

Transcriptome Expression Profiling Reveals the Molecular Response to Salt Stress in Gossypium anomalum Seedlings

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Physiological and Biochemical Evaluation of G. anomalum Seedlings Response to Salt Stress
2.2. Analysis of RNA-Seq Data
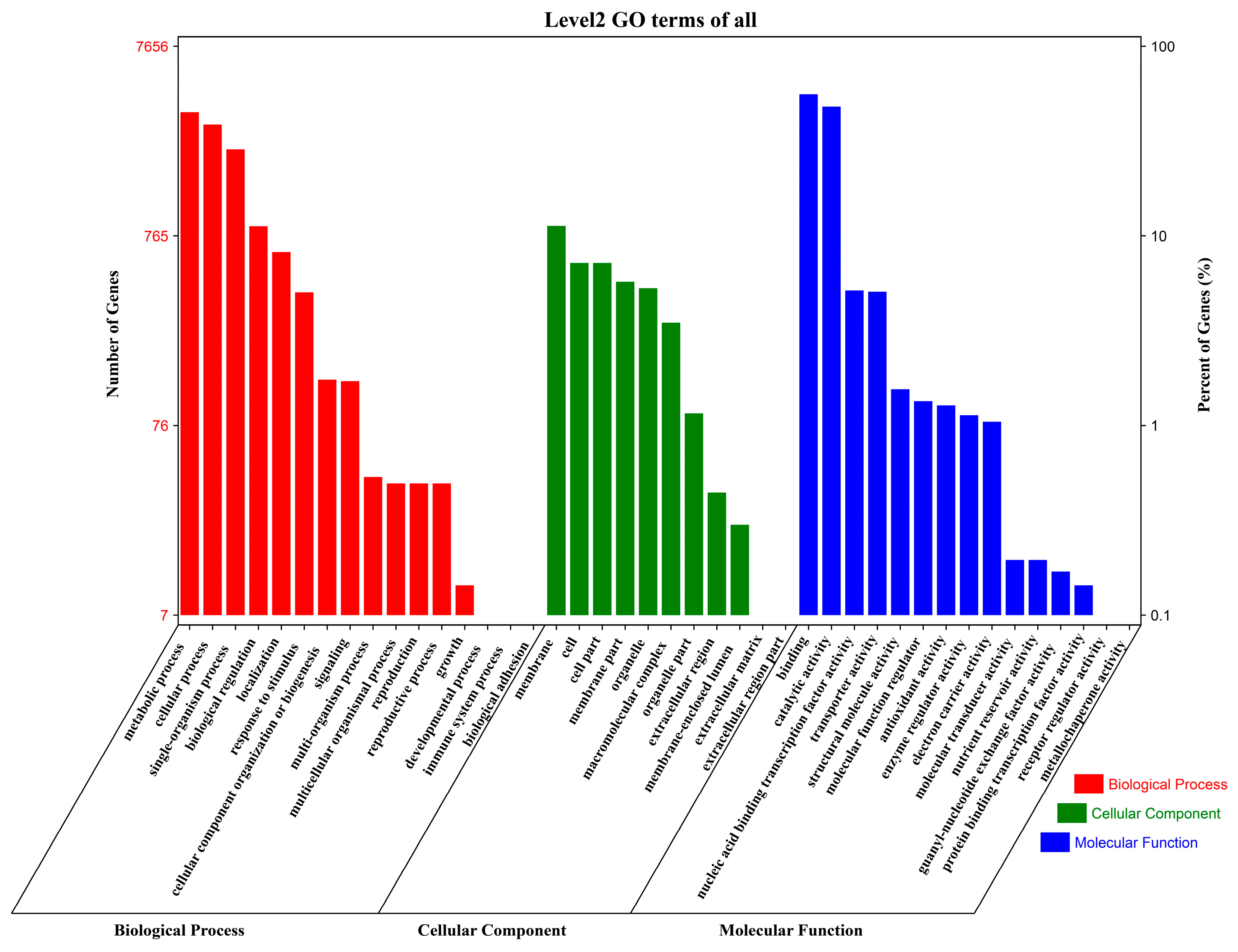
2.3. Gene Ontology (GO) Annotation and Enrichment Analysis
2.4. Kyoto Encyclopedia of Genes and Genomes (KEGG) Annotation and Enrichment Analysis
2.5. Transcriptional Factors (TFs) with Altered Expression Response to Salt Stress
2.6. Validation of RNA-Seq Data by qRT-PCR
3. Discussions
4. Materials and Methods
4.1. Preparation of Plant Material
4.2. Evaluation of Physiological and Biochemical Indexes
4.3. Library Preparation for RNA-Seq
4.4. Differential Expression Analysis
4.5. GO and KEGG Annotation and Enrichment Analysis
4.6. Transcription Factor (TF) Annotation
4.7. Validation of DEGs by qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- FAO. Global Map of Salt Affected Soils Version 1.0. 2021. Available online: https://www.fao.org/soils-portal/data-hub/soil-maps-and-databases/global-map-of-salt-affected-soils/en/ (accessed on 20 October 2021).
