


Peptidome and Transcriptome Analysis of Plant Peptides Involved in Bipolaris maydis Infection of Maize

Abstract
:1. Introduction
2. Results
2.1. Identification of Phenotypes and Characteristics of SCLB-Infected Maize
2.2. Illumina Sequencing and DEGs Analysis
2.3. Functional Classification of DEGs
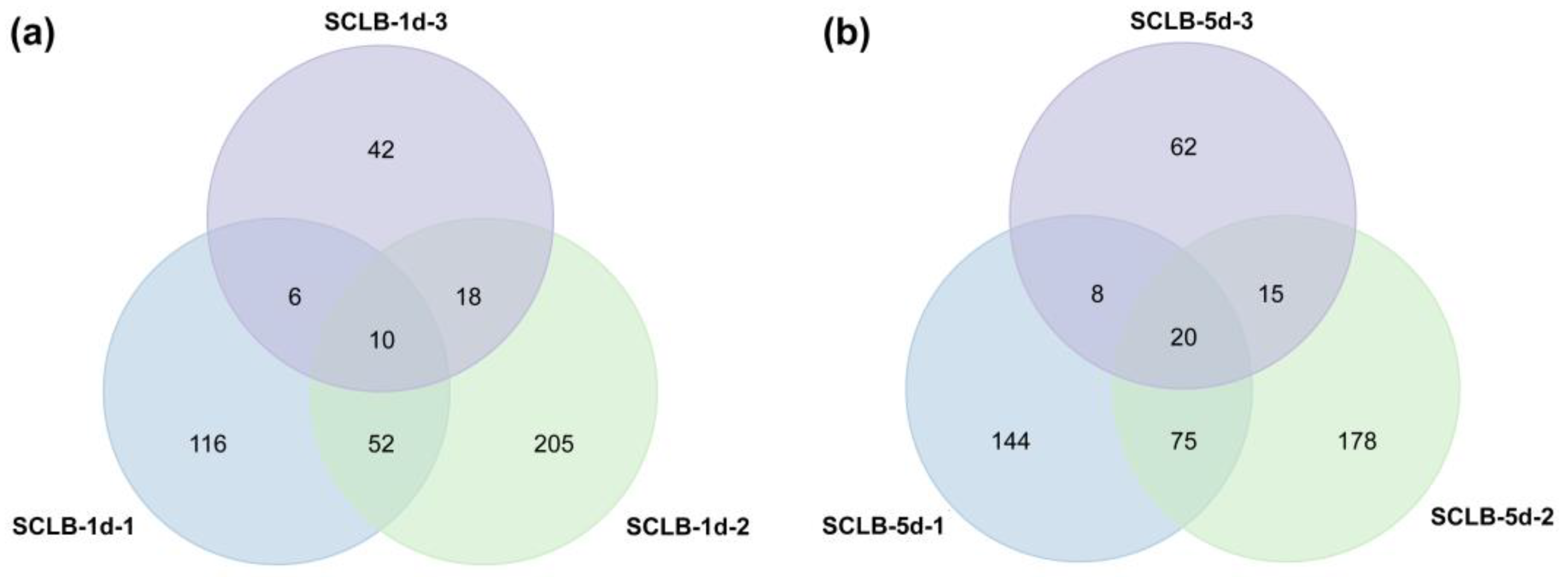
2.4. Identification, Comparison, and Characterization of DEPs
2.5. Functional Classification of DEPs’ Precursor Proteins
3. Discussion
4. Materials and Methods
4.1. Plant Materials and B. maydis Inoculation
4.2. Measurement of ROS Accumulation, MDA Content, and Antioxidant Enzyme Activities
4.3. Total RNA Isolation, mRNA Library Construction, and Sequencing
4.4. Peptide Extraction and Tandem Mass Tag (TMT) Labeling
4.5. Chromatography and MS/MS Analysis
4.6. Database Search and Peptide Identification
4.7. Functional Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Savary, S.; Willocquet, L.; Pethybridge, S.J.; Esker, P.; McRoberts, N.; Nelson, A. The global burden of pathogens and pests on major food crops. Nat. Ecol. Evol. 2019, 3, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Genetic Architecture of Disease Resistance in Maize. Available online: https://portal.nifa.usda.gov/web/crisprojectpages/1008502-genetic-architecture-of-disease-resistance-in-maize.html (accessed on 20 December 2022).
