
Studies on Candidate Genes Related to Flowering Time in a Multiparent Population of Maize Derived from Tropical and Temperate Germplasm


Abstract
:1. Introduction
2. Results
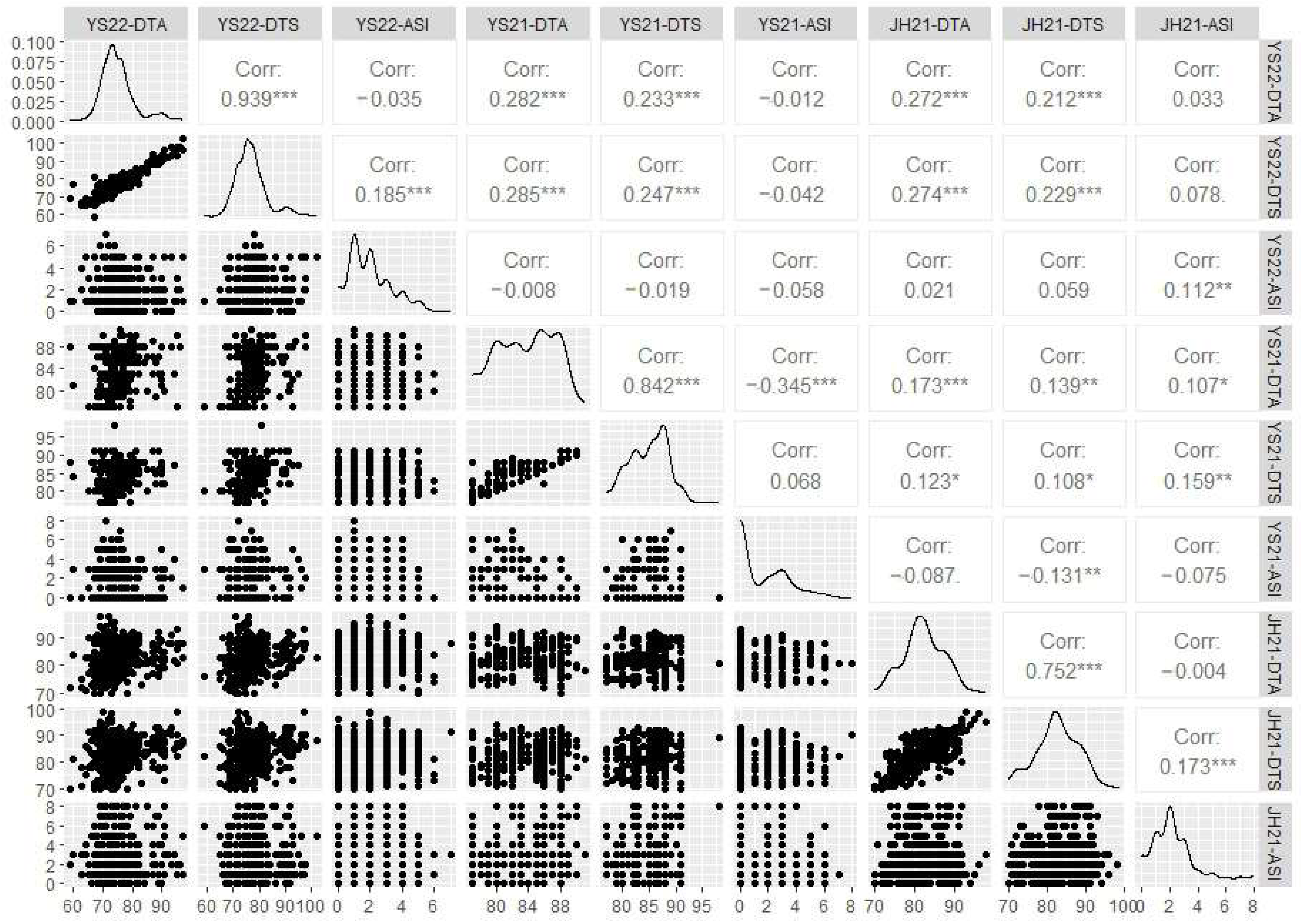
2.1. Phenotypic Analysis of Flowering Time-Related Traits
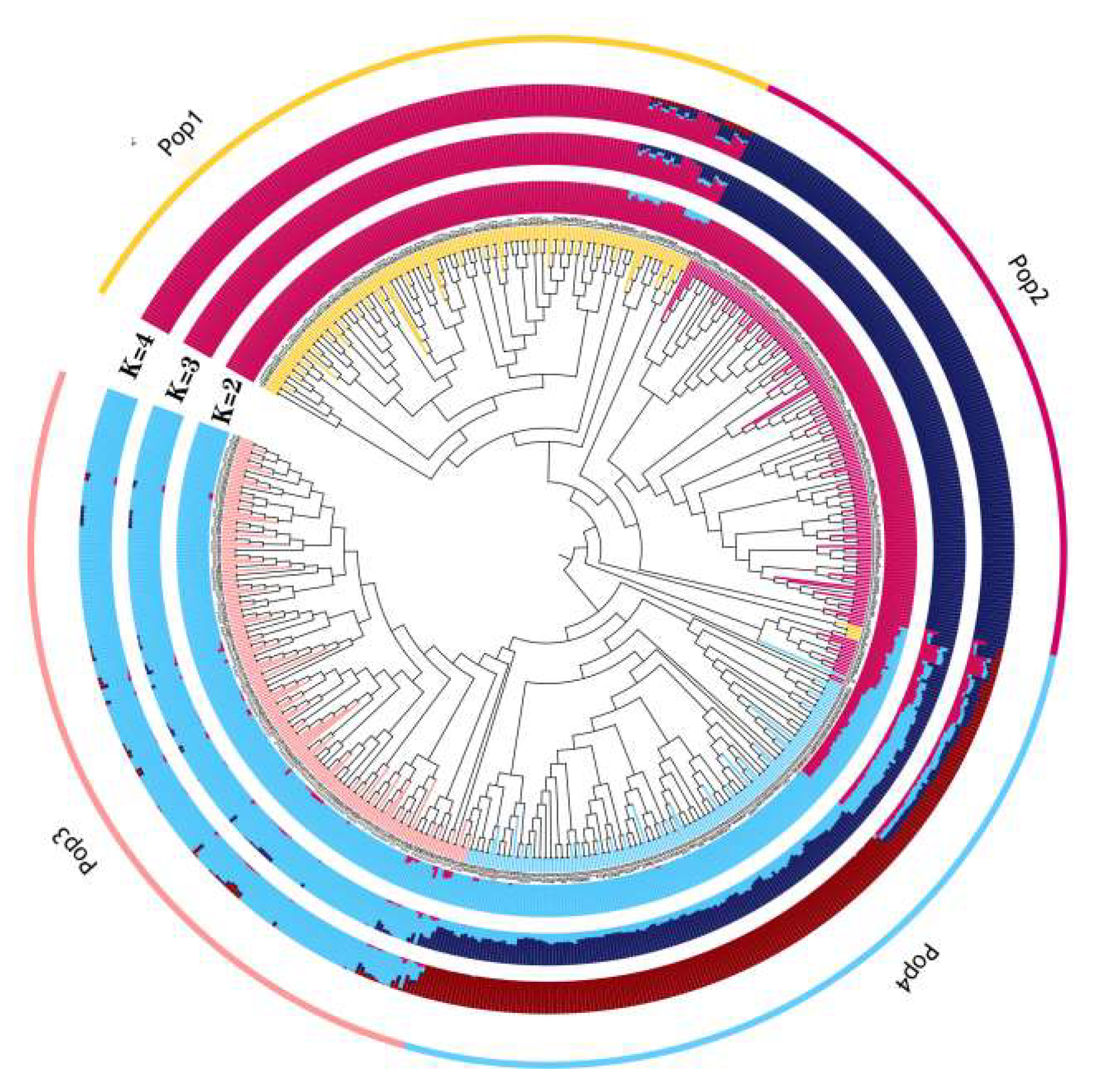
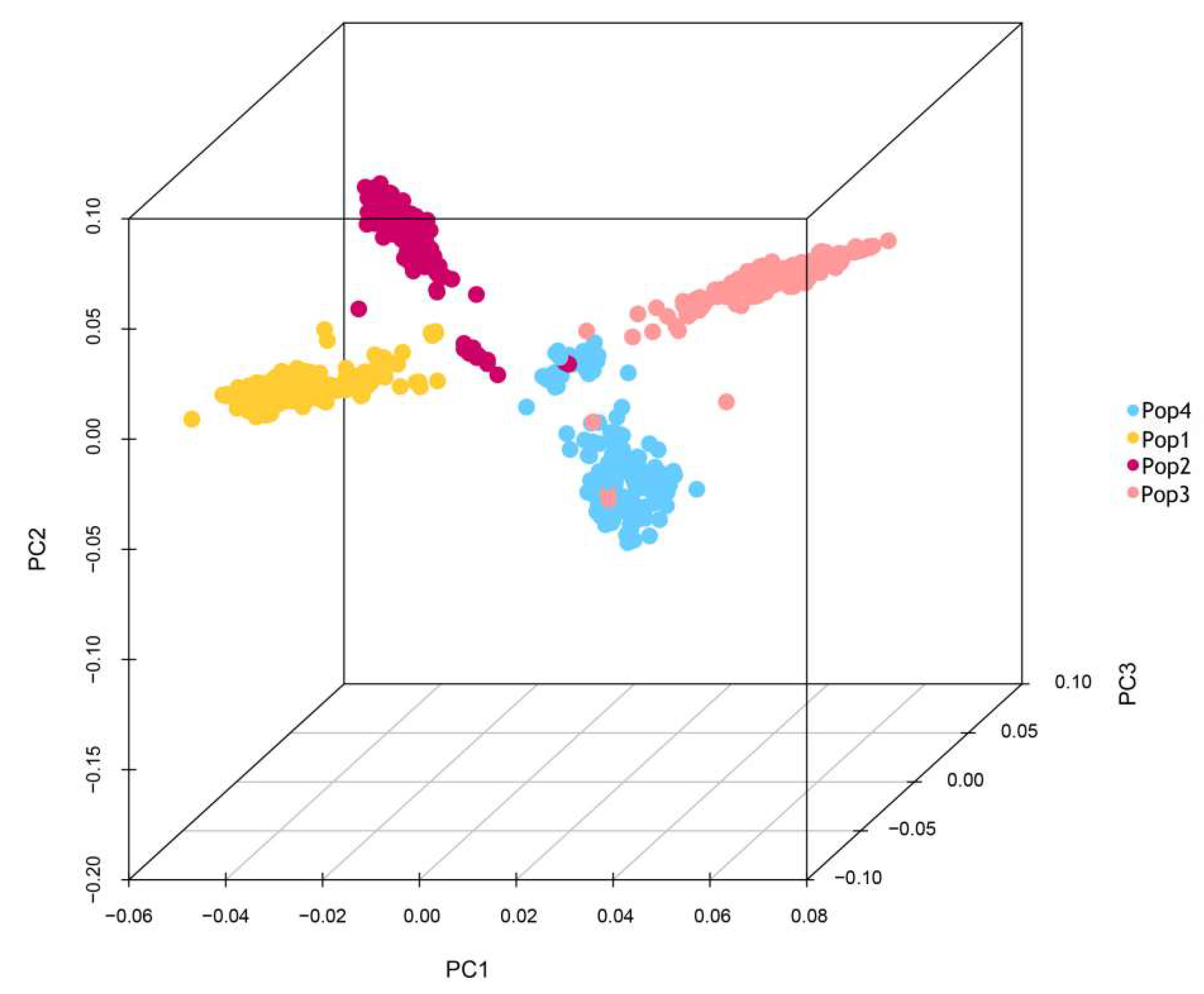
2.2. Population Structure Analysis
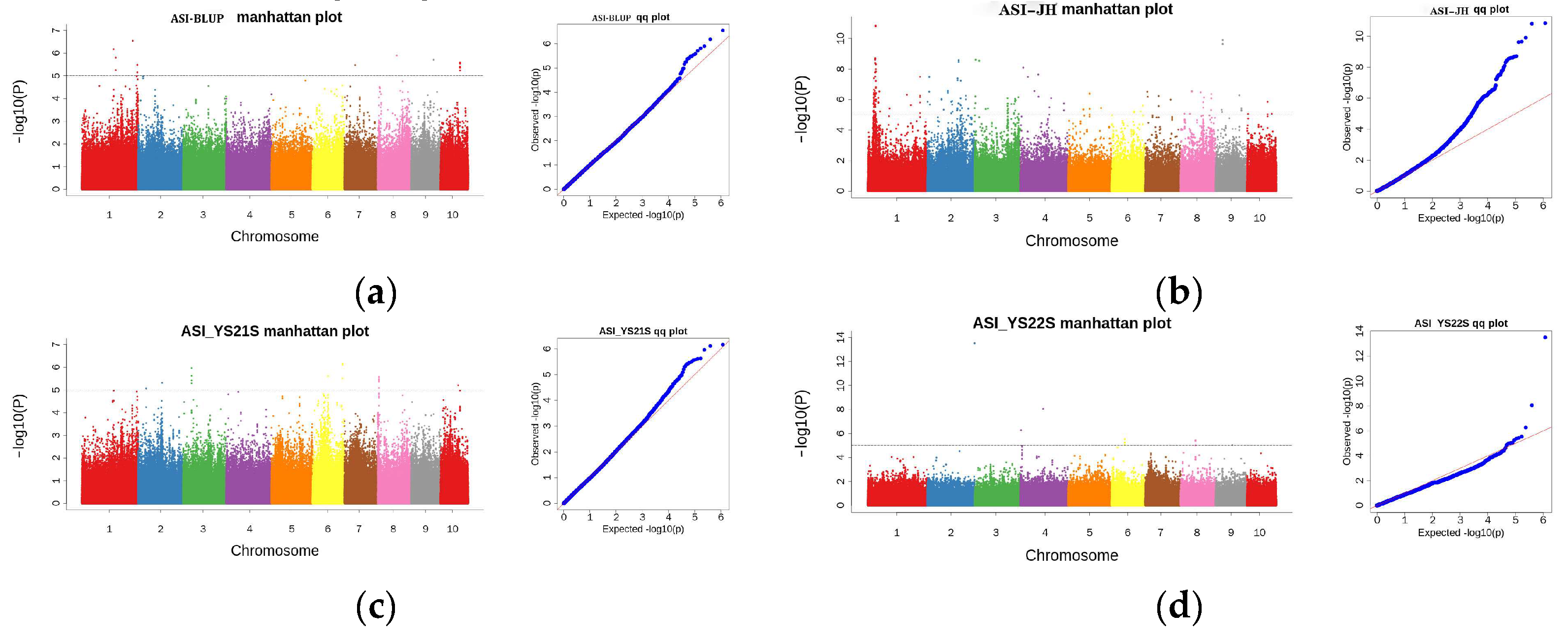
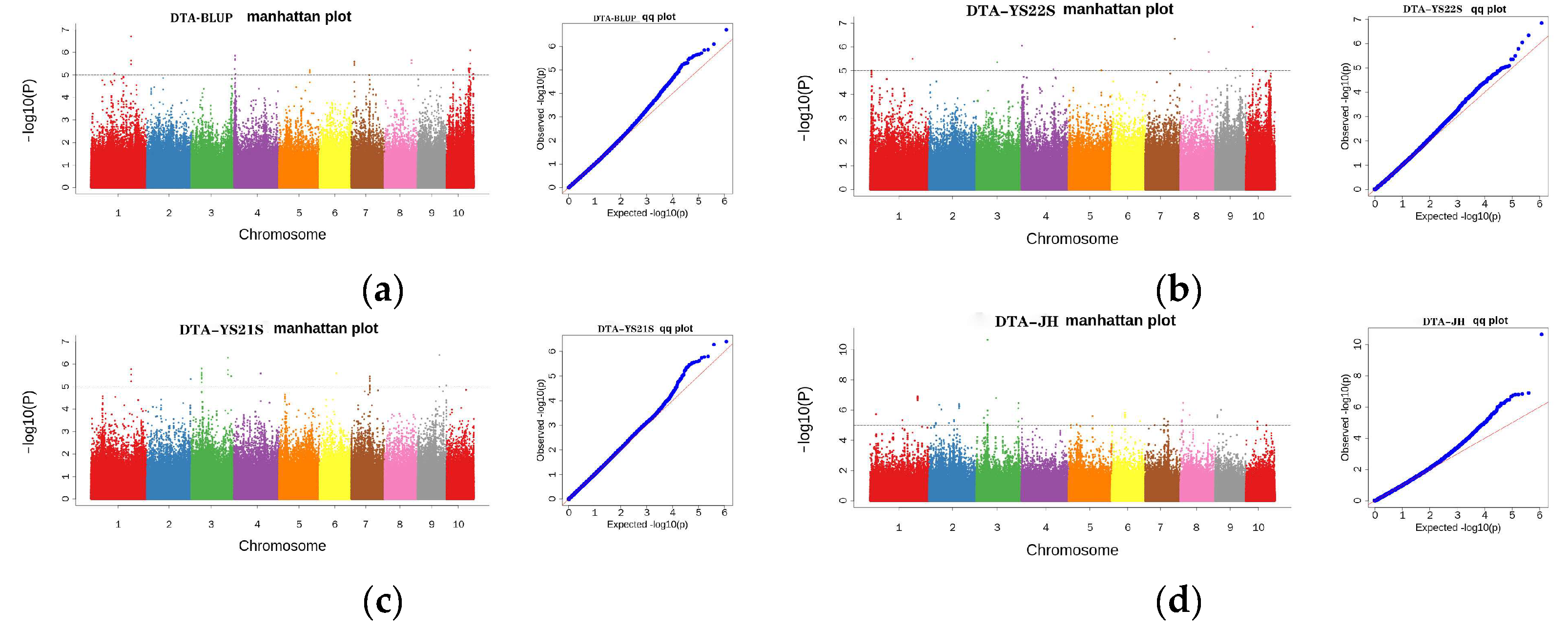
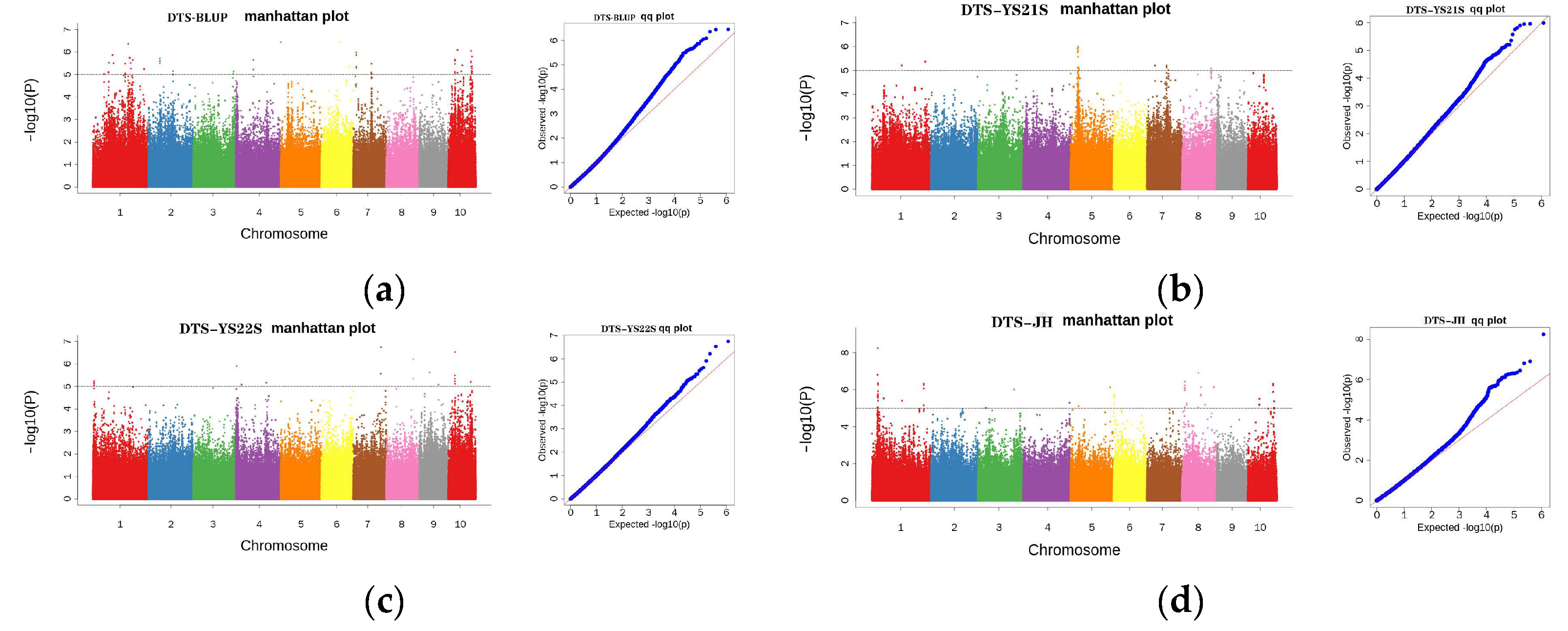
2.3. GWAS Analysis
2.3.1. GWAS Analysis for ASI
2.3.2. GWAS Analysis for DTA
2.3.3. GWAS Analysis for DTS
2.4. Identification and Functional Annotation of Candidate Genes
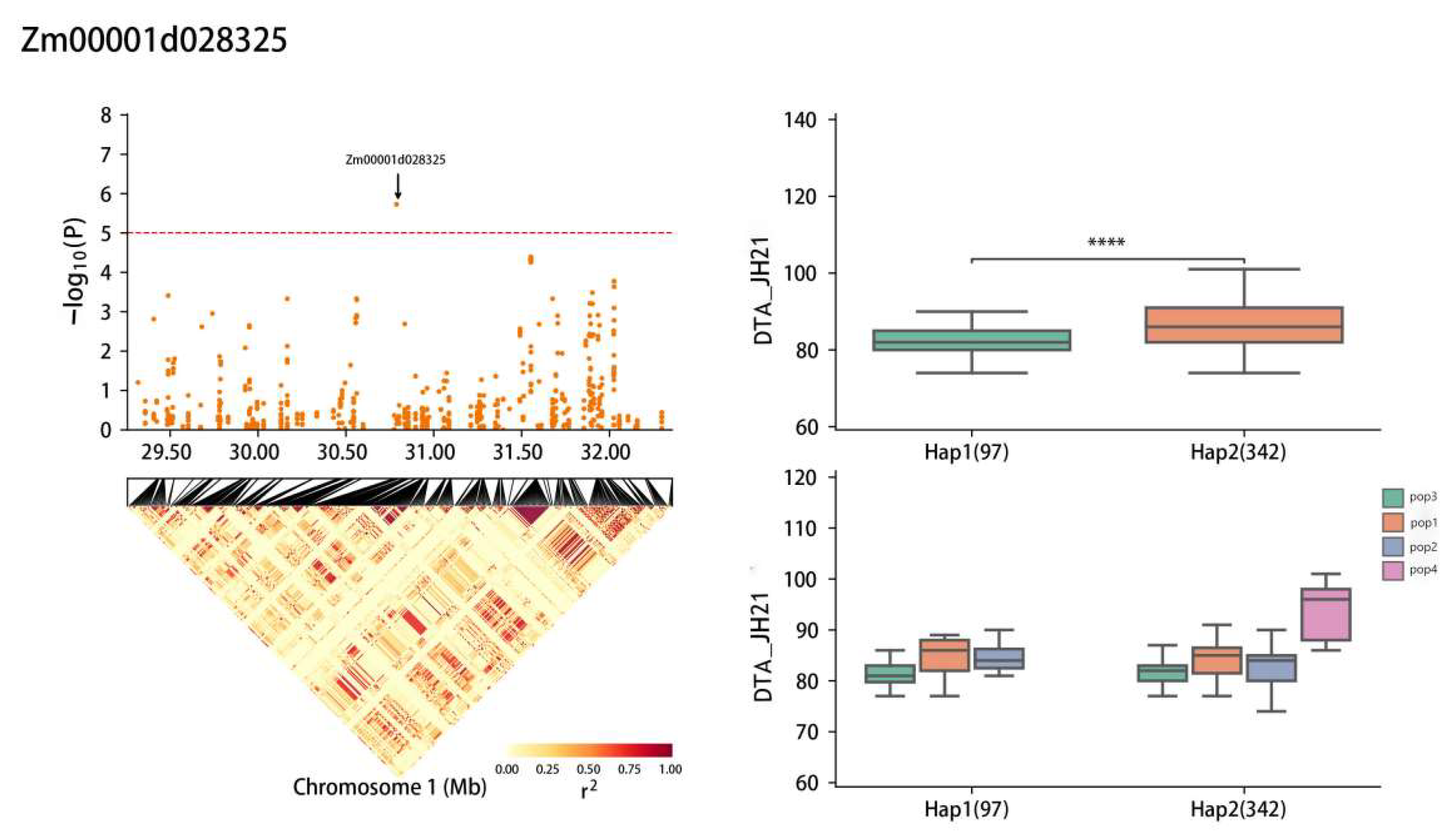
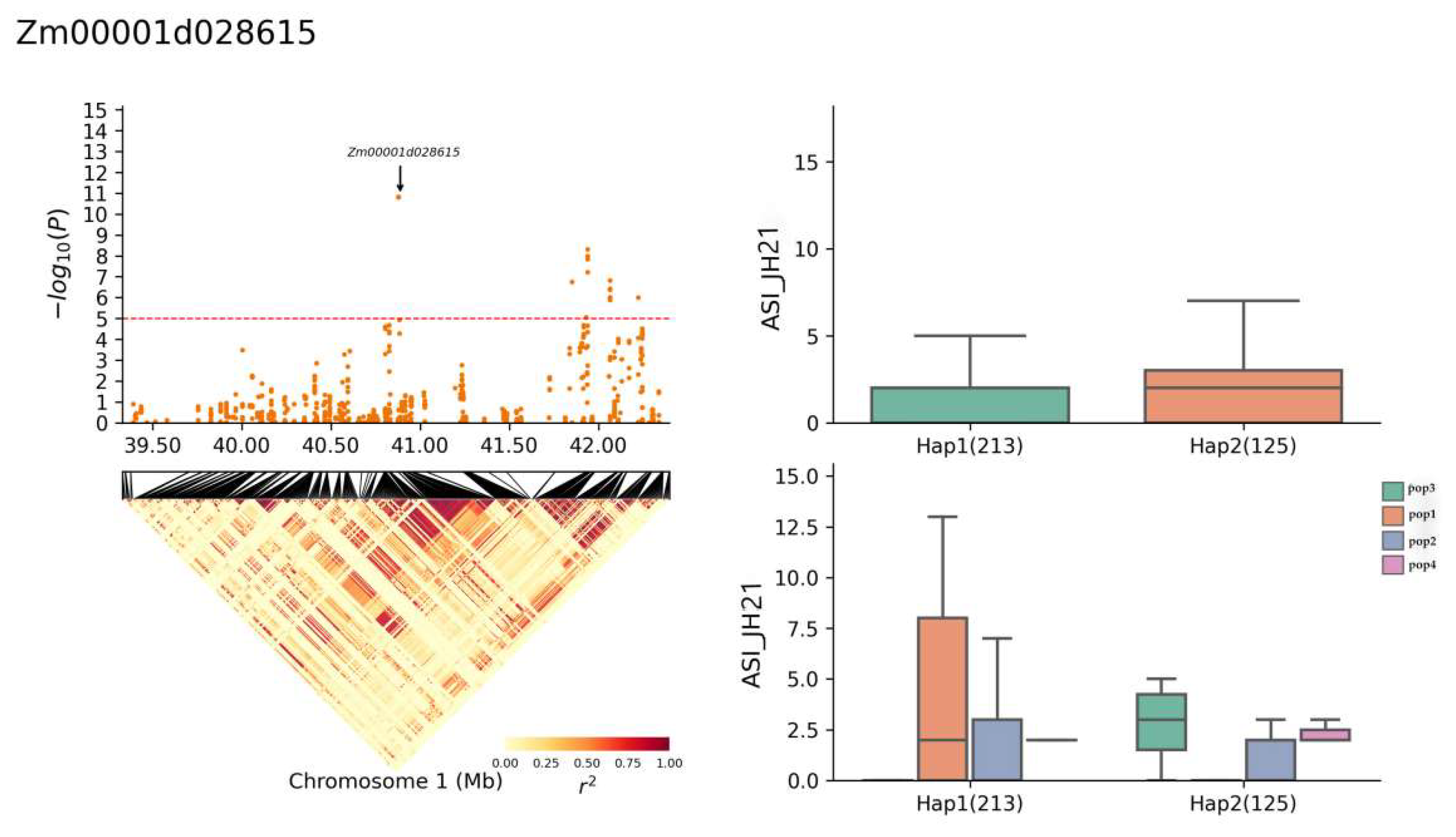
2.5. Haplotype Analysis
2.6. Analysis of Estimated Breeding Value (EBV)
3. Discussion
4. Materials and Methods
4.1. Experimental Materials and Field Trials
4.2. Phenotyping of Flowering Time-Related Traits
4.3. Statistical Analyses of the Phenotypic Data
4.4. Genotyping-by-Sequencing (GBS)
4.5. Population Structure and Linkage Disequilibrium Analysis
4.6. Genome-Wide Association Analysis
4.7. Candidate Gene Identification
4.8. Haplotype Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- IPCC. Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Chang; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2021. [Google Scholar]
- Luo, Y.-C.; Zhang, Z.; Zhang, L.-L.; Zhang, J.; Tao, F.-L. Weakened Maize Phenological Response to Climate Warming in China over 1981–2018 Due to Cultivar Shifts. Adv. Clim. Change Res. 2022, 13, 710–720. [Google Scholar] [CrossRef]
- Zhu, P.; Burney, J.; Chang, J.; Jin, Z.; Mueller, N.D.; Xin, Q.; Xu, J.; Yu, L.; Makowski, D.; Ciais, P. Warming Reduces Global Agricultural Production by Decreasing Cropping Frequency and Yields. Nat. Clim. Change 2022, 12, 1016–1023. [Google Scholar] [CrossRef]
- Zhang, Q.; Han, L.Y.; Zhang, L.Y.; Wang, J.S. On the Risk Characteristics and Management of Dry Climate Disasters in the Context of Climate Warming. Adv. Earth Sci. 2014, 29, 80–91. [Google Scholar]
- Wang, Y.S.; Zhang, T.R.; Bai, Y.M. The research of the assessment method of drought for the impact on agriculture in the northwest-the spring corn in Gansu Province. Adv. Meteorol. Sci. Technol. 2013, 3, 55–58. [Google Scholar]
- Ribaut, J.-M.; Hoisington, D.A.; Deutsch, J.A.; Jiang, C.; Gonzalez-de-Leon, D. Identification of Quantitative Trait Loci under Drought Conditions in Tropical Maize. 1. Flowering Parameters and the Anthesis-Silking Interval. Theor. Appl. Genet. 1996, 92, 905–914. [Google Scholar] [CrossRef]
- Jagadish, S.V.K. Heat Stress during Flowering in Cereals—Effects and Adaptation Strategies. New Phytol. 2020, 226, 1567–1572. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Hou, X.; Sheng, D.; Dong, X.; Gao, Y.; Wang, P.; Huang, S. Maximum Lethal Temperature for Flowering and Seed Set in Maize with Contrasting Male and Female Flower Sensitivities. J Agron. Crop Sci. 2021, 207, 679–689. [Google Scholar] [CrossRef]
