
Identification of QTLs and Putative Candidate Genes for Plant Architecture of Lotus Revealed by Regional Association Mapping

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Phenotypic Characterization
2.2. Molecular Marker Characterization, Genotyping, and Genetic Diversity Analysis
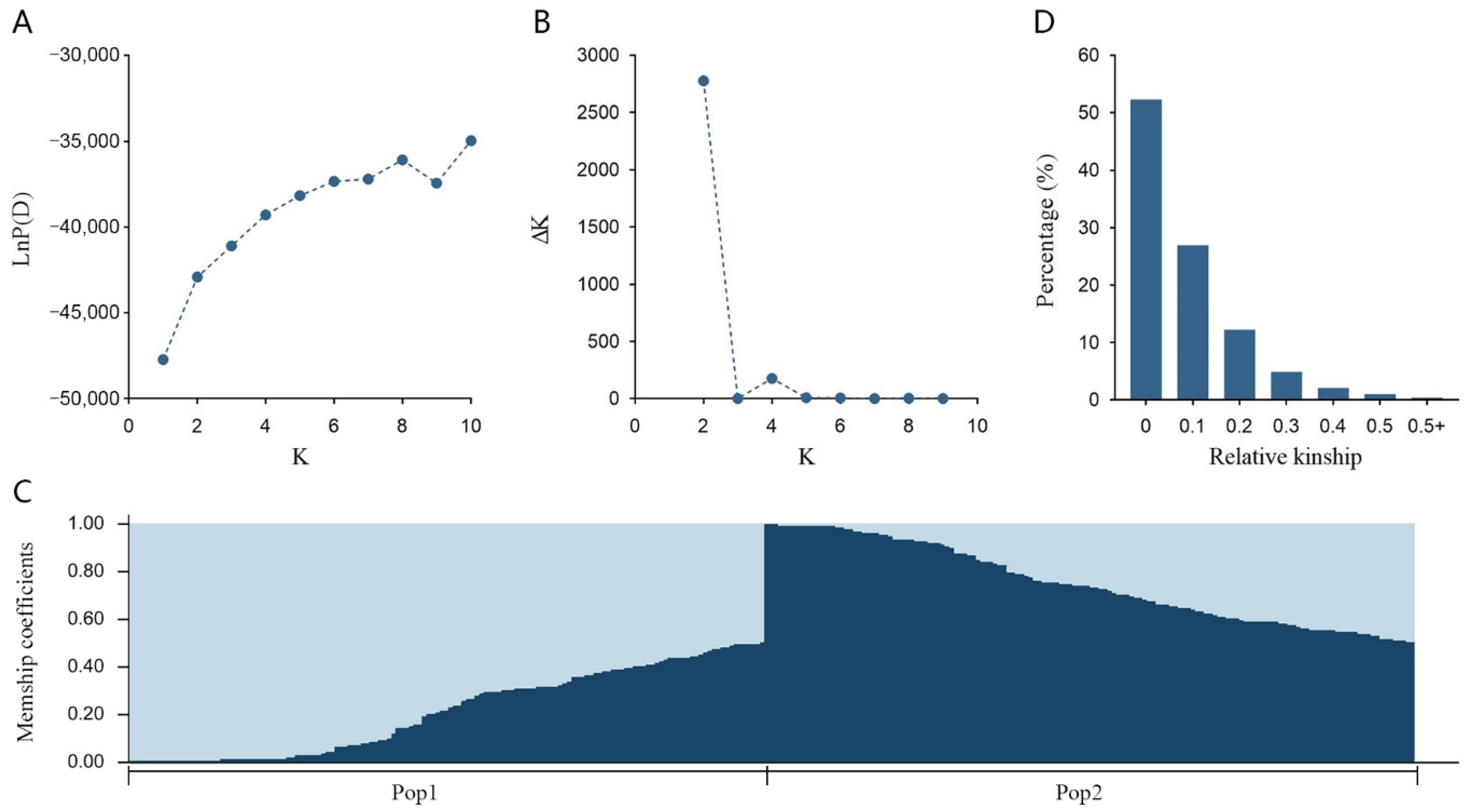
2.3. Population Structure and Kinship Matrix Analysis
2.4. Association Analysis
2.5. Identification of the Key Genes
3. Results
3.1. Phenotypic Variation
3.2. SSR Polymorphisms and Genetic Diversity
3.3. Population Structure and Relative Kinship
3.4. InDel Maker Development in the Candidate Regions
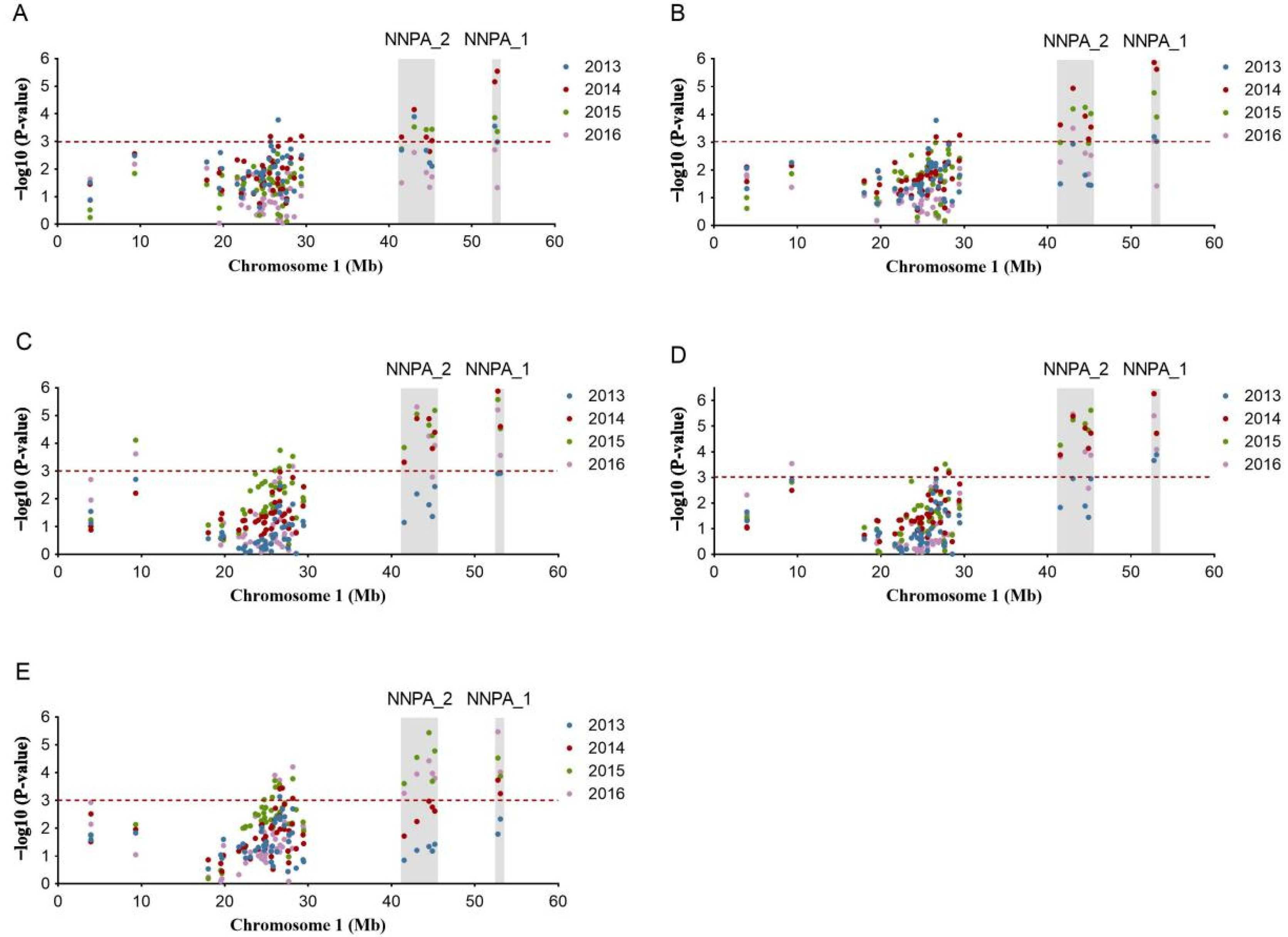
3.5. Marker Trait Associations
3.6. Identification of Key Markers and Genes
4. Discussion
4.1. Phenotypic Diversity and Heredity of PA-Related Traits in Lotus
4.2. Genetic Diversity
4.3. Genetic Structure and the Analysis Model
4.4. Association Mapping Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhang, X.; Chen, L.; Wang, Q. New Lotus Flower Cultivars in China; China Forestry Publishing House: Beijing, China, 2011. [Google Scholar]
- Wang, Q.; Zhang, X. Colored Illustration of Lotus Cultivars in China; China Forestry Publishing House: Beijing, China, 2005. [Google Scholar]
- Abd Rasid, N.; Naim, M.; Man, H.C.; Bakar, N.A.; Mokhtar, M. Evaluation of surface water treated with lotus plant; Nelumbo nucifera. J. Environ. Chem. Eng. 2019, 7, 103048. [Google Scholar] [CrossRef]
- Xu, S.; Wang, Y.; Wang, Y.; Zhao, Y.; Gao, Y. Seasonal influence of reed (Phragmites australis) and lotus (Nelumbo nucifera) on urban wetland of Yi River. Appl. Ecol. Environ. Res. 2019, 17, 7891–7900. [Google Scholar] [CrossRef]
- Zhang, Y.; Cao, G.; Zhang, Z.; Zou, Y.; Zhang, X.; Zhang, H.; Wang, Y.; Marhaba, T. Phytoremediation potential of heavy metal in contaminated sediments by hydrophytes. Fresenius Environ. Bull. 2019, 28, 1395–1403. [Google Scholar]
- Rojas-Vasquez, R.; Gatica-Arias, A. Use of genome editing technologies for genetic improvement of crops of tropical origin. Plant Cell Tissue Organ Cult. (PCTOC) 2020, 140, 215–244. [Google Scholar] [CrossRef]
- Reinhardt, D.; Kuhlemeier, C. Plant architecture. EMBO Rep. 2002, 3, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Brachi, B.; Faure, N.; Horton, M.; Flahauw, E.; Vazquez, A.; Nordborg, M.; Bergelson, J.; Cuguen, J.; Roux, F. Linkage and association mapping of Arabidopsis thaliana flowering time in nature. PLoS Genet. 2010, 6, e1000940. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Gore, M.; Buckler, E.S.; Yu, J. Status and prospects of association mapping in plants. Plant Genome 2008, 1, 5–20. [Google Scholar] [CrossRef]
- Myles, S.; Peiffer, J.; Brown, P.J.; Ersoz, E.S.; Zhang, Z.; Costich, D.E.; Buckler, E.S. Association mapping: Critical considerations shift from genotyping to experimental design. Plant Cell 2009, 21, 2194–2202. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Buckler, E.S. Genetic association mapping and genome organization of maize. Curr. Opin. Biotechnol. 2006, 17, 155–160. [Google Scholar] [CrossRef]