- Zhu, M.; Tong, L.; Xu, M.; Zhong, T. Genetic dissection of maize disease resistance and its applications in molecular breeding. Mol. Breed. 2021, 41, 32. [Google Scholar] [CrossRef]
- Identifying Key Diseases in Corn. Available online: https://www.cropscience.bayer.us/learning-center/articles/corn-diseases-threaten-yields (accessed on 20 December 2022).
- Zheng, H.; Chen, J.; Mu, C.; Makumbi, D.; Xu, Y.; Mahuku, G. Combined linkage and association mapping reveal QTL for host plant resistance to common rust (Puccinia sorghi) in tropical maize. BMC Plant Biol. 2018, 18, 310. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Balint-Kurti, P.; Xu, M. Quantitative disease resistance: Dissection and adoption in maize. Mol. Plant 2017, 10, 402–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, H.L.; Jones, A.; Huber, D.M. Morphological and physiological differences between Bipolaris maydis Races O and T. Mycologia 1977, 69, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Ullstrup, A.J. The impacts of the southern corn leaf blight epidemics of 1970–1971. Annu. Rev. Phytopathol. 1972, 10, 7–50. [Google Scholar] [CrossRef]
- Gregory, P.; Matthews, D.E.; York, D.W.; Earle, E.D.; Gracen, V.E. Southern corn leaf blight disease: Studies on mitochondrial biochemistry and ultrastructure. Mycopathologia 1978, 66, 105–112. [Google Scholar] [CrossRef]
- Carson, M.L.; Stuber, C.W.; Senior, M.L. Identification and mapping of quantitative trait loci conditioning resistance to southern leaf blight of maize caused by Cochliobolus heterostrophus Race O. Phytopathology 2004, 94, 862–867. [Google Scholar] [CrossRef] [Green Version]
- Balint-Kurti, P.J.; Zwonitzer, J.C.; Wisser, R.J.; Carson, M.L.; Oropeza-Rosas, M.A.; Holland, J.B.; Szalma, S.J. Precise mapping of quantitative trait loci for resistance to southern leaf blight, caused by Cochliobolus heterostrophus race O, and flowering time using advanced intercross maize lines. Genetics 2007, 176, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Kump, K.L.; Bradbury, P.J.; Wisser, R.J.; Buckler, E.S.; Belcher, A.R.; Oropeza-Rosas, M.A.; Zwonitzer, J.C.; Kresovich, S.; McMullen, M.D.; Ware, D.; et al. Genome-wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nat. Genet. 2011, 43, 163–168. [Google Scholar] [CrossRef]
- Kaur, M.; Vikal, Y.; Kaur, H.; Pal, L.; Kaur, K.; Chawla, J.S. Mapping quantitative trait loci associated with southern leaf blight resistance in maize (Zea mays L.). J. Phytopathol. 2019, 167, 591–600. [Google Scholar] [CrossRef]
- Ye, Y.; Li, Q.; Fu, G.; Yuan, G.; Miao, J.; Lin, W. Identification of antifungal substance (iturin A2) produced by Bacillus subtilis B47 and its effect on southern corn leaf blight. J. Integr. Agr. 2012, 11, 90–99. [Google Scholar] [CrossRef]
- Lai, Y.-R.; Lin, P.-Y.; Chen, C.-Y.; Huang, C.-J. Feasible management of southern corn leaf blight via induction of systemic resistance by Bacillus cereus C1L in combination with reduced use of dithiocarbamate fungicides. Plant Pathol. J. 2016, 32, 481–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, T.; Su, B.; Chen, X.; Xie, S.; Gu, S.; Wang, Q.; Huang, D.; Jiang, H. An endophytic bacterial strain isolated from Eucommia ulmoides inhibits southern corn leaf blight. Front. Microbiol. 2017, 8, 903. [Google Scholar] [CrossRef] [Green Version]
- Farrokhi, N.; Whitelegge, J.P.; Brusslan, J.A. Plant peptides and peptidomics. Plant Biotechnol. J. 2008, 6, 105–134. [Google Scholar] [CrossRef]