- Hall, A.J.; Lemcoff, J.H.; Trápani, N. Water Stress before and during Flowering in Maize and Its Effects on Yield, Its Components, and Their Determinants; CABI Digital Library: Delémont, Switzerland, 1971. [Google Scholar]
- Dow, E.W.; Daynard, T.B.; Muldoon, J.F.; Major, D.J.; Thurtell, G.W. Resistance to Drought and Density Stress in Canadian And European Maize (Zea mays L.) Hybrids. Can. J. Plant Sci. 1984, 64, 575–585. [Google Scholar] [CrossRef]
- Buckler, E.S.; Holland, J.B.; Bradbury, P.J.; Acharya, C.B.; Brown, P.J.; Browne, C.; Ersoz, E.; Flint-Garcia, S.; Garcia, A.; Glaubitz, J.C.; et al. The Genetic Architecture of Maize Flowering Time. Science 2009, 325, 714–718. [Google Scholar] [CrossRef]
- Coles, N.D.; McMullen, M.D.; Balint-Kurti, P.J.; Pratt, R.C.; Holland, J.B. Genetic Control of Photoperiod Sensitivity in Maize Revealed by Joint Multiple Population Analysis. Genetics 2010, 184, 799–812. [Google Scholar] [CrossRef]
- Steinhoff, J.; Liu, W.; Reif, J.C.; Porta, G.D.; Ranc, N.; Würschum, T. Detection of QTL for Flowering Time in Multiple Families of Elite Maize. Theor. Appl. Genet. 2012, 125, 1539–1551. [Google Scholar] [CrossRef]
- Shi, C.N.; Zhang, Z.H.; Tang, G.L.; Xue, Y.D. QTL analysis of maize flowering time related traits based on RIL population and IF_2 population. J. Henan Agric. Univ. 2016, 50, 147–156. [Google Scholar] [CrossRef]
- Lu, J.; Jiang, Q.; Liu, T.; Zhou, Y.; Liu, M.; Qu, J.; Ma, Y.; Guan, S. Meta-Analysis of Flowering-Related Traits and Mining of Candidate Genes in Maize. Phyton 2022, 91, 1199–1209. [Google Scholar] [CrossRef]
- Colasanti, J.; Yuan, Z.; Sundaresan, V. The Indeterminate Gene Encodes a Zinc Finger Protein and Regulates a Leaf-Generated Signal Required for the Transition to Flowering in Maize. Cell 1998, 93, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Muszynski, M.G.; Dam, T.; Li, B.; Shirbroun, D.M.; Hou, Z.; Bruggemann, E.; Archibald, R.; Ananiev, E.V.; Danilevskaya, O.N. Delayed Flowering1 Encodes a Basic Leucine Zipper Protein That Mediates Floral Inductive Signals at the Shoot Apex in Maize. Plant Physiol. 2006, 142, 1523–1536. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, C.; Chen, X.; Liu, H.; Huang, Y.; Li, S.; Dong, Z.; Zhao, X.; Tian, F.; Jin, W. Dlf1 Promotes Floral Transition by Directly Activating ZmMADS4 and ZmMADS67 in the Maize Shoot Apex. New Phytol. 2020, 228, 1386–1400. [Google Scholar] [CrossRef] [PubMed]