- Wang, Z.; Qiang, H.; Zhao, H.; Xu, R.; Zhang, Z.; Gao, H.; Wang, X.; Liu, G.; Zhang, Y. Association mapping for fiber-related traits and digestibility in alfalfa (Medicago sativa). Front. Plant Sci. 2016, 7, 331. [Google Scholar] [CrossRef] [Green Version]
- Bouchet, S.; Bertin, P.; Presterl, T.; Jamin, P.; Coubriche, D.; Gouesnard, B.; Laborde, J.; Charcosset, A. Association mapping for phenology and plant architecture in maize shows higher power for developmental traits compared with growth influenced traits. Heredity 2017, 118, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado, C.; Mora, F.; Scapim, C.A.; Coan, M. Genome-wide haplotype-based association analysis of key traits of plant lodging and architecture of maize identifies major determinants for leaf angle: Hap LA4. PLoS ONE 2019, 14, e0212925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, J.; Li, L.; Zhang, C.; Wang, C.; Gu, L.; Wang, H.; Wei, H.; Liu, Q.; Huang, L.; Yu, S. Genome-wide association study identified genetic variations and candidate genes for plant architecture component traits in Chinese upland cotton. Theor. Appl. Genet. 2018, 131, 1299–1314. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.; Dai, B.; Wang, T.; Liu, X.; You, C.; Lin, Z. Genetic variations in plant architecture traits in cotton (Gossypium hirsutum) revealed by a genome-wide association study. Crop J. 2019, 7, 209–216. [Google Scholar] [CrossRef]
- Zhao, J.; Mantilla Perez, M.B.; Hu, J.; Salas Fernandez, M.G. Genome-wide association study for nine plant architecture traits in Sorghum. Plant Genome 2016, 9, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Hao, D.; Sitoe, H.M.; Yin, Z.; Hu, Z.; Zhang, G.; Yu, D. Genetic dissection of the relationship between plant architecture and yield component traits in soybean (Glycine max) by association analysis across multiple environments. Plant Breed. 2015, 134, 564–572. [Google Scholar] [CrossRef]
- Yang, D.; Du, F.; Chang, Y.; Cui, J.; Sun, L.; Liu, X.; Yao, D. Association Studies of Plant Architecture Traits and SSR Markers in Nelumbo Adans. Chin. J. Trop. Crops 2020, 41, 2149. [Google Scholar]
- Hu, S.; Wang, M.; Zhang, X.; Chen, W.; Song, X.; Fu, X.; Fang, H.; Xu, J.; Xiao, Y.; Li, Y. Genetic basis of kernel starch content decoded in a maize multi-parent population. Plant Biotechnol. J. 2021, 19, 2192–2205. [Google Scholar] [CrossRef]
- Zhang, H.; Goettel, W.; Song, Q.; Jiang, H.; Hu, Z.; Wang, M.L.; An, Y.-Q.C. Selection of GmSWEET39 for oil and protein improvement in soybean. PLoS Genet. 2020, 16, e1009114. [Google Scholar] [CrossRef]
- Gui, S.; Peng, J.; Wang, X.; Wu, Z.; Cao, R.; Salse, J.; Zhang, H.; Zhu, Z.; Xia, Q.; Quan, Z. Improving Nelumbo nucifera genome assemblies using high-resolution genetic maps and BioNano genome mapping reveals ancient chromosome rearrangements. Plant J. 2018, 94, 721–734. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wu, J.; Li, Q.; Yang, Y.; Zhang, K. Development of Simple Sequence Repeat Markers from Functional Genes and Establishment of Molecular Identity for Tree Peony. J. Plant Biochem. Biotechnol. 2021, 31, 1–15. [Google Scholar] [CrossRef]