- Czyzewicz, N.; Shi, C.-L.; Vu, L.D.; van de Cotte, B.; Hodgman, C.; Butenko, M.A.; de Smet, I. Modulation of Arabidopsis and monocot root architecture by CLAVATA3/EMBRYO SURROUNDING REGION 26 peptide. J. Exp. Bot. 2015, 66, 5229–5243. [Google Scholar] [CrossRef] [Green Version]
- Czyzewicz, N.; Wildhagen, M.; Cattaneo, P.; Stahl, Y.; Pinto, K.G.; Aalen, R.B.; Butenko, M.A.; Simon, R.; Hardtke, C.S.; de Smet, I. Antagonistic peptide technology for functional dissection of CLE peptides revisited. J. Exp. Bot. 2015, 66, 5367–5374. [Google Scholar] [CrossRef] [Green Version]
- Czyzewicz, N.; de Smet, I. The Arabidopsis thaliana CLAVATA3/EMBRYO-SURROUNDING REGION 26 (CLE26) peptide is able to alter root architecture of Solanum lycopersicum and Brassica napus. Plant Signal Behav. 2016, 11, e1118598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsubayashi, Y. Posttranslationally modified small-peptide signals in plants. Annu. Rev. Plant Biol. 2014, 65, 385–413. [Google Scholar] [CrossRef]
- Sun, J.-Q.; Jiang, H.-L.; Li, C.-Y. Systemin/jasmonate-mediated systemic defense signaling in tomato. Mol. Plant 2011, 4, 607–615. [Google Scholar] [CrossRef]
- Nakaminami, K.; Okamoto, M.; Higuchi-Takeuchi, M.; Yoshizumi, T.; Yamaguchi, Y.; Fukao, Y.; Shimizu, M.; Ohashi, C.; Tanaka, M.; Matsui, M.; et al. AtPep3 is a hormone-like peptide that plays a role in the salinity stress tolerance of plants. Proc. Natl. Acad. Sci. USA 2018, 115, 5810–5815. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-L.; Fan, K.-T.; Hung, S.-C.; Chen, Y.-R. The role of peptides cleaved from protein precursors in eliciting plant stress reactions. New Phytol. 2019, 225, 2267–2282. [Google Scholar] [CrossRef] [Green Version]
- Huffaker, A.; Pearce, G.; Ryan, C.A. An endogenous peptide signal in Arabidopsis activates components of the innate immune response. Proc. Natl. Acad. Sci. USA 2006, 103, 10098–10103. [Google Scholar] [CrossRef] [Green Version]
- Hander, T.; Fernández-Fernández, Á.D.; Kumpf, R.P.; Willems, P.; Schatowitz, H.; Rombaut, D.; Staes, A.; Nolf, J.; Pottie, R.; Yao, P.; et al. Damage on plants activates Ca2+-dependent metacaspases for release of immunomodulatory peptides. Science 2019, 363, eaar7486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, P.-S.; Chiang, C.-B.; Wang, Z.; Chiou, T.-J. MicroRNA-mediated signaling and regulation of nutrient transport and utilization. Curr. Opin. Plant Biol. 2017, 39, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Dallas, D.C.; Guerrero, A.; Parker, E.A.; Robinson, R.C.; Gan, J.; German, J.B.; Barile, D.; Lebrilla, C.B. Current peptidomics: Applications, purification, identification, quantification, and functional analysis. Proteomics 2015, 15, 1026–1038. [Google Scholar] [CrossRef] [Green Version]
- Romanova, E.V.; Sweedler, J.V. Peptidomics for the discovery and characterization of neuropeptides and hormones. Trends Pharmacol. Sci. 2015, 36, 579–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forman, R.E.; George, A.L.; Reimann, F.; Gribble, F.M.; Kay, R.G. Peptidomics: A review of clinical applications and methodologies. J. Proteome Res. 2021, 20, 3782–3797. [Google Scholar] [CrossRef]
- Van, J.A.D.; Scholey, J.W.; Konvalinka, A. Insights into diabetic kidney disease using urinary proteomics and bioinformatics. J. Am. Soc. Nephrol. 2017, 28, 1050–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davenport, A.P.; Scully, C.C.G.; de Graaf, C.; Brown, A.J.H.; Maguire, J.J. Advances in therapeutic peptides targeting G protein-coupled receptors. Nat. Rev. Drug Discov. 2020, 19, 389–413. [Google Scholar] [CrossRef]