- Salvi, S.; Sponza, G.; Morgante, M.; Tomes, D.; Niu, X.; Fengler, K.A.; Meeley, R.; Ananiev, E.V.; Svitashev, S.; Bruggemann, E.; et al. Conserved Noncoding Genomic Sequences Associated with a Flowering-Time Quantitative Trait Locus in Maize. Proc. Natl. Acad. Sci. USA 2007, 104, 11376–11381. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Liang, J.; Abou-Elwafa, S.F.; Cheng, H.; Dou, D.; Ren, Z.; Xie, J.; Chen, Z.; Gao, F.; Ku, L.; et al. ZmCCT Regulates Photoperiod-Dependent Flowering and Response to Stresses in Maize. BMC Plant Biol. 2021, 21, 453. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.A.; Muslin, E.H.; Dorweiler, J.E. A Maize Constans-like Gene, Conz1, Exhibits Distinct Diurnal Expression Patterns in Varied Photoperiods. Planta 2008, 227, 1377–1388. [Google Scholar] [CrossRef]
- Bomblies, K.; Doebley, J.F. Pleiotropic Effects of the Duplicate Maize Floricaula/Leafy Genes Zfl1 and Zfl2 on Traits Under Selection During Maize Domestication. Genetics 2006, 172, 519–531. [Google Scholar] [CrossRef]
- Meng, X.; Muszynski, M.G.; Danilevskaya, O.N. The FT -Like ZCN8 Gene Functions as a Floral Activator and Is Involved in Photoperiod Sensitivity in Maize. Plant Cell 2011, 23, 942–960. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Wang, X.; Zhao, M.; Huang, C.; Li, C.; Li, D.; Yang, C.J.; York, A.M.; Xue, W.; Xu, G.; et al. Stepwise Cis-Regulatory Changes in ZCN8 Contribute to Maize Flowering-Time Adaptation. Curr. Biol. 2018, 28, 3005–3015.e4. [Google Scholar] [CrossRef]
- Su, H.; Cao, L.; Ren, Z.; Sun, W.; Zhu, B.; Ma, S.; Sun, C.; Zhang, D.; Liu, Z.; Zeng, H.; et al. ZmELF6-ZmPRR37 Module Regulates Maize Flowering and Salt Response. Plant Biotechnol. J. 2023, 22, 929–945. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gao, F.; Liu, Y.; Abou-Elwafa, S.F.; Qi, J.; Pan, H.; Hu, X.; Ren, Z.; Zeng, H.; Liu, Z.; et al. ZmGI2 Regulates Flowering Time through Multiple Flower Development Pathways in Maize. Plant Sci. 2023, 332, 111701. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Chen, Z.; Dong, Y.; Ku, L.; Abou-Elwafa, S.F.; Ren, Z.; Cao, Y.; Dou, D.; Liu, Z.; Liu, H.; et al. Identification of ZmNF-YC2 and Its Regulatory Network for Maize Flowering Time. J. Exp. Bot. 2021, 72, 7792–7807. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Sun, H.; Xu, D.; Chen, Q.; Liang, Y.; Wang, X.; Xu, G.; Tian, J.; Wang, C.; Li, D.; et al. ZmCCT9 Enhances Maize Adaptation to Higher Latitudes. Proc. Natl. Acad. Sci. USA 2018, 115, E334–E341. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, Z.; Jiang, H.; Luo, J.; Guo, T.; Tian, F.; Rossi, V.; He, Y. ZmCCT10 -relayed Photoperiod Sensitivity Regulates Natural Variation in the Arithmetical Formation of Male Germinal Cells in Maize. New Phytol. 2023, 237, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, Y.; Song, G.; Yang, D.; Xia, Z.; Sun, C.; Zhao, Y.; Hou, M.; Zhang, M.; Qi, Z.; et al. Gene Expression and Expression Quantitative Trait Loci Analyses Uncover Natural Variations Underlying the Improvement of Important Agronomic Traits during Modern Maize Breeding. Plant J. 2023, 115, 772–787. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.; Ruan, Y.N.; Li, Q.J.; Han, Y. Single nucleotide polymorphism and its application in crop genetic breeding. J. Liaoning Univ. Nat. Sci. Ed. 2009, 36, 280–283. [Google Scholar]