- Huang, L.; Yang, M.; Li, L.; Li, H.; Yang, D.; Shi, T.; Yang, P. Whole genome re-sequencing reveals evolutionary patterns of sacred lotus (Nelumbo nucifera). J. Integr. Plant Biol. 2018, 60, 2–15. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, F.-L.; Zheng, X.-W.; Hu, M.-L.; Dong, C.; Diao, Y.; Wang, Y.-W.; Xie, K.-Q.; Hu, Z.-L. Comparative population genomics reveals genetic divergence and selection in lotus, Nelumbo nucifera. BMC Genom. 2020, 21, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Gui, S.; Zhu, Z.; Wang, X.; Ke, W.; Ding, Y. Genome-wide identification of SSR and SNP markers based on whole-genome re-sequencing of a Thailand wild sacred lotus (Nelumbo nucifera). PLoS ONE 2015, 10, e0143765. [Google Scholar] [CrossRef] [PubMed]
- Remington, D.L.; Thornsberry, J.M.; Matsuoka, Y.; Wilson, L.M.; Whitt, S.R.; Doebley, J.; Kresovich, S.; Goodman, M.M.; Buckler, E.S. Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. USA 2001, 98, 11479–11484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Han, Y.; VanBuren, R.; Ming, R.; Xu, L.; Han, Y.; Liu, Y. Genetic linkage maps for Asian and American lotus constructed using novel SSR markers derived from the genome of sequenced cultivar. BMC Genom. 2012, 13, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Yang, J.-X.; Mao, T.-Y.; Zhu, H.-H.; Xiang, L.; Zhang, J.; Chen, L.-Q. Detection of highly differentiated genomic regions between lotus (Nelumbo nucifera Gaertn.) with contrasting plant architecture and their functional relevance to plant architecture. Front. Plant Sci. 2018, 9, 1219. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A Rapid DNA Isolation Procedure for Small Quantities of Fresh Leaf Tissue. Phytochem Bull. 1987, 19, 11–15. [Google Scholar]
- Cao, H. Genetic Diversity Analysis of Nelumbo nucifera Cultivars Based on Morphological and SSR Markers; Huazhong Agricultural University: Wuhan, China, 2016. [Google Scholar]
- Shriner, D.; Vaughan, L.K.; Padilla, M.A.; Tiwari, H.K. Problems with genome-wide association studies. Science 2007, 316, 1840–1842. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Li, J.; Li, S.; Qin, G.; Novák, O.; Pěnčík, A.; Ljung, K.; Aoyama, T.; Liu, J.; Murphy, A. ADP1 affects plant architecture by regulating local auxin biosynthesis. PLoS Genet. 2014, 10, e1003954. [Google Scholar] [CrossRef] [Green Version]
- Mackay, I.; Powell, W. Methods for linkage disequilibrium mapping in crops. Trends Plant Sci. 2007, 12, 57–63. [Google Scholar] [CrossRef]
- Ingvarsson, P.K. Multilocus patterns of nucleotide polymorphism and the demographic history of Populus tremula. Genetics 2008, 180, 329–340. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Quan, Z.; Hu, J.; Wang, G.; Liu, S.; He, Y.; Ke, W.; Ding, Y. Genetic diversity and differentiation of lotus (Nelumbo nucifera) accessions assessed by simple sequence repeats. Ann. Appl. Biol. 2011, 159, 428–441. [Google Scholar] [CrossRef]
