurt, C.; Chaudhary, H.J.; Khalil, I.H.; Yildiz, M.; Comertpay, G.; et al. Identification of genetic basis associated with agronomic traits in a global safflower panel using genome-wide association study. Turk. J. Agric. For. 2021, 45, 834–849. [Google Scholar]
- Lu, M.; An, H.M.; Li, L.L. Genome survey sequencing for the characterization of the genetic background of Rosa roxburghii Tratt and leaf ascorbate metabolism genes. PLoS ONE 2016, 11, e0147530. [Google Scholar] [CrossRef]
- Ozdemir, B.; Okay, F.Y.; Sarikamis, G.; Ozmen, C.Y.; Kibar, U.; Ergul, A. Crosstalk between flowering and cold tolerance genes in almonds (Amygdalus spp.). Turk. J. Agric. For. 2021, 45, 484–494. [Google Scholar] [CrossRef]
- Unamba, C.I.N.; Nag, A.; Sharma, R.K. Next generation sequencing technologies: The doorway to the unexplored genomics of non-model plants. Front. Plant Sci. 2015, 6, 1074. [Google Scholar] [CrossRef] [PubMed]
- Ekblom, R.; Galindo, J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity 2011, 107, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.Q.; Chen, J.B.; Zhang, J.; Liu, J.F.; Yu, J.J.; Cai, D.B.; Yao, L.G.; Duan, P.F. First genome survey and repeatome analysis of Chrysopogon zizanioides based on next-generation sequencing. Biologia 2020, 75, 1273–1282. [Google Scholar] [CrossRef]
- Ma, L.; Wang, X.; Yan, M.; Liu, F.; Zhang, S.X.; Wang, X.M. Genome survey sequencing of common vetch (Vicia sativa L.) and genetic diversity analysis of Chinese germplasm with genomic SSR markers. Mol. Biol. Rep. 2022, 49, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Bi, Q.X.; Zhao, Y.; Cui, Y.F.; Wang, L.B. Genome survey sequencing and genetic background characterization of yellow horn based on next-generation sequencing. Mol. Biol. Rep. 2019, 46, 4303–4312. [Google Scholar] [CrossRef] [PubMed]
- Greilhuber, J.; Leitch, I.J. Genome size and the phenotype. In Plant Genome Diversity; Springer: Vienna, Austria, 2013; Volume 2, pp. 323–344. [Google Scholar]
- Leitch, I.J.; Leitch, A.R. Genome size diversity and evolution in land plants. In Plant Genome Diversity; Springer: Vienna, Austria, 2013; pp. 307–322. [Google Scholar]
- Pati, K.; Zhang, F.; Batley, J. First report of genome size and ploidy of the underutilized leguminous tuber crop Yam Bean (Pachyrhizus erosus and P. tuberosus) by flow cytometry. Plant Genet. Resour. 2019, 17, 456–459. [Google Scholar] [CrossRef]
- Gschwend, A.R.; Wai, C.M.; Zee, F.; Arumuganathan, A.K.; Ming, R. Genome size variation among sex types in dioecious and trioecious Caricaceae species. Euphytica 2013, 189, 461–469. [Google Scholar] [CrossRef]
- Liu, B.H.; Shi, Y.J.; Yuan, J.Y.; Hu, X.S.; Zhang, H.; Li, N.; Li, Z.Y.; Chen, Y.X.; Mu, D.S.; Fan, W. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv 2012, arXiv:1308.2012v2. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, J.W.; Yang, Q.; Li, B.; Zhou, W.; Wang, Z.Z. Genome survey sequencing and genetic diversity of cultivated Akebia trifoliata assessed via phenotypes and SSR markers. Mol. Biol. Rep. 2021, 48, 241–250. [Google Scholar] [CrossRef]