- Lu, X.C. Current status and prospect of molecular markers application on classification of chilli germplasm resources in China. Mod. Horticultur. 2013, 15–16. [Google Scholar] [CrossRef]
- Li, Z.; Liu, X.; Xu, X.; Liu, J.; Sang, Z.; Yu, K.; Yang, Y.; Dai, W.; Jin, X.; Xu, Y. Favorable Haplotypes and Associated Genes for Flowering Time and Photoperiod Sensitivity Identified by Comparative Selective Signature Analysis and GWAS in Temperate and Tropical Maize. Crop J. 2020, 8, 227–242. [Google Scholar] [CrossRef]
- Li, Y.; Li, C.; Bradbury, P.J.; Liu, X.; Lu, F.; Romay, C.M.; Glaubitz, J.C.; Wu, X.; Peng, B.; Shi, Y.; et al. Identification of Genetic Variants Associated with Maize Flowering Time Using an Extremely Large Multi-genetic Background Population. Plant J. 2016, 86, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.G.; Zhang, X.; Beyene, Y.; Semagn, K.; Olsen, M.; Prasanna, B.M.; Buckler, E.S. Genome-wide Association for Plant Height and Flowering Time across 15 Tropical Maize Populations under Managed Drought Stress and Well-Watered Conditions in Sub-Saharan Africa. Crop Sci. 2016, 56, 2365–2378. [Google Scholar] [CrossRef]
- Bezrutczyk, M.; Hartwig, T.; Horschman, M.; Char, S.N.; Yang, J.; Yang, B.; Frommer, W.B.; Sosso, D. Impaired Phloem Loading in Zmsweet13a,b,c Sucrose Transporter Triple Knock-out Mutants in Zea mays. New Phytol. 2018, 218, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Austin, D.F.; Lee, M. Comparative Mapping in F2∶3 and F6∶7 Generations of Quantitative Trait Loci for Grain Yield and Yield Components in Maize. Theor. Appl. Genet. 1996, 92, 817–826. [Google Scholar] [CrossRef]
- Li, J.Z.; Zhang, Z.W.; Li, Y.L.; Wang, Q.L.; Zhou, Y.G. QTL Consistency and Meta-Analysis for Grain Yield Components in Three Generations in Maize. Theor. Appl. Genet. 2011, 122, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-Wide Complex Trait Analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Risch, N.; Merikangas, K. The Future of Genetic Studies of Complex Human Diseases. Science 1996, 273, 1516–1517. [Google Scholar] [CrossRef] [PubMed]
- Habier, D.; Fernando, R.L.; Dekkers, J.C.M. Genomic Selection Using Low-Density Marker Panels. Genetics 2009, 182, 343–353. [Google Scholar] [CrossRef]
- Alexandrov, N.N.; Brover, V.V.; Freidin, S.; Troukhan, M.E.; Tatarinova, T.V.; Zhang, H.; Swaller, T.J.; Lu, Y.-P.; Bouck, J.; Flavell, R.B.; et al. Insights into Corn Genes Derived from Large-Scale cDNA Sequencing. Plant Mol. Biol. 2009, 69, 179–194. [Google Scholar] [CrossRef]
- Xing, R.X.; Zhu, J.J.; Qi, X.T.; Xie, C.X.; Jiang, H.Y.; Liu, C.L. Progress on the regulation mechanism of maize flowering. Anhui Agric. Sci. 2022, 50, 23–26+29. [Google Scholar]