- Hu, J.; Pan, L.; Liu, H.; Wang, S.; Wu, Z.; Ke, W.; Ding, Y. Comparative analysis of genetic diversity in sacred lotus (Nelumbo nucifera Gaertn.) using AFLP and SSR markers. Mol. Biol. Rep. 2012, 39, 3637–3647. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Jiang, H.; Li, C.; Ma, J.; Chen, L. Genetic diversity analysis of tea plant in Baiyingshan Mountain of Yunnan. J. Tea Sci. 2018, 38, 69–77. [Google Scholar]
- Zhao, J.; Wang, B.; Jia, X.; Tong, Y.; He, Y.; Ge, T. Development of SSR markers to assess genetic diversity in Osmunda japonica Thunb. Plant Sci. J. 2015, 33, 801–807. [Google Scholar]
- Flint-Garcia, S.A.; Thornsberry, J.M.; Buckler, E.S., IV. Structure of linkage disequilibrium in plants. Annu. Rev. Plant Biol. 2003, 54, 357–374. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Yang, M.; Zhu, L.; Xu, L.; Liu, Y. Population structure and association mapping of flower-related traits in lotus (Nelumbo Adans.) accessions. Sci. Hortic. 2014, 175, 214–222. [Google Scholar] [CrossRef]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]
- Nie, G.; Huang, L.; Zhang, X.; Taylor, M.; Jiang, Y.; Yu, X.; Liu, X.; Wang, X.; Zhang, Y. Marker-trait association for biomass yield of potential bio-fuel feedstock Miscanthus sinensis from Southwest China. Front. Plant Sci. 2016, 7, 802. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhou, Z.; Ding, J.; Wu, Y.; Zhou, B.; Wang, R.; Ma, J.; Wang, S.; Zhang, X.; Xia, Z. Combined linkage and association mapping reveals QTL and candidate genes for plant and ear height in maize. Front. Plant Sci. 2016, 7, 833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peiffer, J.A.; Romay, M.C.; Gore, M.A.; Flint-Garcia, S.A.; Zhang, Z.; Millard, M.J.; Gardner, C.A.; McMullen, M.D.; Holland, J.B.; Bradbury, P.J. The genetic architecture of maize height. Genetics 2014, 196, 1337–1356. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wei, Y.; Moore, K.J.; Michaud, R.; Viands, D.R.; Hansen, J.L.; Acharya, A.; Brummer, E.C. Association mapping of biomass yield and stem composition in a tetraploid alfalfa breeding population. Plant Genome 2011, 4, 24. [Google Scholar] [CrossRef] [Green Version]
- Du, Q.; Pan, W.; Xu, B.; Li, B.; Zhang, D. Polymorphic simple sequence repeat (SSR) loci within cellulose synthase (PtoCesA) genes are associated with growth and wood properties in P opulus tomentosa. New Phytol. 2013, 197, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Ray, J.D.; Dhanapal, A.P.; Singh, S.K.; Hoyos-Villegas, V.; Smith, J.R.; Purcell, L.C.; King, C.A.; Boykin, D.; Cregan, P.B.; Song, Q. Genome-wide association study of ureide concentration in diverse maturity group IV soybean [Glycine max (L.) Merr.] accessions. G3 Genes Genomes Genet. 