- Phetsanthad, A.; Vu, N.Q.; Yu, Q.; Buchberger, A.R.; Chen, Z.; Keller, C.; Li, L. Recent advances in mass spectrometry analysis of neuropeptides. Mass Spectrom. Rev. 2021, e21734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, C.-Y.; Chen, W.-C.; Shi, Y.-C.; Wang, C.-M.; Lin, S.; He, H.-F. Regulation of neuropeptide Y in body microenvironments and its potential application in therapies: A review. Cell Biosci. 2021, 11, 151. [Google Scholar] [CrossRef] [PubMed]
- Lease, K.A.; Walker, J.C. The Arabidopsis unannotated secreted peptide database, a resource for plant peptidomics. Plant Physiol. 2006, 142, 831–838. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-L.; Lee, C.-Y.; Cheng, K.-T.; Chang, W.-H.; Huang, R.-N.; Nam, H.; Chen, Y.-R. Quantitative peptidomics study reveals that a wound-induced peptide from PR-1 regulates immune signaling in tomato. Plant Cell 2014, 26, 4135–4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellinger, R.; Koehbach, J.; Soltis, D.E.; Carpenter, E.J.; Wong, G.K.; Gruber, C.W. Peptidomics of circular cysteine-rich plant peptides: Analysis of the diversity of cyclotides from Viola tricolor by transcriptome and proteome mining. J. Proteome Res. 2015, 14, 4851–4862. [Google Scholar] [CrossRef]
- Ziemannm, S.; van der Linde, K.; Lahrmann, U.; Acar, B.; Kaschani, F.; Colby, T.; Kaiser, M.; Ding, Y.; Schmelz, E.; Huffaker, A.; et al. An apoplastic peptide activates salicylic acid signalling in maize. Nat. Plants 2018, 4, 172–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Vinocur, B.; Altman, A. Plant responses to drought, salinity and extreme temperatures: Towards genetic engineering for stress tolerance. Planta 2003, 218, 1–14. [Google Scholar] [CrossRef]
- Grayson, M. Agriculture and drought. Nature 2013, 501, S1. [Google Scholar] [CrossRef] [Green Version]
- Stotz, H.U.; Mitrousia, G.K.; de Wit, P.J.G.M.; Fitt, B.D.L. Effector-triggered defence against apoplastic fungal pathogens. Trends Plant Sci. 2014, 19, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Gong, Z.; Xiong, L.; Shi, H.; Yang, S.; Herrera-Estrella, L.R.; Xu, G.; Chao, D.Y.; Li, J.; Wang, P.-Y.; Qin, F.; et al. Plant abiotic stress response and nutrient use efficiency. Sci. China Life Sci. 2020, 63, 635–674. [Google Scholar] [CrossRef]
- Raghavendra, A.S.; Gonugunta, V.K.; Christmann, A.; Grill, E. ABA perception and signalling. Trends Plant Sci. 2010, 15, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Golldack, D.; Li, C.; Mohan, H.; Probst, N. Tolerance to drought and salt stress in plants: Unraveling the signaling networks. Front. Plant Sci. 2014, 5, 151. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Tsuda, K.; Parker, J.E. Effector-triggered immunity: From pathogen perception to robust defense. Annu. Rev. Plant Biol. 2015, 66, 487–511. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhu, Y.-X.; Balint-Kurti, P.J.; Wang, G.-F. Fine-tuning immunity: Players and regulators for plant NLRs. Trends Plant Sci. 2020, 25, 695–713. [Google Scholar] [CrossRef] [PubMed]
- Tavormina, P.; De Coninck, B.; Nikonorova, N.; De Smet, I.; Cammue, B.P.A. The plant peptidome: An expanding repertoire of structural features and biological functions. Plant Cell 2015, 27, 2095–2118. [Google Scholar] [CrossRef] [Green Version]
- Mittler, R.; Zandalinas, S.I.; Fichman, Y.; Van Breusegem, F. Reactive oxygen species signalling in plant stress responses. Nat. Rev. Mol. Cell Biol. 2022, 23, 663–679. [Google Scholar] [CrossRef]