- Wang, Y.; Zhang, W.-Z.; Song, L.-F.; Zou, J.-J.; Su, Z.; Wu, W.-H. Transcriptome Analyses Show Changes in Gene Expression to Accompany Pollen Germination and Tube Growth in Arabidopsis. Plant Physiol. 2008, 148, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Nziengui, H.; Bouhidel, K.; Pillon, D.; Der, C.; Marty, F.; Schoefs, B. Reticulon-like Proteins in Arabidopsis thaliana: Structural Organization and ER Localization. FEBS Lett. 2007, 581, 3356–3362. [Google Scholar] [CrossRef] [PubMed]
- Essis, D.; Dijkuis, F.J. The Influence of the Time Lag between Pollen Shedding and Silking on the Yield of Maize; CABI Digital Library: Delémont, Switzerland, 1967. [Google Scholar]
- Cai, W.; Ma, J.; Chi, W.; Zou, M.; Guo, J.; Lu, C.; Zhang, L. Cooperation of LPA3 and LPA2 Is Essential for Photosystem II Assembly in Arabidopsis. Plant Physiol. 2010, 154, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Altmann, M.; Altmann, S.; Rodriguez, P.A.; Weller, B.; Elorduy Vergara, L.; Palme, J.; Marín-de La Rosa, N.; Sauer, M.; Wenig, M.; Villaécija-Aguilar, J.A.; et al. Extensive Signal Integration by the Phytohormone Protein Network. Nature 2020, 583, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Pandey, N.; Ranjan, A.; Pant, P.; Tripathi, R.K.; Ateek, F.; Pandey, H.P.; Patre, U.V.; Sawant, S.V. CAMTA 1 Regulates Drought Responses in Arabidopsis thaliana. BMC Genom. 2013, 14, 216. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Wang, R.L.; Li, C.; Zhang, C.; Wang, Z.J.; Kang, H.B.; Yang, C.Y. Correlation of flowering interval between male and female ears with yield and drought tolerance in maize self-incompatible lines. Crop Res. 2020, 34, 427–430+434. [Google Scholar] [CrossRef]
- Li, D.; Wang, L.; Wang, M.; Xu, Y.; Luo, W.; Liu, Y.; Xu, Z.; Li, J.; Chong, K. Engineering OsBAK1 Gene as a Molecular Tool to Improve Rice Architecture for High Yield. Plant Biotechnol. J. 2009, 7, 791–806. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zuo, S.; Schwessinger, B.; Chern, M.; Canlas, P.E.; Ruan, D.; Zhou, X.; Wang, J.; Daudi, A.; Petzold, C.J.; et al. An XA21-Associated Kinase (OsSERK2) Regulates Immunity Mediated by the XA21 and XA3 Immune Receptors. Mol. Plant 2014, 7, 874–892. [Google Scholar] [CrossRef]
- Lidder, P.; Gutiérrez, R.A.; Salomé, P.A.; McClung, C.R.; Green, P.J. Circadian Control of Messenger RNA Stability. Association with a Sequence-Specific Messenger RNA Decay Pathway. Plant Physiol. 2005, 138, 2374–2385. [Google Scholar] [CrossRef]
- Zhang, F.L.; Bolanos, J. Progress on the study of anthesis-silking interval in maize. J. Hebei Agric. Univ. 2002, 25 (Suppl. S1), 24–25. [Google Scholar]
- Liu, C.; Shen, H.B.; Shi, Y.S.; Song, C.Y.; Wang, T.Y.; Li, Y. Relationship between anthesis interval time (ASI) and yield and drought tolerance of maize male and female ears after water stress. Xinjiang Agric. Sci. 2008, 609–612. [Google Scholar]
- Li, X.H.; Gao, G.L. Differences in drought tolerance at flowering and improvement of major maize inbred lines in China. J. Crops 2002, 595–600. [Google Scholar]