2015, 5, 2391–2403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thudi, M.; Upadhyaya, H.D.; Rathore, A.; Gaur, P.M.; Krishnamurthy, L.; Roorkiwal, M.; Nayak, S.N.; Chaturvedi, S.K.; Basu, P.S.; Gangarao, N. Genetic dissection of drought and heat tolerance in chickpea through genome-wide and candidate gene-based association mapping approaches. PLoS ONE 2014, 9, e96758. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhu, H.; Liu, Y.; Kuang, J.; Zhou, K.; Liang, F.; Liu, Z.; Wang, D.; Ke, W. Construction of a high-density, high-quality genetic map of cultivated lotus (Nelumbo nucifera) using next-generation sequencing. BMC Genom. 2016, 17, 1–11. [Google Scholar] [CrossRef] [Green Version]
- White, S.W.; Zheng, J.; Zhang, Y.-M.; Rock, C.O. The structural biology of type II fatty acid biosynthesis. Annu. Rev. Biochem. 2005, 74, 791–831. [Google Scholar] [CrossRef]
- Lim, G.-H.; Singhal, R.; Kachroo, A.; Kachroo, P. Fatty acid-and lipid-mediated signaling in plant defense. Annu. Rev. Phytopathol. 2017, 55, 505–536. [Google Scholar] [CrossRef]
- Wu, G.-Z.; Xue, H.-W. Arabidopsis β-ketoacyl-[acyl carrier protein] synthase I is crucial for fatty acid synthesis and plays a role in chloroplast division and embryo development. Plant Cell 2010, 22, 3726–3744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.; Lin, L.; Zhang, B.; Xiang, X.; Wu, J.; Pan, Z.; Zhu, S. OsKASI, a β-ketoacyl-[acyl carrier protein] synthase I, is involved in root development in rice (Oryza sativa L.). Planta 2015, 242, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Cortés, A.J.; Chavarro, M.C.; Blair, M.W. SNP marker diversity in common bean (Phaseolus vulgaris L.). Theor. Appl. Genet. 2011, 123, 827–845. [Google Scholar] [CrossRef] [PubMed]
- Blair, M.W.; Cortés, A.J.; This, D. Identification of an ERECTA gene and its drought adaptation associations with wild and cultivated common bean. Plant Sci. 2016, 242, 250–259. [Google Scholar] [CrossRef]
- López-Hernández, F.; Cortés, A.J. Last-generation genome–environment associations reveal the genetic basis of heat tolerance in common bean (Phaseolus vulgaris L.). Front. Genet. 2019, 10, 954. [Google Scholar] [CrossRef] [Green Version]
- Burbano-Erazo, E.; León-Pacheco, R.I.; Cordero-Cordero, C.C.; López-Hernández, F.; Cortés, A.J.; Tofiño-Rivera, A.P. Multi-environment yield components in advanced common bean (Phaseolus vulgaris L.)× tepary bean (P. acutifolius A. Gray) interspecific lines for heat and drought tolerance. Agronomy 2021, 11, 1978. [Google Scholar] [CrossRef]
- Cortés, A.J.; López-Hernández, F.; Osorio-Rodriguez, D. Predicting thermal adaptation by looking into populations’ genomic past. Front. Genet. 2020, 11, 564515. [Google Scholar] [CrossRef]
- Buitrago-Bitar, M.A.; Cortés, A.J.; López-Hernández, F.; Londoño-Caicedo, J.M.; Muñoz-Florez, J.E.; Muñoz, L.C.; Blair, M.W. Allelic diversity at abiotic stress responsive genes in relationship to ecological drought indices for cultivated tepary bean, Phaseolus acutifolius A. Gray, and its wild relatives. Genes 2021, 12, 556. [Google Scholar] [CrossRef]
