
A Walk on the Wild Side: Genome Editing of Tuber-Bearing Solanum bulbocastanum

 , , , , , and
, , , , , and 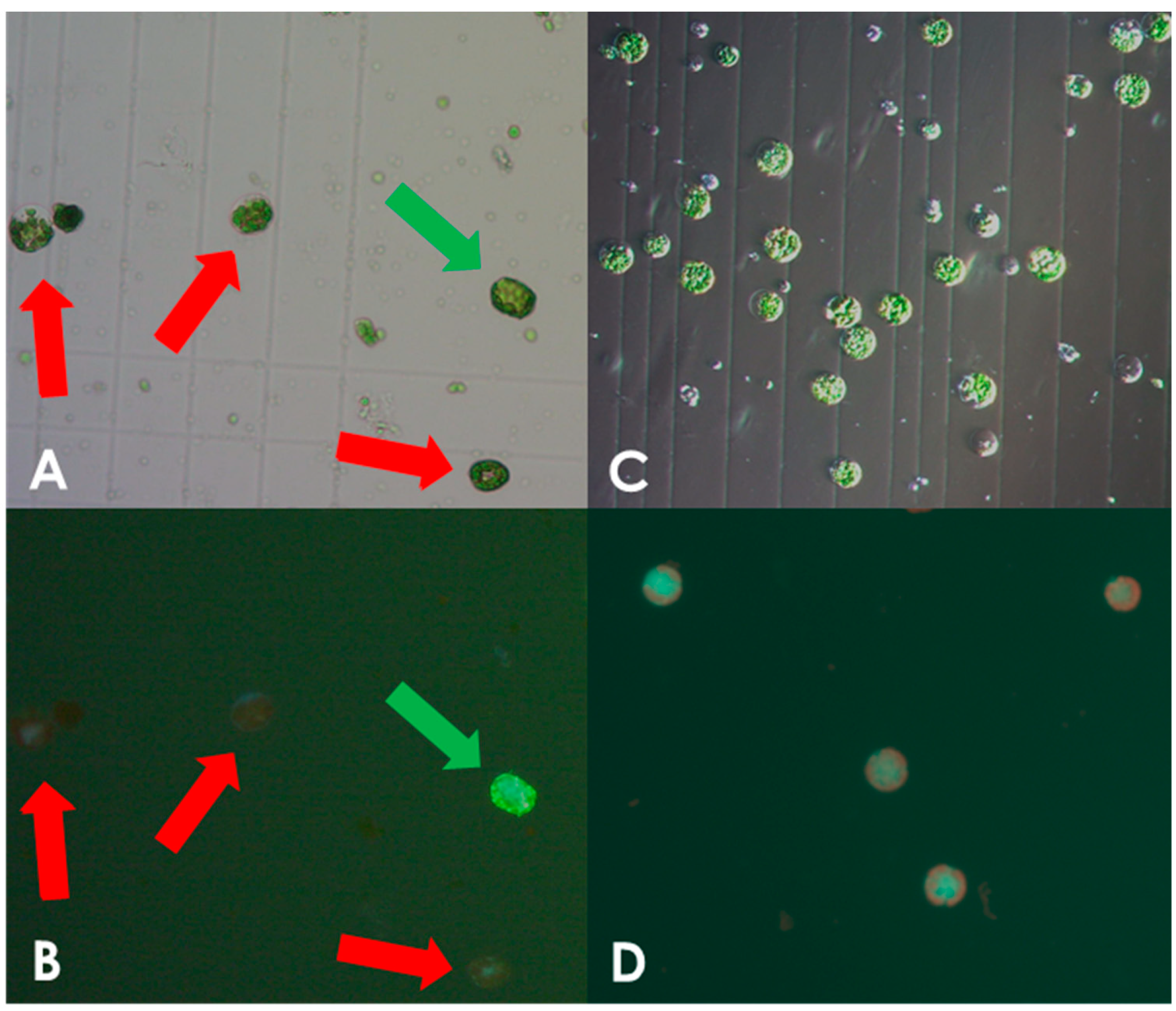{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
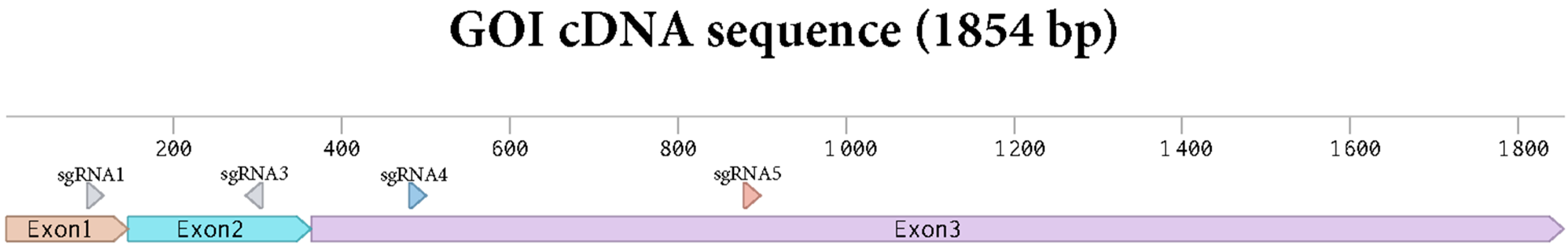
2.2. Design of sgRNAs
2.3. Protoplast Isolation and Test for Viability
2.4. Preparation of Ribonucleoproteins (RNPs) and Genome Editing of Protoplasts
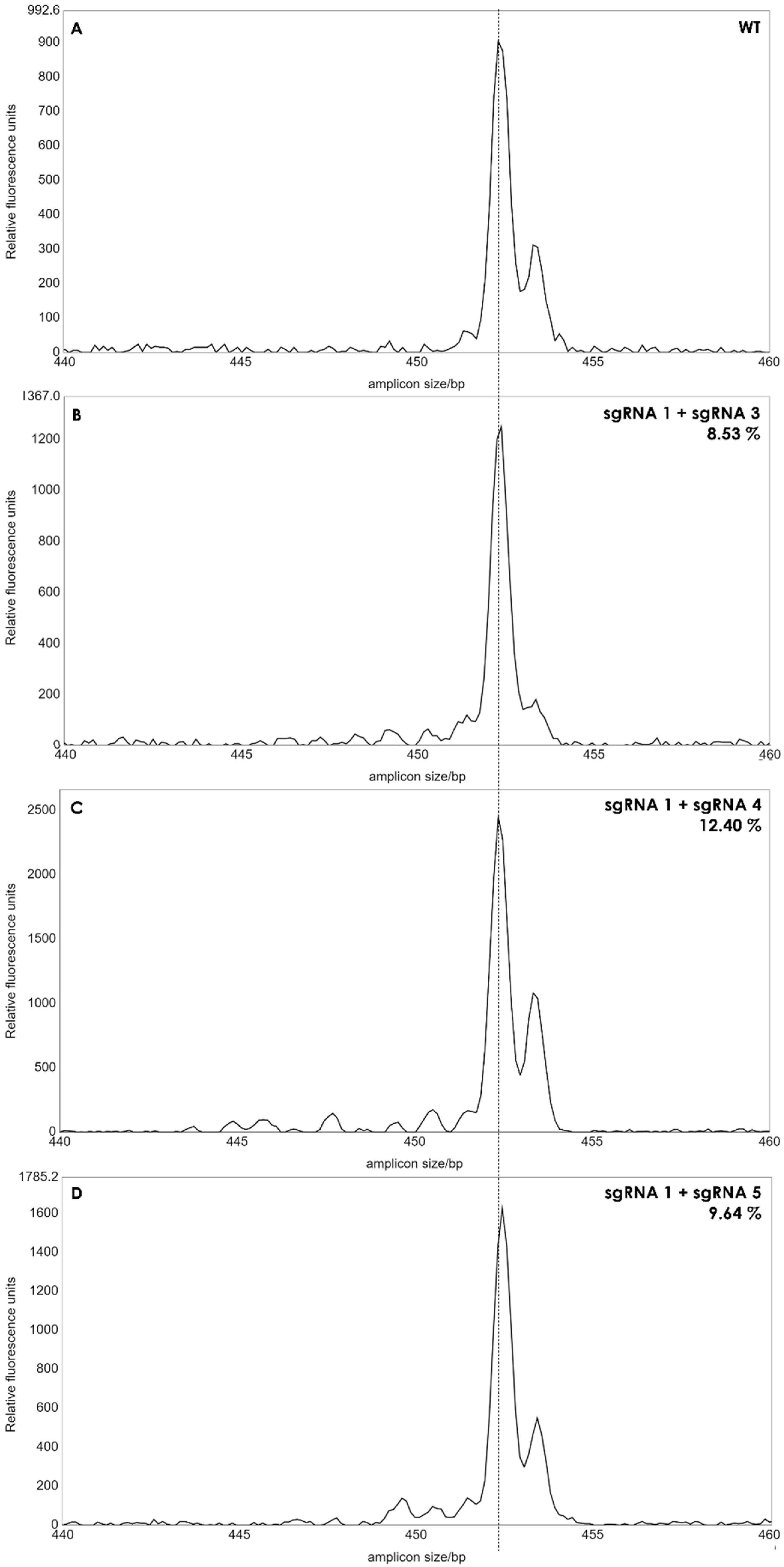
2.5. Indel Detection by Amplicon Analysis (IDAA) for Editing Efficiency in Protoplasts and Regenerated Plants
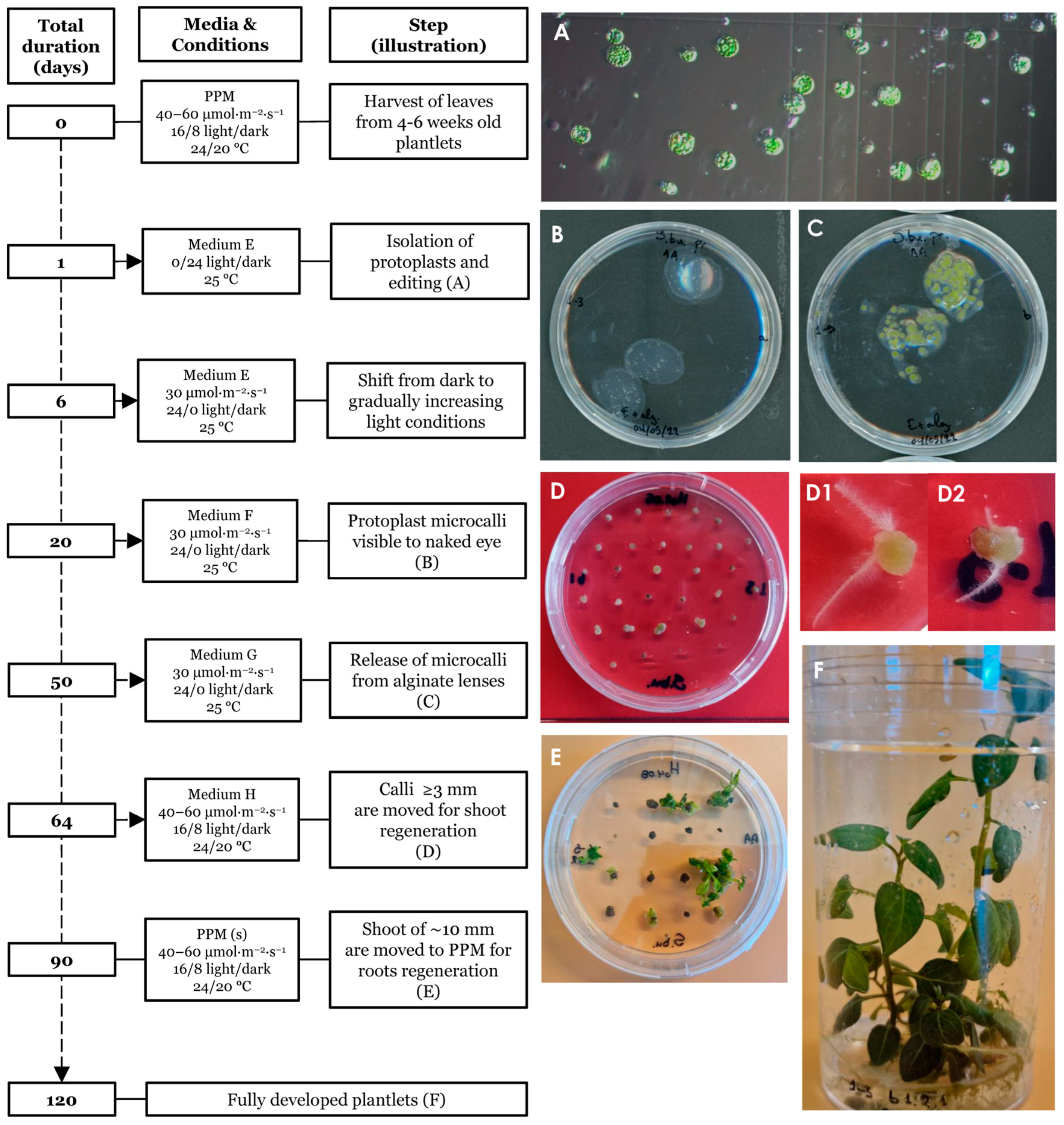
2.6. Regeneration of Plants from Cas9-gRNA RNP Edited Protoplasts
2.7. Genotyping by Sequencing
3. Results
3.1. Optimizing Isolation of Leaf Protoplasts from S. bulbocastanum