- Yang, Z.; Gao, C.; Zhang, Y.; Yan, Q.; Hu, W.; Yang, L.; Wang, Z.; Li, F. Recent progression and future perspectives in cotton genomic breeding. J. Integr. Plant Biol. 2023, 65, 548–569. [Google Scholar] [PubMed]
- Ahmad, I.; Zhu, G.; Zhou, G.; Song, X.; Hussein Ibrahim, M.E.; Ibrahim Salih, E.G.; Hussain, S.; Younas, M.U. Pivotal role of phytohormones and their responsive genes in plant growth and their signaling and transduction pathway under salt stress in Cotton. Int. J. Mol. Sci. 2022, 23, 7339. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, Z.; Zhang, J.; Zong, R. Soil salinity variations and cotton growth under long-term mulched drip irrigation in Saline-Alkali land of Arid Oasis. Irrig. Sci. 2021, 40, 103–113. [Google Scholar] [CrossRef]
- Shi, X.; Wang, C.; Zhao, J.; Wang, K.; Chen, F.; Chu, Q. Increasing inconsistency between climate suitability and production of cotton (Gossypium hirsutum L.) in China. Ind. Crops Prod. 2021, 171, 113959. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, J.D.; Fang, L.; Zhang, Z.Y.; Ma, W.; Niu, Y.C.; Ju, L.Z.; Deng, J.Q.; Zhao, T.; Lian, J.M.; et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 2019, 51, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, M. Salt Tolerance of cotton: Some new advances. Crit. Rev. Plant Sci. 2002, 21, 1–30. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, X.; Xu, Y.; Yang, H.; Tong, Z.; Tian, R.; Xu, S.; Yu, L.; Guo, Y.; Shi, P.; et al. Molecular mechanisms of adaptive evolution in wild animals and plants. Sci. China Life Sci. 2023, 66, 453–495. [Google Scholar] [CrossRef]
- Grover, C.E.; Yuan, D.; Arick, M.A.; Miller, E.R.; Hu, G.; Peterson, D.G.; Wendel, J.F.; Udall, J.A. The Gossypium anomalum genome as a resource for cotton improvement and evolutionary analysis of hybrid incompatibility. G3 2021, 11, jkab319. [Google Scholar] [CrossRef]
- Fryxell, P.A. A revised taxonomic interpretation of Gossypium, L. (Malvacea). Rheedea 1992, 2, 108–165. [Google Scholar]
- Nikalje, G.C.; Variyar, P.S.; Joshi, M.V.; Nikam, T.D.; Suprasanna, P. Temporal and spatial changes in ion homeostasis, antioxidant defense and accumulation of flavonoids and glycolipid in a halophyte Sesuvium portulacastrum (L.) L. PLoS ONE 2018, 13, e0193394. [Google Scholar] [CrossRef]
- Peng, Z.; Rehman, A.; Li, X.; Jiang, X.; Tian, C.; Wang, X.; Li, H.; Wang, Z.; He, S.; Du, X. Comprehensive evaluation and transcriptome analysis reveal the salt tolerance mechanism in semi-wild cotton (Gossypium purpurascens). Int. J. Mol. Sci. 2023, 24, 12853. [Google Scholar] [CrossRef]
- Xu, Z.Z.; Chen, J.D.; Meng, S.; Xu, P.; Zhai, C.J.; Huang, F.; Guo, Q.; Zhao, L.; Quan, Y.G.; Shangguan, Y.X.; et al. Genome sequence of Gossypium anomalum facilitates interspecific introgression breeding. Plant Commun. 2022, 3, 100350. [Google Scholar] [PubMed]
- James, R.A.; Rivelli, A.R.; Munns, R.; Caemmerer, S.V. Factors affecting CO2 assimilation, leaf injury and growth in salt-stressed durum wheat. Funct. Plant Biol. 2002, 29, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [PubMed]