- Cortés, A.J.; López-Hernández, F. Harnessing crop wild diversity for climate change adaptation. Genes 2021, 12, 783. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Year | Min | Max | Mean | SD | CV (%) | p-Value | H2 |
|---|---|---|---|---|---|---|---|---|
| Petiole height (cm) | 2013 | 31.30 | 163.80 | 94.25 | 23.84 | 25.29 | 0.013 * | 0.94 |
| 2014 | 30.40 | 163.20 | 95.43 | 23.94 | 25.09 | |||
| 2015 | 37.14 | 167.20 | 99.50 | 23.21 | 23.33 | |||
| 2016 | 41.96 | 145.14 | 96.88 | 19.86 | 20.50 | |||
| Peduncle height (cm) | 2013 | 45.10 | 165.60 | 109.73 | 23.57 | 21.48 | 0.029 * | 0.92 |
| 2014 | 43.25 | 179.70 | 111.39 | 26.03 | 23.37 | |||
| 2015 | 38.00 | 197.20 | 109.96 | 25.58 | 23.26 | |||
| 2016 | 53.17 | 157.29 | 106.63 | 21.35 | 20.02 | |||
| Leaf length (cm) | 2013 | 16.76 | 60.00 | 37.63 | 8.26 | 21.94 | <0.000 ** | 0.98 |
| 2014 | 16.72 | 62.73 | 40.76 | 8.36 | 20.51 | |||
| 2015 | 18.07 | 63.35 | 41.39 | 8.61 | 20.79 | |||
| 2016 | 19.15 | 60.65 | 40.69 | 8.15 | 20.04 | |||
| Leaf width (cm) | 2013 | 11.90 | 51.22 | 29.23 | 7.23 | 24.72 | <0.000 ** | 0.99 |
| 2014 | 13.86 | 52.50 | 32.51 | 7.15 | 21.99 | |||
| 2015 | 14.64 | 53.05 | 33.29 | 7.53 | 22.61 | |||
| 2016 | 15.80 | 49.00 | 32.26 | 6.87 | 21.28 | |||
| Flower diameter (cm) | 2013 | 10.70 | 30.60 | 19.00 | 3.79 | 19.95 | 0.021 * | 0.93 |
| 2014 | 10.28 | 31.80 | 18.76 | 3.95 | 21.07 | |||
| 2015 | 9.90 | 29.60 | 19.10 | 3.86 | 20.21 | |||
| 2016 | 10.50 | 28.60 | 17.96 | 3.35 | 18.66 |
| Marker | Trait | Based on Phenotypic Data of 2013 | Based on Phenotypic Data of 2014 | Based on Phenotypic Data of 2015 | Based on Phenotypic Data of 2016 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p-Value | Q-Value | R2 (%) | p-Value | Q-Value | R2 (%) | p-Value | Q-Value | R2 (%) | p-Value | Q-Value | R2 (%) | ||
| SSR35 | Flower diameter | 2.00 × 104 | 1.29 × 102 | 14.62 | 6.48 × 104 | 7.60 × 103 | 14.69 | ||||||
| SSR41 | Petiole height | 4.83 × 104 | 1.56 × 102 | 13.85 | 1.65 × 104 | 5.33 × 103 | 13.38 | ||||||
| Peduncle height | 8.78 × 104 | 3.78 × 102 | 13.14 | 4.90 × 104 | 7.91 × 103 | 12.27 | |||||||
| SSR67 | Petiole height | 2.84 × 106 | 3.66 × 104 | 12.54 | 4.34 × 104 | 1.12 × 102 | 8.92 | ||||||
| Peduncle height | 9.34 × 104 | 3.01 × 102 | 8.50 | 2.37 × 106 | 1.53 × 104 | 12.72 | 1.23 × 104 | 3.18 × 103 | 10.13 | ||||
| Leaf length | 2.47 × 105 | 7.96 × 104 | 10.73 | 2.97 × 105 | 7.66 × 104 | 11.55 | 2.73 × 104 | 5.88 × 103 | 9.62 | ||||
| Leaf width | 1.30 × 104 | 1.68 × 102 | 10.43 | 1.90 × 105 | 4.89 × 104 | 10.95 | 1.92 × 105 | 4.13 × 104 | 11.97 | 8.05 × 105 | 3.46 × 103 | 10.82 | |
| Flower diameter | 5.67 × 104 | 1.46 × 102 | 8.03 | 1.33 × 104 | 3.44 × 103 | 10.24 | 9.55 × 105 | 4.11 × 103 | 10.76 | ||||
| SSR70 | Flower diameter | 3.69 × 104 | 5.29 × 103 | 11.92 | |||||||||
| SSR88 | Petiole height | 1.71 × 104 | 4.40 × 103 | 16.59 | 2.25 × 104 | 2.90 × 102 | 18.16 | ||||||
| Peduncle height | 3.98 × 104 | 7.33 × 103 | 15.64 | ||||||||||