- Lei, Z.G.; Li, Z.F.; Wang, Y.J.; Liang, X.Y.; Han, D.X.; Yang, J.; Li, M.D. Study on the relationship between female and male flowering interval and yield of maize self-fertilised lines at different densities. Xinjiang Agric. Sci. 2016, 53, 1595–1601. [Google Scholar]
- Zhang, F.L.; Chen, J.T. A study on the potential causes of the flowering interval between male and female ears of maize affecting the number of grains in the ears. Maize Sci. 2002, 53–55. [Google Scholar]
- Zhang, F.L.; Kirubi, D. Studies on the relationship between flowering interval and yield in male and female ears of maize. J. Crops 2002, 76–78. [Google Scholar]
- Mock, J.J.; Pearce, R.B. An ideotype of maize. Euphytica 1975, 24, 613–623. [Google Scholar] [CrossRef]
- Messmer, R.; Fracheboud, Y.; Bänziger, M.; Vargas, M.; Stamp, P.; Ribaut, J.-M. Drought Stress and Tropical Maize: QTL-by-Environment Interactions and Stability of QTLs across Environments for Yield Components and Secondary Traits. Theor. Appl. Genet. 2009, 119, 913–930. [Google Scholar] [CrossRef]
- Guo, X.Y.; Hu, X.; Zhu, Y.F.; Wang, A.G.; Liu, P.F.; Chen, Z.H. Genetic analysis of tropical maize Suwan1 population imported with different types of temperate germplasm. Maize Sci. 2019, 27, 9–13+21. [Google Scholar] [CrossRef]
- Sun, C.T.; Chen, H.M.; Tan, J.; Li, X.Z.; Huang, B.H.; Fan, X.M. Cooperation of 25 good temperate maize inbred lines with 4 tropical maize inbred lines for agronomic traits. Maize Sci. 2007, 36–41. [Google Scholar]
- Wu, J.C.; Xu, C.X.; Cheng, H.M.; Tan, J.; Han, X.R.; Huang, B.H.; Fan, X.M. Studies on fitness and heterosis groups of 24 high-quality protein maize inbred lines with representative inbred lines of the four major dominance groups of temperate maize in China. Chin. Agric. Sci. 2007, 1288–1296. [Google Scholar]
- Fan, X.-M.; Bi, Y.-Q.; Zhang, Y.D.; Jeffers, D.P.; Yao, W.; Chen, H.M.; Zhao, L.; Kang, M.S. Use of the Suwan1 Heterotic Group in Maize Breeding Programs in Southwestern China. Agron. J. 2015, 107, 2353–2362. [Google Scholar] [CrossRef]
- Stewart, C.N.; Via, L.E. A Rapid CTAB DNA Isolation Technique Useful for RAPD Fingerprinting and Other PCR Applications. Biotechniques 1993, 14, 748–750. [Google Scholar] [PubMed]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.-L. Development of High-Density Genetic Maps for Barley and Wheat Using a Novel Two-Enzyme Genotyping-by-Sequencing Approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Guan, H.; Jing, X.; Li, Y.; Wang, B.; Li, Y.; Liu, X.; Zhang, D.; Liu, C.; Xie, X.; et al. Genomic Insights into Historical Improvement of Heterotic Groups during Modern Hybrid Maize Breeding. Nat. Plants 2022, 8, 750–763. [Google Scholar] [CrossRef] [PubMed]