3.2. Genome Editing of Protoplasts
3.3. Regeneration of Plants from Edited Protoplasts
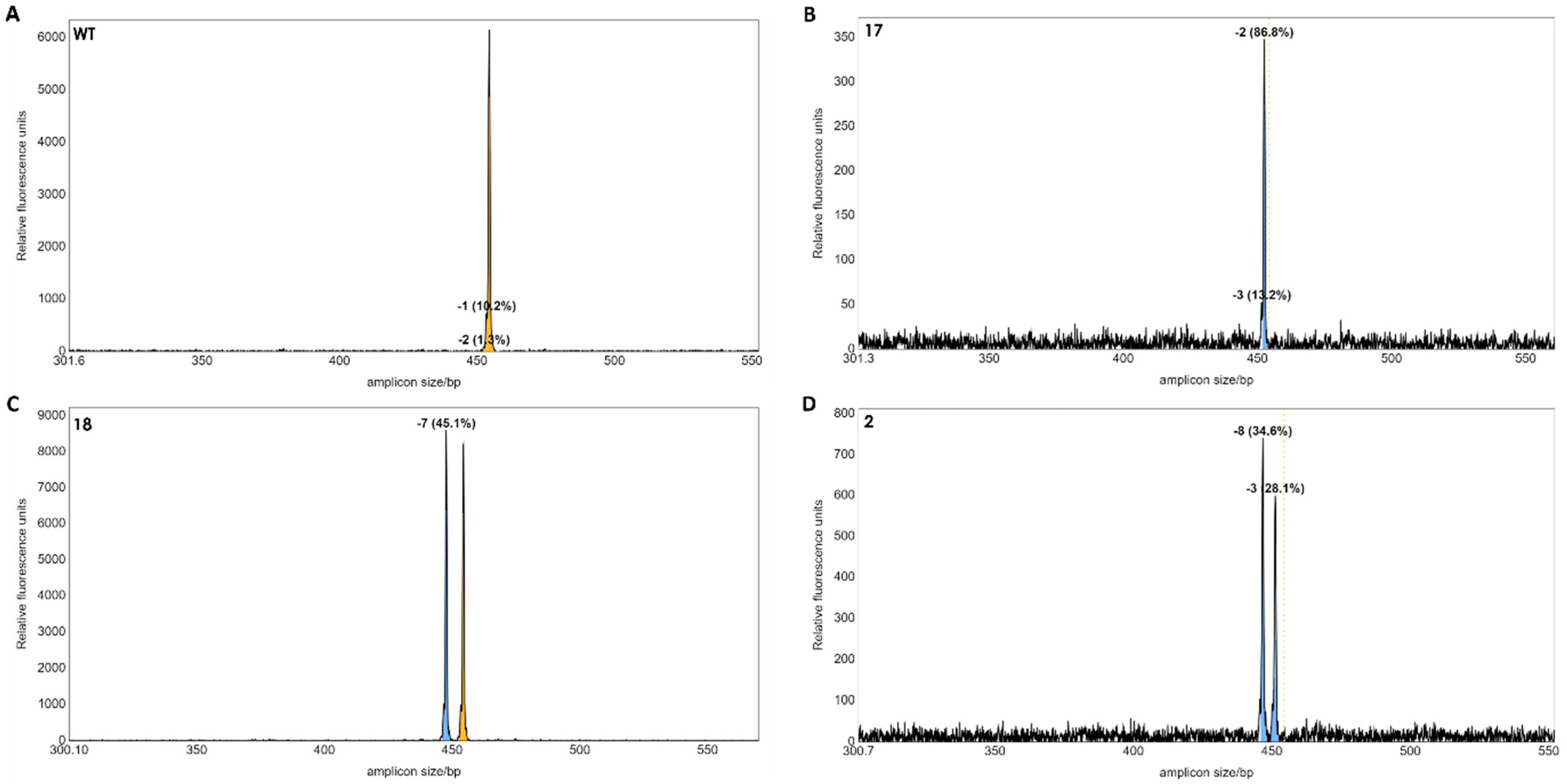
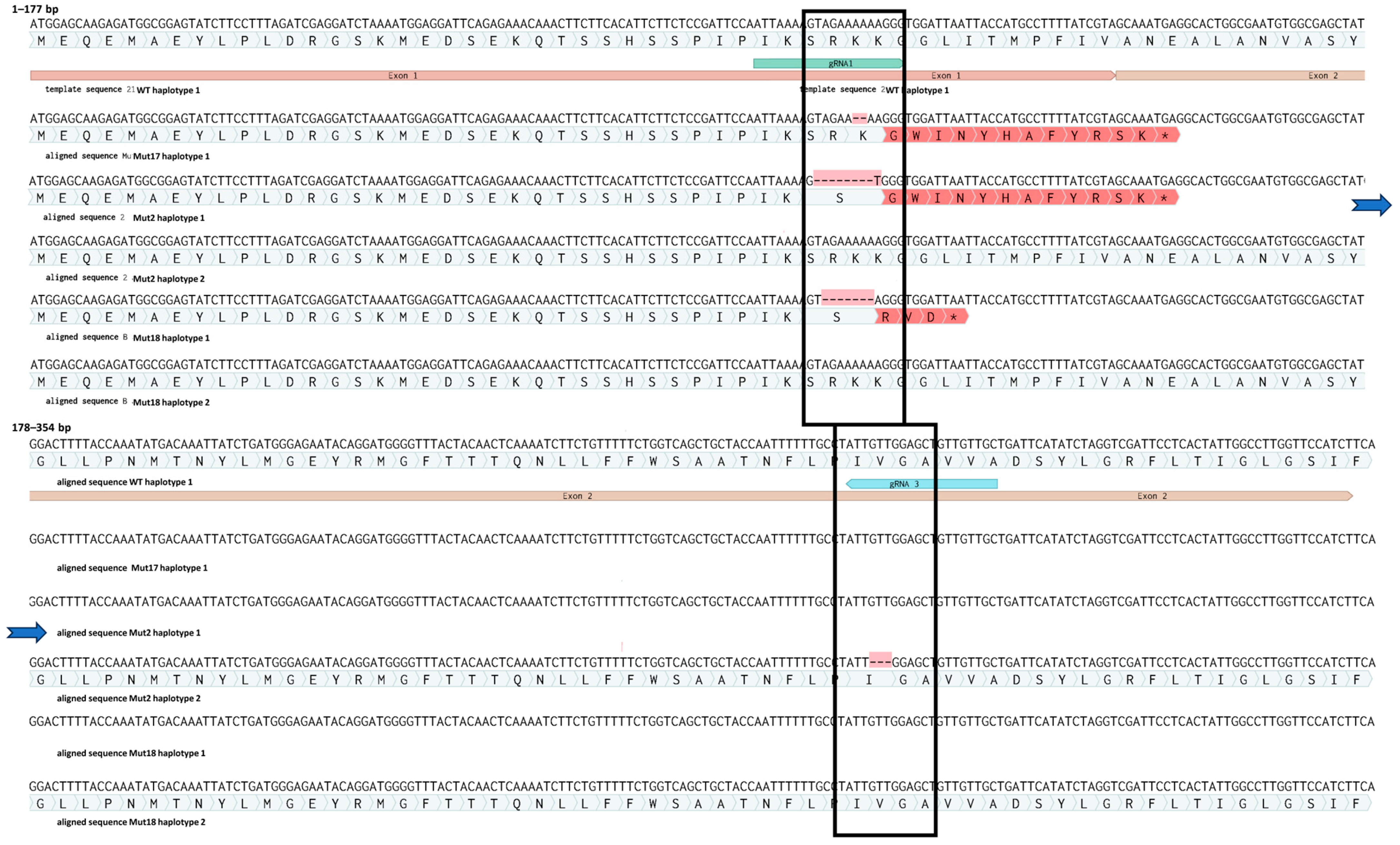
3.4. Confirmation of Gene Editing in Regenerated Plants by IDAA and Sanger Sequencing
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Aldenderfer, M. The Pleistocene/Holocene Transition in Peru and Its Effects upon Human Use of the Landscape. Quat. Int. 1999, 53, 11–19. [Google Scholar] [CrossRef]
- Rumold, C.U.; Aldenderfer, M.S. Late Archaic-Early Formative Period Microbotanical Evidence for Potato at Jiskairumoko in the Titicaca Basin of Southern Peru. Proc. Natl. Acad. Sci. USA 2016, 113, 13672–13677. [Google Scholar] [CrossRef] [PubMed]