| SSR122 | Petiole height | 8.57 × 104 | 2.21 × 102 | 7.71 | |||||||||
| Peduncle height | 9.08 × 104 | 1.95 × 102 | 8.24 | ||||||||||
| SSR165 | Petiole height | 3.75 × 104 | 8.06 × 103 | 9.08 | |||||||||
| SSR319 | Leaf width | 5.72 × 104 | 9.23 × 103 | 6.05 | |||||||||
| NNIndel_66 | Petiole height | 6.56 × 104 | 1.06 × 102 | 5.31 | |||||||||
| Flower diameter | 7.63 × 104 | 8.20 × 103 | 5.77 | ||||||||||
| NNIndel_68 | Leaf length | 9.70 × 104 | 1.04 × 102 | 5.50 | |||||||||
| Flower diameter | 1.93 × 104 | 4.15 × 103 | 6.91 | 1.23 × 104 | 2.65 × 103 | 7.42 | |||||||
| NNIndel_69 | Flower diameter | 3.27 × 104 | 4.22 × 103 | 6.47 | |||||||||
| NNIndel_71 | Leaf length | 8.06 × 104 | 9.45 × 103 | 5.65 | |||||||||
| Flower diameter | 2.60 × 104 | 3.72 × 103 | 6.66 | 1.87 × 104 | 3.01 × 103 | 7.07 | |||||||
| NNIndel_72 | Flower diameter | 3.80 × 104 | 1.22 × 102 | 5.77 | |||||||||
| NNIndel_73 | Petiole height | 1.66 × 104 | 1.07 × 102 | 7.14 | |||||||||
| Peduncle height | 1.64 × 104 | 2.11 × 102 | 7.16 | 6.38 × 104 | 8.23 × 103 | 5.34 | |||||||
| Leaf length | 1.80 × 104 | 2.58 × 103 | 6.89 | ||||||||||
| Leaf width | 4.71 × 104 | 7.60 × 103 | 5.58 | ||||||||||
| NNIndel_74 | Flower diameter | 3.59 × 104 | 1.54 × 102 | 5.81 | |||||||||
| NNIndel_78 | Leaf length | 6.68 × 104 | 8.61 × 103 | 7.73 | |||||||||
| Leaf width | 3.02 × 104 | 4.87 × 103 | 8.46 | ||||||||||
| NNIndel_80 | Leaf length | 7.73 × 105 | 1.42 × 103 | 7.58 | 2.40 × 104 | 6.19 × 103 | 6.77 | ||||||
| Leaf width | 2.89 × 104 | 5.33 × 103 | 6.63 | ||||||||||
| NNIndel_81 | Petiole height | 1.27 × 104 | 1.64 × 102 | 7.37 | 6.92 × 105 | 2.98 × 103 | 7.00 | 2.96 × 104 | 1.91 × 102 | 6.42 | |||
| Peduncle height | 1.14 × 105 | 4.90 × 104 | 8.39 | 6.32 × 105 | 2.72 × 103 | 7.70 | 3.14 × 104 | 4.05 × 102 | 6.55 | ||||
| Leaf length | 1.29 × 105 | 8.32 × 104 | 8.31 | 8.90 × 106 | 3.83 × 104 | 9.40 | 4.89 × 106 | 6.31 × 104 | 10.10 | ||||
| Leaf width | 4.06 × 106 | 2.62 × 104 | 9.21 | 5.66 × 106 | 2.43 × 104 | 9.78 | 3.37 × 106 | 4.34 × 104 | 10.43 | ||||
| Flower diameter | 2.84 × 105 | 1.22 × 103 | 8.52 | 1.12 × 104 | 2.90 × 103 | 7.50 | |||||||
| NNIndel_88 | Flower diameter | 9.31 × 104 | 9.24 × 103 | 7.52 | |||||||||
| NNIndel_94 | Petiole height | 6.92 × 104 | 9.92 × 103 | 5.27 | |||||||||
| Peduncle height | 2.38 × 104 | 6.14 × 103 | 6.08 | ||||||||||
| Leaf length | 4.83 × 104 | 8.90 × 103 | 5.56 | 1.42 × 104 | 2.29 × 103 | 7.08 | 4.62 × 104 | 8.51 × 103 | 6.23 | ||||
| Leaf width | 1.31 × 104 | 2.42 × 103 | 6.54 | 5.56 × 105 | 1.02 × 103 | 7.86 | 1.56 × 104 | 3.36 × 103 | 7.14 | ||||
| Flower diameter | 2.46 × 104 | 3.96 × 103 | 6.71 | 5.52 × 104 | 7.12 × 103 | 6.15 | |||||||
| NNIndel_95 | Petiole height | 6.95 × 104 | 8.96 × 103 | 5.27 | 3.70 × 104 | 1.19 × 102 | 6.24 | ||||||
| Peduncle height | 1.15 × 104 | 3.69 × 103 | 6.63 | 5.57 × 105 | 3.59 × 103 | 7.81 | |||||||
| Leaf length | 1.31 × 105 | 5.61 × 104 | 8.30 | 2.22 × 105 | 7.15 × 104 | 8.63 | 5.49 × 105 | 2.36 × 103 | 8.02 | ||||