- Barba-Espín, G.; Clemente-Moreno, M.J.; Alvarez, S.; García-Legaz, M.F.; Hernández, J.A.; Díaz-Vivancos, P. Salicylic acid negatively affects the response to salt stress in pea plants. Plant Biol. (Stuttg.) 2011, 13, 909–917. [Google Scholar] [CrossRef]
- Kato, T.; Takeda, K. Associations among characters related to yield sink capacity in space-planted rice. Crop Sci. 1996, 36, 1135–1139. [Google Scholar]
- Moradi, F.; Ismail, A.M. Responses of photosynthesis, chlorophyll fluorescence and ROS-scavenging systems to salt stress during seedling and reproductive stages in rice. Ann. Bot. 2007, 99, 1161–1173. [Google Scholar]
- Xu, P.; Liu, Z.; Fan, X.; Gao, J.; Zhang, X.; Zhang, X.; Shen, X. De novo transcriptome sequencing and comparative analysis of differentially expressed genes in Gossypium aridum under salt stress. Gene 2013, 525, 26–34. [Google Scholar] [CrossRef]
- Wei, Y.; Xu, Y.; Lu, P.; Wang, X.; Li, Z.; Cai, X.; Zhou, Z.; Wang, Y.; Zhang, Z.; Lin, Z.; et al. Salt stress responsiveness of a wild cotton species (Gossypium klotzschianum) based on transcriptomic analysis. PLoS ONE 2017, 12, e0178313. [Google Scholar]
- Zhang, F.; Zhu, G.; Du, L.; Shang, X.; Cheng, C.; Yang, B.; Hu, Y.; Cai, C.; Guo, W. Genetic regulation of salt stress tolerance revealed by RNA-Seq in cotton diploid wild species, Gossypium davidsonii. Sci. Rep. 2016, 6, 20582. [Google Scholar]
- Hammoudi, V. Oxidative stress is H2O2 under the bridge without MED8. Plant Cell 2021, 33, 1855–1856. [Google Scholar]
- Ahluwalia, O.; Singh, P.C.; Bhatia, R. A review on drought stress in plants: Implications, mitigation and the role of plant growth promoting rhizobacteria. Resour. Environ. Sustain. 2021, 5, 100032. [Google Scholar] [CrossRef]
- Nounjan, N.; Nghia, P.T.; Theerakulpisut, P. Exogenous proline and trehalose promote recovery of rice seedlings from salt-stress and differentially modulate antioxidant enzymes and expression of related genes. J. Plant Physiol. 2012, 169, 596–604. [Google Scholar] [PubMed]
- Xie, Z.; Duan, L.; Tian, X.; Wang, B.; Eneji, A.E.; Li, Z. Coronatine alleviates salinity stress in cotton by improving the antioxidative defense system and radical-scavenging activity. J. Plant Physiol. 2008, 165, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.; Suzuki, N.; Ciftci-Yilmaz, S.; Mittler, R. Reactive oxygen species homeostasis and signalling during drought and salinity stresses. Plant Cell Environ. 2010, 33, 453–467. [Google Scholar] [PubMed]
- Pan, J.; Zhang, M.; Kong, X.; Xing, X.; Liu, Y.; Zhou, Y.; Liu, Y.; Sun, L.; Li, D. ZmMPK17, a novel maize group D MAP kinase gene, is involved in multiple stress responses. Planta 2011, 235, 661–676. [Google Scholar]
- Zhang, D.; Jiang, S.; Pan, J.; Kong, X.; Zhou, Y.; Liu, Y.; Li, D. The overexpression of a maize mitogen-activated protein kinase gene (ZmMPK5) confers salt stress tolerance and induces defense responses in tobacco. Plant Biol. 2013, 16, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zou, D.; Li, Y.; Sun, X.; Wang, N.N.; Gong, S.Y.; Zheng, Y.; Li, X.B. GhMPK17, a cotton mitogen-activated protein kinase, is involved in plant response to high salinity and osmotic stresses and ABA signaling. PLoS ONE 2014, 9, e95642. [Google Scholar]