- Van der Linde, K.; Hemetsberger, C.; Kastner, C.; Kaschani, F.; van der Hoorn, R.A.L.; Kumlehn, J.; Doehlemann, G. A maize cystatin suppresses host immunity by inhibiting apoplastic cysteine proteases. Plant Cell 2012, 24, 1285–1300. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lu, S.; Liu, K.; Wang, S.; Huang, L.; Guo, L. Proteomics: A powerful tool to study plant responses to biotic stress. Plant Methods 2019, 15, 135. [Google Scholar] [CrossRef]
- Peng, B.; Li, H.; Peng, X. Proteomics approach to understand bacterial antibiotic resistance strategies. Expert Rev. Proteomic 2019, 16, 829–839. [Google Scholar] [CrossRef]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [Green Version]
- Daudi, A.; O’Brien, J.A. Detection of hydrogen peroxide by DAB staining in Arabidopsis leaves. Bio-protocol 2012, 2, e263. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.E.; Cui, J.M.; Li, G.X.; Yuan, M.; Zhang, Z.W.; Yuan, S.; Zhang, H.Y. Effect of salicylic acid on the antioxidant system and photosystem II in wheat seedlings. Biol. Plant. 2016, 60, 139–147. [Google Scholar] [CrossRef]
- Li, D.; Zhang, Y.; Hu, X.; Hu, X.; Shen, X.; Ma, L.; Su, Z.; Wang, T.; Dong, J. Transcriptional profiling of Medicago truncatula under salt stress identified a novel CBF transcription factor MtCBF4 that plays an important role in abiotic stress responses. BMC Plant Biol. 2011, 11, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Xia, X.; Wang, J.; Zhu, L.; Wang, J.; Wang, G.; Chen, Y.; Kim, Y.M. Oxidative stress and genotoxicity of nitenpyram to earthworms (Eisenia foetida). Chemosphere 2021, 264, 128493. [Google Scholar] [CrossRef]
- Andrews, S. FASTQC. A quality control tool for high throughput sequence data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 April 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.; Guo, X.; Yin, M.; Tariq, M.; Lai, Y.; Kanwal, S.; Zhou, J.; Li, N.; Lv, Y.; Pulido-Quetglas, C.; et al. Capturing the interactome of newly transcribed RNA. Nat. Methods 2018, 15, 213–220. [Google Scholar] [CrossRef]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. AgriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res 2017, 45, W122–W129. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Raw Reads | Clean Reads | Q20 (%) | Q30 (%) | GC (%) |
|---|---|---|---|---|---|
| CK-5d-1 | 52040214 | 50857364 | 98.26 | 94.87 | 55.07 |
| CK-5d-2 | 50225462 | 48705260 | 98.08 | 94.43 | 54.84 |
| SCLB-5d-1 | 50900144 | 49825876 | 98.34 | 95.07 | 55.36 |
| SCLB-5d-2 | 53742962 | 52147524 | 98.26 | 94.90 | 57.64 |
| Peptide Sequence | Accession | Precursor Protein | Positions | MH + [Da] | FC |
|---|---|---|---|---|---|
| SRINPLVRLK | B6SSH9 | Extracellular ribonuclease LE | (143–152) | 1654.089 | 0.22 |
| AKGIEPDFGLYGLK | B6SSH9 | Extracellular ribonuclease LE | (153–166) | 2195.304 | 0.44 |
| LKAKGIEPDFGLYGLK | B6SSH9 | Extracellular ribonuclease LE | (151–166) | 2665.646 | 0.48 |
| EKDYFETALSFR | B6SSH9 | Extracellular ribonuclease LE | (131–142) | 1964.053 | 0.65 |
| AYPTSDVVIETHKEEEL | P27787 | Ferredoxin-1, chloroplastic | (131–147) | 2418.28 | 1.32 |
| LVLPGELAKHAVSEGTKAVTKFTSS | Q43261 | Histone H2B.3 * | (214–238) | 3487.071 | 1.44 |
| LVLFEHFGGDPSKISF | A0A1D6JWI7 | Beta-galactosidase, EC 3.2.1.23 | (767–782) | 2251.253 | 1.42 |
| LVLLEEFGGDLPGVKLVTRTA | B6T0D0 | Beta-galactosidase, EC 3.2.1.23 | (703–723) | 2685.596 | 1.40 |
| LVLLEEFGGDLPGVKLVT | B6T0D0 | Beta-galactosidase, EC 3.2.1.23 | (703–720) | 2357.41 | 2.05 |
| GLGGLFAKKSS | B4FS10 | Uncharacterized protein | (107–117) | 1752.099 | 2.18 |
| Peptide Sequence | Accession | Precursor Protein | Positions | MH + [Da] | FC |