- Pei, S.; Liu, T.; Ren, X.; Li, W.; Chen, C.; Xie, Z. Benchmarking Variant Callers in Next-Generation and Third-Generation Sequencing Analysis. Brief. Bioinform. 2021, 22, bbaa148. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast Model-Based Estimation of Ancestry in Unrelated Individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A Fast and Effective Tool for Linkage Disequilibrium Decay Analysis Based on Variant Call Format Files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Environment/Traits | Max | Mix | Average Value | Standard Deviation | Variance | Coefficient of Variation |
|---|---|---|---|---|---|---|
| YS22-DTA | 59 | 97 | 75.03 | 5.774 | 33.337 | 0.076955884 |
| YS22-DTS | 59 | 102 | 76.87 | 5.885 | 34.635 | 0.076557825 |
| YS22-ASI | 0 | 7 | 1.9316 | 1.33785 | 1.79 | 0.692612342 |
| YS21-DTA | 74 | 101 | 85.41 | 5.919 | 35.039 | 0.069301019 |
| YS21-DTS | 77 | 101 | 86.07 | 5.197 | 27.007 | 0.060381085 |
| YS21-ASI | 0 | 15 | 1.83 | 2.554 | 6.524 | 1.395628415 |
| JH21-DTA | 70 | 98 | 82.37 | 4.955 | 24.553 | 0.060155396 |
| JH21-DTS | 70 | 99 | 82.72 | 5.468 | 29.9 | 0.066102515 |
| JH21-ASI | 0 | 8 | 1.986 | 1.28866 | 1.661 | 0.648872105 |
| Traits | Genetic Variance (Vg) | Residual Variance (Ve) | Number of Environments (L) | Heritability H2 = Vg/(Vg + Ve/L) |
|---|---|---|---|---|
| DTA | 5.27 | 25.57 | 3 | 0.382068632 |
| DTS | 6.296 | 25.932 | 3 | 0.421419009 |
| ASI | 0.209 | 4.6203 | 3 | 0.119490023 |
| Gene ID | Chr. | Environments/Traits | Physical Position | Functional Annotation |
|---|---|---|---|---|
| Zm00001d020229 | 7 | YS21-DTA, YS21-DTS, BULP-DTS | 101,183,127–101,183,962 | Reticulon-like protein B1 |
| Zm00001d021470 | 7 | YS22-DTA, YS22-DTS | 153,075,349–153,079,559 | Glutathione S-transferase F10 |
| Zm00001d028295 | 1 | JH21-DTS, JH21-ASI | 29,497,242–29,504,081 | Protein LOW PSII ACCUMULATION 3, chloroplastic |
| Zm00001d028325 | 1 | JH21-DTS, JH21-DTA | 30,793,737–30,799,897 | Cytochrome P450 90B1 |
| Zm00001d028615 | 1 | JH21-ASI | 40,886,065–40,889,048 | Probable protein phosphatase 2C 31 |
| Zm00001d033665 | 1 | JH21-DTS, JH21-ASI | 269,986,885–269,988,386 | 30S ribosomal protein S13, chloroplastic |
| Zm00001d033666 | 1 | JH21-DTS, JH21-ASI | 269,989,007–269,992,773 | Probable plastid–lipid-associated protein 4, chloroplastic |
| Zm00001d039455 | 3 | JH21-ASI | 4,833,807–4,837,335 | PRA1 family protein F3 |
| Zm00001d039456 | 3 | JH21-ASI | 4,838,566–4,841,548 | |
| Zm00001d047209 | 9 | JH21-ASI | 122112583–122121985 | Calmodulin-binding transcription activator 1 |
| Zm00001d051680 | 4 | YS22-DTA, YS22-DTS | 166,593,637–166,595,477 | Ferredoxin–NADP reductase, chloroplastic |
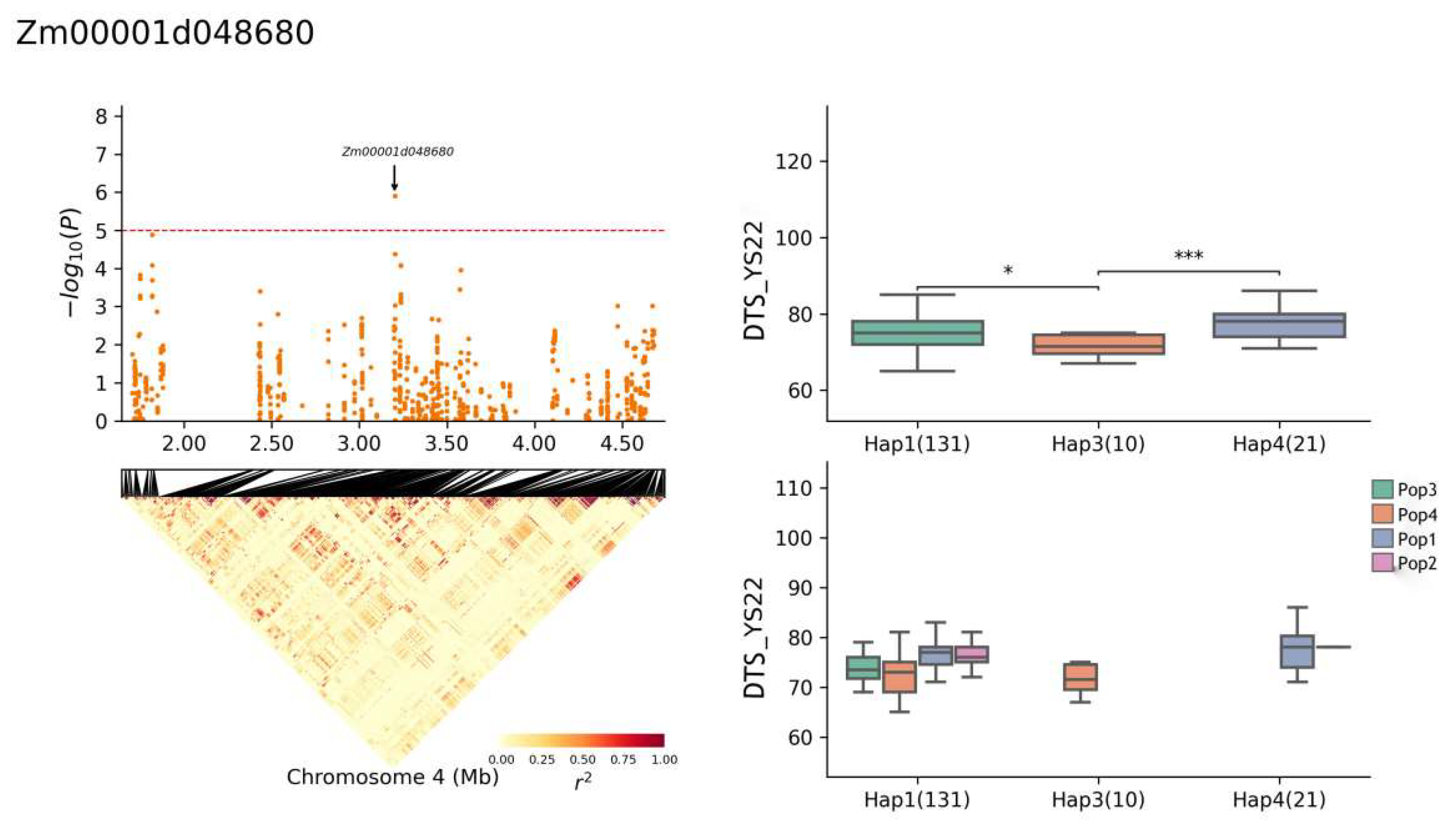
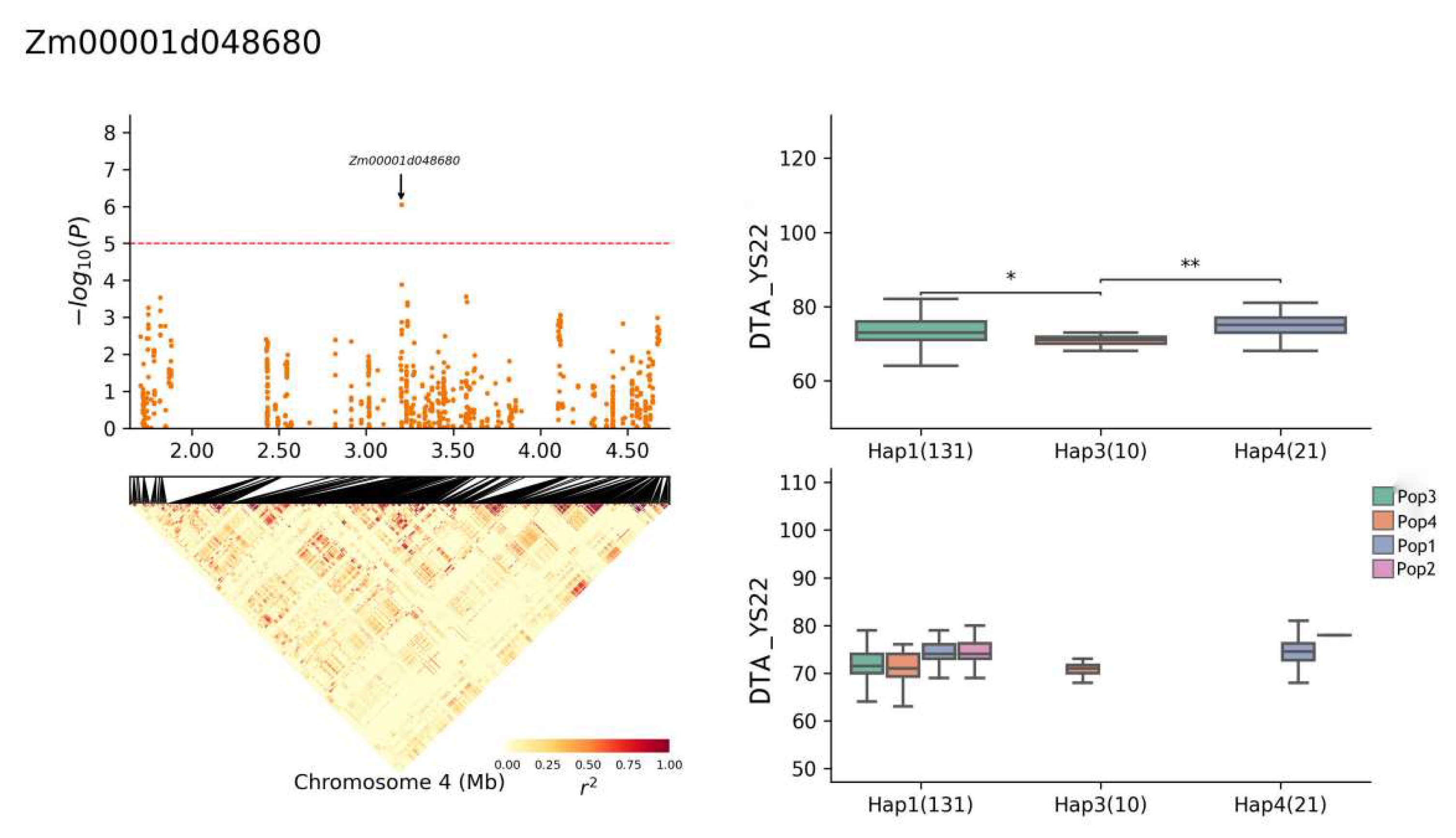
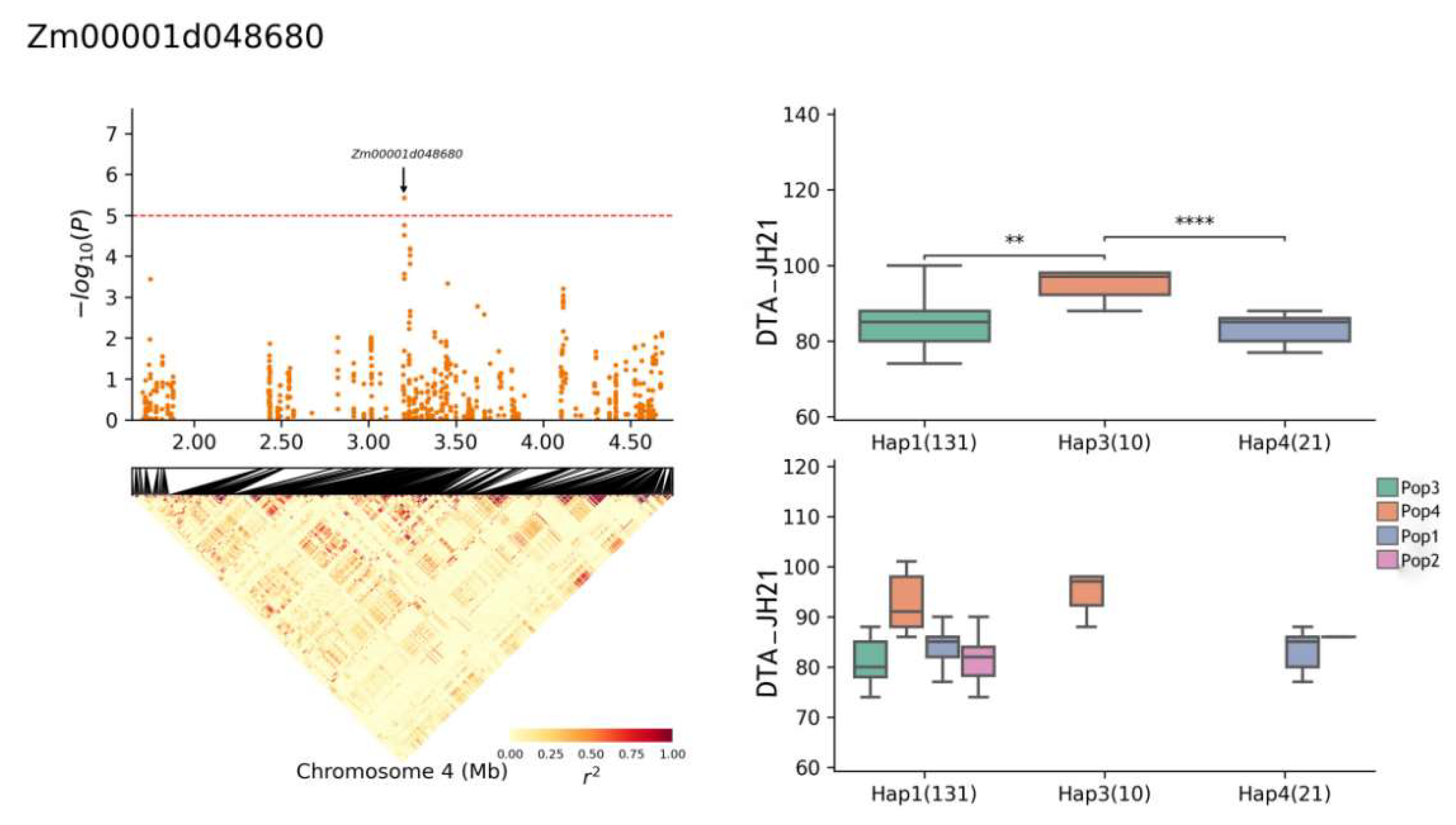
| Zm00001d048680 | 4 | YS22-DTA, YS22-DTS, JH21-DTA, BLUP-DTA | 3,197,914–3,204,309 | Cysteine-rich receptor-like protein kinase 10 |
| Gene ID | Position | Haplotype | Hap_Sample_Num 1 |
|---|---|---|---|