- Spooner, D.M.; McLean, K.; Ramsay, G.; Waugh, R.; Bryan, G.J. A Single Domestication for Potato Based on Multilocus Amplified Fragment Length Polymorphism Genotyping. Proc. Natl. Acad. Sci. USA 2005, 102, 14694–14699. [Google Scholar] [CrossRef] [PubMed]
- Das Dangol, S.; Barakate, A.; Stephens, J.; Çalıskan, M.E.; Bakhsh, A. Genome Editing of Potato Using CRISPR Technologies: Current Development and Future Prospective. Plant Cell Tissue Organ Cult. 2019, 139, 403–416. [Google Scholar] [CrossRef]
- Hofvander, P.; Andreasson, E.; Andersson, M. Potato Trait Development Going Fast-Forward with Genome Editing. Trends Genet. 2022, 38, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, J.K.; Buckseth, T.; Challam, C.; Zinta, R.; Bhatia, N.; Dalamu, D.; Naik, S.; Poonia, A.K.; Singh, R.K.; Luthra, S.K.; et al. CRISPR/Cas Genome Editing in Potato: Current Status and Future Perspectives. Front. Genet. 2022, 13, 827808. [Google Scholar] [CrossRef] [PubMed]
- Marshall, M. How CRISPR Could Yield the next Blockbuster Crop. Nature 2024, 625, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Kanstrup, C.; Nour-Eldin, H.H. The Emerging Role of the Nitrate and Peptide Transporter Family: NPF in Plant Specialized Metabolism. Curr. Opin. Plant Biol. 2022, 68, 102243. [Google Scholar] [CrossRef] [PubMed]
- Johansen, I.E.; Liu, Y.; Jørgensen, B.; Bennett, E.P.; Andreasson, E.; Nielsen, K.L.; Blennow, A.; Petersen, B.L. High Efficacy Full Allelic CRISPR/Cas9 Gene Editing in Tetraploid Potato. Sci. Rep. 2019, 9, 17715. [Google Scholar] [CrossRef] [PubMed]