|---|---|---|---|---|---|
| FISYVGDGFKLL | A0A1D6F9C2 | Oxygen-evolving enhancer protein 2-1 chloroplastic * | (90–101) | 1817.061 | 0.63 |
| AYGEAANVFGKTKKNTD | A0A1D6F9C2 | Oxygen-evolving enhancer protein 2-1 chloroplastic * | (73–89) | 2730.56 | 0.62 |
| ALGDVLAKLG | Q41048 | Oxygen-evolving enhancer protein 3-1, chloroplastic * | (208–217) | 1414.903 | 0.55 |
| ALGDVLAKLA | A0A1D6EXK9 | Oxygen-evolving enhancer protein 3-1, chloroplastic | (207–216) | 1428.919 | 0.64 |
| DLDHAAKIKSTPEAEKYFAATKD | A0A1D6EXK9; B6TI20 | Oxygen-evolving enhancer protein 3-1, chloroplastic, OEE3 | (184–206); (185–207) | 3695.103 | 0.69 |
| AAKLIRTQLASAK | Q2QLY5 | 5-methyltetrahydropteroyltriglutamate-homocysteine methyltransferase 1, EC 2.1.1.14 | (754–766) | 2058.337 | 0.51 |
| ALAKYFIGSVL | A0A1D6L4E9 | Exoglucanase1 | (101–111) | 1640.019 | 0.69 |
| AALVSAFASKGLD | A0A1D6H658 | Peroxidase, EC 1.11.1.7 | (169–181) | 1708.005 | 0.64 |
| AASEDTSASGDELIEDLK | B6TCN7 | Threonine endopeptidase | (48–65) | 2309.176 | 0.69 |
| SRINPLVRLK | B6SSH9 | Extracellular ribonuclease LE | (143–152) | 1654.089 | 0.72 |
| GDDLVDVLK | B4FAJ3 | Uncharacterized protein | (179–187) | 1431.846 | 0.71 |
| ATSTTDLPASYGVALGTGNYVVPVRLGTPAERF | A0A1D6LRY4 | Microtubule-associated protein MAP65-1 | (142–174) | 3609.911 | 1.29 |
| AQLDATYFAMEKLG | A0A1D6IE31 | Glucan endo-1,3-beta-D-glucosidase, EC 3.2.1.39 | (244–257) | 2032.083 | 1.31 |
| AVYQRSGGAPGGDADGGVDDDHDEL | B4FWJ8 | Luminal-binding protein 3, BiP3 * | (639–663) | 2702.213 | 1.34 |
| YILATSSNGYDPNFF | B6U534 | PSI-G (Photosystem I reaction center subunit V, chloroplastic) | (131–145) | 1937.948 | 1.50 |
| AKGIEPDFGLYGLKAITKVF | B6SSH9 | Extracellular ribonuclease LE | (153–172) | 3083.868 | 1.51 |
| AKANSLAQLGKYTSDG | B4FTI5 | Fructose-bisphosphate aldolase, chloroplastic, EC 4.1.2.13 (Chloroplastic aldolase, AldP) | (357–372) | 2311.322 | 1.51 |
| AKANSLAQLGKYTSDGEAAE | B4FTI5 | Fructose-bisphosphate aldolase, chloroplastic, EC 4.1.2.13 (Chloroplastic aldolase, AldP) | (357–376) | 2711.482 | 1.54 |
| ASTEEAVEAPKGFVAPQLD | B4FAW3 | Photosystem I reaction center subunit II (Photosystem I reaction center subunit II-1 chloroplastic) | (47–65) | 2417.296 | 1.58 |
| AVGDLAFKALTAGLGVATLY | A0A1D6FI52 | Uncharacterized protein | (2–21) | 2409.416 | 2.68 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheng, P.; Xu, M.; Zheng, Z.; Liu, X.; Ma, W.; Ding, T.; Zhang, C.; Chen, M.; Zhang, M.; Cheng, B.; et al. Peptidome and Transcriptome Analysis of Plant Peptides Involved in Bipolaris maydis Infection of Maize. Plants 2023, 12, 1307. https://doi.org/10.3390/plants12061307
Sheng P, Xu M, Zheng Z, Liu X, Ma W, Ding T, Zhang C, Chen M, Zhang M, Cheng B, et al. Peptidome and Transcriptome Analysis of Plant Peptides Involved in Bipolaris maydis Infection of Maize. Plants. 2023; 12(6):1307. https://doi.org/10.3390/plants12061307
Chicago/Turabian StyleSheng, Pijie, Minyan Xu, Zhenzhen Zheng, Xiaojing Liu, Wanlu Ma, Ting Ding, Chenchen Zhang, Meng Chen, Mengting Zhang, Beijiu Cheng, and et al. 2023. "Peptidome and Transcriptome Analysis of Plant Peptides Involved in Bipolaris maydis Infection of Maize" Plants 12, no. 6: 1307. https://doi.org/10.3390/plants12061307
APA StyleSheng, P., Xu, M., Zheng, Z., Liu, X., Ma, W., Ding, T., Zhang, C., Chen, M., Zhang, M., Cheng, B., & Zhang, X. (2023). Peptidome and Transcriptome Analysis of Plant Peptides Involved in Bipolaris maydis Infection of Maize. Plants, 12(6), 1307. https://doi.org/10.3390/plants12061307