| Zm00001d028325 | Chr1: 29,316,772–32,297,235 | CCACA(Hap1) | 97 |
| TTGTC(Hap2) | 342 | ||
| CCGCA(Hap3) | 3 | ||
| Zm00001d028615 | Chr1: 39,389,512–42,337,172 | TCGCAAGGG(Hap1) | 213 |
| GGTACGATA(Hap2) | 125 | ||
| Zm00001d048680 | Chr4: 1,703,082–4,679,821 | CTAGTATGATCGCTA(Hap1) | 131 |
| TCATTGAGATAACAC(Hap2) | 3 | ||
| CCATTGTGGTCGCAC(Hap3) | 10 | ||
| TCGTTGTAATCACAC(Hap4) | 21 |
| Gene ID | Environments/Traits | Average | GWAS-PVE |
|---|---|---|---|
| Zm00001d028325 | JH21-DTA | 82.5 (Hap1) | 5.90% |
| 87.0 (Hap2) | |||
| Zm00001d028615 | JH21-ASI | 2.1 (Hap1) | 12.43% |
| 1.9 (Hap2) | |||
| Zm00001d048680 | YS22-DTS | 75.8 (Hap1) | 4.67% |
| 71.7 (Hap3) | |||
| 77.5 (Hap4) | |||
| YS22-DTA | 74.1 (Hap1) | 5.06% | |
| 69.7 (Hap3) | |||
| 74.8 (Hap4) | |||
| JH21-DTA | 85.5 (Hap1) | 3.82% | |
| 94.8 (Hap3) | |||
| 83.9 (Hap4) |
| Subpopulation | EBV 1 (DTA 2) | EBV (DTS 2) | EBV (ASI 2) | |||
|---|---|---|---|---|---|---|
| + | − | + | − | + | − | |
| Pop3 | 63 | 126 | 79 | 110 | 67 | 122 |
| Pop4 | 87 | 64 | 92 | 59 | 85 | 66 |
| Pop1 | 87 | 88 | 76 | 99 | 69 | 106 |
| Pop2 | 84 | 79 | 72 | 88 | 65 | 95 |
| Environment | Population | Average Day |
|---|---|---|
| JH21 | pop1 | 2.67 |
| pop2 | 2.17 | |
| pop3 | 2.45 | |
| pop4 | 3.85 | |
| YS21 | pop1 | 1.94 |
| pop2 | 1.77 | |
| pop3 | 1.33 | |
| pop4 | 2.98 | |
| YS22 | pop1 | 2.15 |
| pop2 | 1.93 | |
| pop3 | 2.03 | |
| pop4 | 2.13 |
| Line | Pedigree | Heterotic Group | Ecotype |
|---|---|---|---|
| CML384 | P502-C1-771-2-2-1-3-B | Reid | Tropical |
| CML444 | P43-C9-1-1-1-1-1 | nonReid | Tropical |
| CML171 | Pool25QPM | nonReid | Tropical |
| Shen137 | Derived from US hybrid (6JK111) | nonReid | Temperate |
| Ye107 | Form American | Reid | Temperate |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ran, F.; Wang, Y.; Jiang, F.; Yin, X.; Bi, Y.; Shaw, R.K.; Fan, X. Studies on Candidate Genes Related to Flowering Time in a Multiparent Population of Maize Derived from Tropical and Temperate Germplasm. Plants 2024, 13, 1032. https://doi.org/10.3390/plants13071032
Ran F, Wang Y, Jiang F, Yin X, Bi Y, Shaw RK, Fan X. Studies on Candidate Genes Related to Flowering Time in a Multiparent Population of Maize Derived from Tropical and Temperate Germplasm. Plants. 2024; 13(7):1032. https://doi.org/10.3390/plants13071032
Chicago/Turabian StyleRan, Fengyun, Yizhu Wang, Fuyan Jiang, Xingfu Yin, Yaqi Bi, Ranjan K. Shaw, and Xingming Fan. 2024. "Studies on Candidate Genes Related to Flowering Time in a Multiparent Population of Maize Derived from Tropical and Temperate Germplasm" Plants 13, no. 7: 1032. https://doi.org/10.3390/plants13071032
APA StyleRan, F., Wang, Y., Jiang, F., Yin, X., Bi, Y., Shaw, R. K., & Fan, X. (2024). Studies on Candidate Genes Related to Flowering Time in a Multiparent Population of Maize Derived from Tropical and Temperate Germplasm. Plants, 13(7), 1032. https://doi.org/10.3390/plants13071032