- Zhang, L.; Xi, D.; Li, S.; Gao, Z.; Zhao, S.; Shi, J.; Wu, C.; Guo, X. A cotton group C MAP kinase gene, GhMPK2, positively regulates salt and drought tolerance in tobacco. Plant Mol. Biol. 2011, 77, 17–31. [Google Scholar] [CrossRef]
- Xing, Y.; Chen, W.H.; Jia, W.; Zhang, J. Mitogen-activated protein kinase kinase 5 (MKK5)-mediated signaling cascade regulates expression of iron superoxide dismutase gene in Arabidopsis under salinity stress. J. Exp. Bot. 2015, 66, 5971–5981. [Google Scholar]
- Sicilia, A.; Testa, G.; Santoro, D.F.; Cosentino, S.L.; Lo Piero, A.R. RNASeq analysis of giant cane reveals the leaf transcriptome dynamics under long-term salt stress. BMC Plant Biol. 2019, 19, 355. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Qin, Y.; Hu, X.; Li, G.; Ding, H.; Xiong, X.; Wang, W. Transcriptome analysis uncovers the gene expression profile of salt-stressed potato (Solanum tuberosum L.). Sci. Rep. 2020, 10, 5411. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Sun, H.; Hakim; Yang, X.; Zhang, X. A novel cotton WRKY gene, GhWRKY6-like, improves salt tolerance by activating the ABA signaling pathway and scavenging of reactive oxygen species. Physiol. Plant. 2018, 162, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Q.; Liu, M.; Zhou, H.; Ma, C.; Wang, P. Regulation of plant responses to salt stress. Int. J. Mol. Sci. 2021, 22, 4609. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Abiotic stress signaling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, Z.; Gao, J.; Wang, P.; Hu, T.; Wang, Z.; Hou, Y.J.; Wan, Y.; Liu, W.; Xie, S.; et al. Arabidopsis duodecuple mutant of PYL ABA receptors reveals PYL repression of ABA-independent SnRK2 activity. Cell Rep. 2018, 23, 3340–3351.e5. [Google Scholar] [PubMed]
- Yamaguchi-Shinozaki, K.; Shinozaki, K. Organization of cis-acting regulatory elements in osmotic- and cold-stress-responsive promoters. Trends Plant Sci. 2005, 10, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Chinnusamy, V.; Rodrigues, A.; Rubio, S.; Antoni, R.; Park, S.Y.; Cutler, S.R.; Sheen, J.; Rodriguez, P.L.; Zhu, J.K. In vitro reconstitution of an abscisic acid signaling pathway. Nature 2009, 462, 660–664. [Google Scholar] [CrossRef]
- Cutler, S.R.; Rodriguez, P.L.; Finkelstein, R.R.; Abrams, S.R. Abscisic acid: Emergence of a core signaling network. Annu. Rev. Plant Biol. 2010, 61, 651–679. [Google Scholar] [CrossRef]
- Jung, J.; Won, S.Y.; Suh, S.C.; Kim, H.; Wing, R.; Jeong, Y.; Hwang, I.; Kim, M. The barley ERF-type transcription factor HvRAF confers enhanced pathogen resistance and salt tolerance in Arabidopsis. Planta 2007, 225, 575–588. [Google Scholar] [CrossRef]