- Claros, M.G.; Bautista, R.; Guerrero-Fernández, D.; Benzerki, H.; Seoane, P.; Fernández-Pozo, N. Why assembling plant genome sequences is so challenging. Biology 2012, 1, 439–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panhwar, S.K.; Liu, Q.; Khan, F.; Siddiqui, P.J. Maximum sustainable yield estimates of Ladypees, Sillago sihama (Forsskål), fishery in Pakistan using the ASPIC and CEDA packages. J. Ocean Univ. China 2012, 11, 93–98. [Google Scholar] [CrossRef]
- Li, G.Q.; Song, L.X.; Jin, C.Q.; Li, M.; Gong, S.P.; Wang, Y.F. Genome survey and SSR analysis of Apocynum venetum. Biosci. Rep. 2019, 39, BSR20190146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.F.; Xiao, F.M.; Xu, H.N.; Zhang, T.; Jiang, X.M. Genome survey in Cinnamomum camphora (L.) Presl. J. Plant Genet. Resour. 2014, 15, 150–153. [Google Scholar]
- Wang, C.; Yan, H.; Li, J.; Zhou, S.; Liu, T.; Zhang, X.; Huang, L. Genome survey sequencing of purple elephant grass (Pennisetum purpureum Schum ‘Zise’) and identification of its SSR markers. Mol. Breed. 2018, 38, 94. [Google Scholar] [CrossRef]
- Yao, X.; Song, Y.; Yang, J.B.; Tan, Y.H.; Corlett, R.T. Phylogeny and biogeography of the hollies (Ilex L., Aquifoliaceae). J. Syst. Evol. 2021, 59, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Ming, R.; VanBuren, R.; Wai, C.M.; Tang, H.; Schatz, M.C.; Bowers, J.E.; Lyons, E.; Wang, M.-L.; Chen, J.; Biggers, E.; et al. The pineapple genome and the evolution of CAM photosynthesis. Nat. Genet. 2015, 47, 1435. [Google Scholar] [CrossRef] [Green Version]
- Hall, M.R.; Kocot, K.M.; Baughman, K.W.; Fernandez-Valverde, S.L.; Gauthier, M.E.A.; Hatleberg, W.L.; Krishnan, A.; McDougall, C.; Motti, C.A.; Shoguchi, E.; et al. The crown-of-thorns starfish genome as a guide for biocontrol of this coral reef pest. Nature 2017, 544, 231–234. [Google Scholar] [CrossRef] [Green Version]
- Roach, M.J.; Schmidt, S.A.; Borneman, A.R. Purge Haplotigs: Allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinform. 2018, 19, 460. [Google Scholar] [CrossRef]
- Feng, Y.Y.; Guo, L.L.; Jin, H.; Lin, C.C.; Zhou, C.H.; Fang, X.S.; Wang, J.H.; Song, Z.Q. Quantitative trait loci analysis of phenolic acids contents in Salvia miltiorrhiza based on genomic simple sequence repeat markers. BMC Bioinform. 2019, 133, 365–372. [Google Scholar] [CrossRef]
- Wu, Z.G.; Jiang, W.; Mantri, N.; Bao, X.Q.; Chen, S.L.; Tao, Z.M. Transciptome analysis reveals flavonoid biosynthesis regulation and simple sequence repeats in yam (Dioscorea alata L.) tubers. BMC Genom. 2015, 16, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa, S.N.; Almeida, C. Genome survey and development of 15 microsatellite molecular markers in Syagrus coronata (Martius) Beccari (Arecaceae) by next-generation sequencing. Braz. J. Bot. 2019, 42, 195–200. [Google Scholar] [CrossRef]
- Wang, S.; Chen, S.; Liu, C.; Liu, Y.; Zhao, X.; Yang, C.; Qu, G.Z. Genome survey sequencing of Betula platyphylla. Forests 2019, 10, 826. [Google Scholar] [CrossRef]
- Qiu, B.X.; Fang, S.B.; Ikhwanuddin, M.; Wong, L.L.; Ma, H.Y. Genome survey and development of polymorphic microsatellite loci for Sillago sihama based on Illumina sequencing technology. Mol. Biol. Rep. 2020, 47, 3011–3017. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.Y.; Huang, Z.H.; Wang, Q.W.; Wu, J.Y.; Feng, X.M.; Qi, Y.W. Genome survey sequence and the development of simple sequence repeat (SSR) markers in Erianthus arundinaceus. Sugar Tech. 2021, 23, 77–85. [Google Scholar] [CrossRef]