| Leaf width | 1.17 × 105 | 5.04 × 104 | 8.39 | 8.01 × 106 | 2.58 × 104 | 9.49 | 9.99 × 105 | 3.22 × 103 | 7.52 | ||||
| Flower diameter | 3.65 × 106 | 4.71 × 104 | 10.28 | 3.73 × 105 | 2.41 × 103 | 8.45 | |||||||
| NNIndel_96 | Peduncle height | 7.71 × 104 | 9.05 × 103 | 6.94 | |||||||||
| Leaf length | 1.54 × 104 | 3.31 × 103 | 8.31 | 5.25 × 105 | 1.13 × 103 | 10.06 | |||||||
| Leaf width | 7.28 × 105 | 1.56 × 103 | 8.93 | 1.44 × 105 | 3.72 × 104 | 11.24 | |||||||
| Flower diameter | 2.05 × 104 | 3.77 × 103 | 8.92 | 1.06 × 104 | 3.43 × 103 | 9.70 | |||||||
| NNIndel_97 | Petiole height | 8.50 × 104 | 9.97 × 103 | 9.72 | |||||||||
| NNIndel_99 | Petiole height | 2.79 × 104 | 1.20 × 102 | 6.70 | 6.87 × 106 | 4.43 × 104 | 8.76 | 1.37 × 104 | 1.77 × 102 | 7.04 | |||
| Peduncle height | 6.32 × 104 | 4.07 × 102 | 6.02 | 1.35 × 106 | 1.74 × 104 | 10.04 | 1.66 × 105 | 2.14 × 103 | 8.82 | ||||
| Leaf length | 1.32 × 106 | 1.70 × 104 | 10.08 | 2.66 × 106 | 3.43 × 104 | 10.42 | 6.20 × 106 | 4.00 × 104 | 9.89 | ||||
| Leaf width | 2.16 × 104 | 1.39 × 102 | 6.92 | 5.35 × 107 | 6.90 × 105 | 10.79 | 5.40 × 107 | 6.97 × 105 | 11.79 | 3.90 × 106 | 2.52 × 104 | 10.30 | |
| Flower diameter | 1.85 × 104 | 2.39 × 102 | 6.31 | 2.95 × 105 | 9.52 × 104 | 8.49 | 3.34 × 106 | 4.31 × 104 | 10.55 | ||||
| NNIndel_101 | Petiole height | 9.30 × 104 | 1.00 × 102 | 5.05 | 3.62 × 104 | 1.55 × 102 | 6.25 | ||||||
| Peduncle height | 2.87 × 104 | 6.18 × 103 | 5.94 | 9.26 × 105 | 2.99 × 103 | 7.39 | |||||||
| Leaf length | 4.03 × 105 | 1.04 × 103 | 7.44 | 6.56 × 106 | 4.23 × 104 | 9.65 | 1.19 × 104 | 3.85 × 103 | 7.36 | ||||
| Leaf width | 1.87 × 105 | 6.02 × 104 | 8.03 | 2.38 × 106 | 1.53 × 104 | 10.52 | 1.34 × 104 | 3.45 × 103 | 7.28 | ||||
| Flower diameter | 1.64 × 105 | 1.06 × 103 | 8.99 | 1.58 × 104 | 2.91 × 103 | 7.21 | |||||||
| NNIndel_104 | Petiole height | 6.48 × 104 | 1.19 × 102 | 5.32 | |||||||||
| Peduncle height | 5.56 × 104 | 7.97 × 103 | 5.45 | ||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, M.; Zhang, J.; Yang, C.; Cui, Z.; Chen, L. Identification of QTLs and Putative Candidate Genes for Plant Architecture of Lotus Revealed by Regional Association Mapping. Plants 2023, 12, 1221. https://doi.org/10.3390/plants12061221
Zhao M, Zhang J, Yang C, Cui Z, Chen L. Identification of QTLs and Putative Candidate Genes for Plant Architecture of Lotus Revealed by Regional Association Mapping. Plants. 2023; 12(6):1221. https://doi.org/10.3390/plants12061221
Chicago/Turabian StyleZhao, Mei, Jibin Zhang, Chuxuan Yang, Zhenhua Cui, and Longqing Chen. 2023. "Identification of QTLs and Putative Candidate Genes for Plant Architecture of Lotus Revealed by Regional Association Mapping" Plants 12, no. 6: 1221. https://doi.org/10.3390/plants12061221
APA StyleZhao, M., Zhang, J., Yang, C., Cui, Z., & Chen, L. (2023). Identification of QTLs and Putative Candidate Genes for Plant Architecture of Lotus Revealed by Regional Association Mapping. Plants, 12(6), 1221. https://doi.org/10.3390/plants12061221