- Yun, K.Y.; Park, M.R.; Mohanty, B.; Herath, V.; Xu, F.; Mauleon, R.; Wijaya, E.; Bajic, V.B.; Bruskiewich, R.; de Los Reyes, B.G. Transcriptional regulatory network triggered by oxidative signals configures the early response mechanisms of japonica rice to chilling stress. BMC Plant Biol. 2010, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Yang, W.W.; Liao, P.; Guo, Y.W.; Kumar, A.; Gao, W. Transcriptome analysis reveals differentially expressed ERF transcription factors associated with salt response in cotton. Plant Sci. 2019, 281, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Yu, F.; Gao, Z.; An, H.; Cao, X.; Guo, X. GhWRKY3, a novel cotton (Gossypium hirsutum L.) WRKY gene, is involved in diverse stress responses. Mol. Biol. Rep. 2011, 38, 49–58. [Google Scholar] [PubMed]
- Guo, Q.; Zhao, L.; Fan, X.; Xu, P.; Xu, Z.; Zhang, X.; Meng, S.; Shen, X. Transcription factor GarWRKY5 is involved in salt stress response in diploid cotton species (Gossypium aridum L.). Int. J. Mol. Sci. 2019, 20, 5244. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, Z.; Ding, Y.; Liu, L.; Han, X.; Zhan, J.; Wei, X.; Diao, Y.; Qin, W.; Wang, P.; et al. Over-expression of an R2R3 MYB Gene, GhMYB73, increases tolerance to salt stress in transgenic Arabidopsis. Plant Sci. 2019, 286, 28–36. [Google Scholar] [PubMed]
- Xu, P.; Guo, Q.; Meng, S.; Zhang, X.; Xu, Z.; Guo, W.; Shen, X. Genome-wide association analysis reveals genetic variations and candidate genes associated with salt tolerance related traits in Gossypium hirsutum. BMC Genom. 2021, 22, 26. [Google Scholar]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Parallel mapping of transcriptomes to detect InDels, gene fusions, and more. Genome Biol. 2013, 14, R36. [Google Scholar]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-Seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar]
- Aubert, J.; Bar-Hen, A.; Daudin, J.J.; Robin, S. Determination of the differentially expressed genes in microarray experiments using local FDR. BMC Bioinform. 2004, 5, 125. [Google Scholar]
- Pérez-Rodríguez, P.; Riaño-Pachón, D.M.; Corrêa, L.G.; Rensing, S.A.; Kersten, B.; Mueller-Roeber, B. PlnTFDB: Updated content and new features of the plant transcription factor database. Nucleic Acids Res. 2010, 38, D822–D827. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, H.; Guo, Q.; Ji, W.; Wang, H.; Tao, J.; Xu, P.; Chen, X.; Ali, W.; Wu, X.; Shen, X.; et al. Transcriptome Expression Profiling Reveals the Molecular Response to Salt Stress in Gossypium anomalum Seedlings. Plants 2024, 13, 312. https://doi.org/10.3390/plants13020312
Yu H, Guo Q, Ji W, Wang H, Tao J, Xu P, Chen X, Ali W, Wu X, Shen X, et al. Transcriptome Expression Profiling Reveals the Molecular Response to Salt Stress in Gossypium anomalum Seedlings. Plants. 2024; 13(2):312. https://doi.org/10.3390/plants13020312
Chicago/Turabian StyleYu, Huan, Qi Guo, Wei Ji, Heyang Wang, Jingqi Tao, Peng Xu, Xianglong Chen, Wuzhimu Ali, Xuan Wu, Xinlian Shen, and et al. 2024. "Transcriptome Expression Profiling Reveals the Molecular Response to Salt Stress in Gossypium anomalum Seedlings" Plants 13, no. 2: 312. https://doi.org/10.3390/plants13020312
APA StyleYu, H., Guo, Q., Ji, W., Wang, H., Tao, J., Xu, P., Chen, X., Ali, W., Wu, X., Shen, X., Xie, Y., & Xu, Z. (2024). Transcriptome Expression Profiling Reveals the Molecular Response to Salt Stress in Gossypium anomalum Seedlings. Plants, 13(2), 312. https://doi.org/10.3390/plants13020312