- Wang, R.K.; Fan, J.S.; Chang, P.; Zhu, L.; Zhao, M.R.; Li, L.L. Genome survey sequencing of Acer truncatum Bunge to identify genomic information, simple sequence repeat (SSR) markers and complete chloroplast genome. Forests 2019, 10, 87. [Google Scholar] [CrossRef] [Green Version]
- Kandaiah, V.; Singaram, N.; Kandasamy, K.I. Optimized flow cytometric protocol and genome size estimation of Sabah snake grass (Clinacanthus nutans). J. Med. Plants Res. 2021, 15, 531–539. [Google Scholar]
- International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar] [CrossRef] [Green Version]
- Doležel, J.; Bartoš, J.A.N. Plant DNA flow cytometry and estimation of nuclear genome size. Ann. Bot. 2005, 95, 99–110. [Google Scholar] [CrossRef] [Green Version]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [Green Version]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, E.; Cho, Y.H.; Lee, S.J.; Choi, E.; Kim, J.; Kim, J.H.; Chi, Y.M.; Park, H. Genome survey and microsatellite motif identification of Pogonophryne albipinna. Biosci. Rep. 2021, 41, BSR20210824. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Size (Mb) Mean ± SD | CV (%) of Samples | CV (%) of Standard | |
|---|---|---|---|
| Female | 656 ± 6 | 4.00 | 4.19 |
| Male | 665 ± 12 | 4.09 | 4.64 |
| Average | 660 ± 10 | 4.05 | 4.42 |
| Number of Raw Reads | Raw Base (Gbp) | Number of Clean Reads | Error Rate (%) | Q20 (%) | Q30 (%) | GC Content (%) | Average Quality |
|---|---|---|---|---|---|---|---|
| 298,755,430 | 44.813 | 297,637,262 | 0.354 | 95.275 | 87.84 | 37.52 | 34.75 |
| Total Length (bp) | Total Number | Max Length (bp) | Min Length (bp) | N20 Length (bp) | N50 Length (bp) | N80 Length (bp) | |
|---|---|---|---|---|---|---|---|
| Scaffold | 685,140,399 | 250,019 | 112,652 | 500 | 15,137 | 5738 | 1663 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, P.; Li, J.; Huang, J.; Li, F.; Zhang, Q.; Zhang, M. Genome Survey Sequencing and Genetic Background Characterization of Ilex chinensis Sims (Aquifoliaceae) Based on Next-Generation Sequencing. Plants 2022, 11, 3322. https://doi.org/10.3390/plants11233322
Zhou P, Li J, Huang J, Li F, Zhang Q, Zhang M. Genome Survey Sequencing and Genetic Background Characterization of Ilex chinensis Sims (Aquifoliaceae) Based on Next-Generation Sequencing. Plants. 2022; 11(23):3322. https://doi.org/10.3390/plants11233322
Chicago/Turabian StyleZhou, Peng, Jiao Li, Jing Huang, Fei Li, Qiang Zhang, and Min Zhang. 2022. "Genome Survey Sequencing and Genetic Background Characterization of Ilex chinensis Sims (Aquifoliaceae) Based on Next-Generation Sequencing" Plants 11, no. 23: 3322. https://doi.org/10.3390/plants11233322
APA StyleZhou, P., Li, J., Huang, J., Li, F., Zhang, Q., & Zhang, M. (2022). Genome Survey Sequencing and Genetic Background Characterization of Ilex chinensis Sims (Aquifoliaceae) Based on Next-Generation Sequencing. Plants, 11(23), 3322. https://doi.org/10.3390/plants11233322