- Kazachkova, Y.; Zemach, I.; Panda, S.; Bocobza, S.; Vainer, A.; Rogachev, I.; Dong, Y.; Ben-Dor, S.; Veres, D.; Kanstrup, C.; et al. The GORKY Glycoalkaloid Transporter Is Indispensable for Preventing Tomato Bitterness. Nat. Plants 2021, 7, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Nicolia, A.; Proux-Wéra, E.; Åhman, I.; Onkokesung, N.; Andersson, M.; Andreasson, E.; Zhu, L.H. Targeted Gene Mutation in Tetraploid Potato through Transient TALEN Expression in Protoplasts. J. Biotechnol. 2015, 204, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.P.; Petersen, B.L.; Johansen, I.E.; Niu, Y.; Yang, Z.; Chamberlain, C.A.; Met, Ö.; Wandall, H.H.; Frödin, M. INDEL Detection, the ‘Achilles Heel’ of Precise Genome Editing: A Survey of Methods for Accurate Profiling of Gene Editing Induced Indels. Nucleic Acids Res. 2020, 48, 11959. [Google Scholar] [CrossRef] [PubMed]
- Spooner, D.M.; Hijmans, R.J. Potato Systematics and Germplasm Collecting, 1989–2000. Am. J. Potato Res. 2001, 78, 237–268. [Google Scholar] [CrossRef]
- Carlsen, F.M.; Johansen, I.E.; Yang, Z.; Liu, Y.; Westberg, I.N.; Kieu, N.P.; Jørgensen, B.; Lenman, M.; Andreasson, E.; Petersen, B.L.; et al. Strategies for Efficient Gene Editing in Protoplasts of Solanum Tuberosum Theme: Determining GRNA Efficiency Design by Utilizing Protoplast (Research). Front. Genome Ed. 2021, 3, 795644. [Google Scholar] [CrossRef] [PubMed]
- EU Rethinks Genome Editing. Nat. Plants 2023, 9, 1169–1170. [CrossRef] [PubMed]
- Zheng, Z.; Ye, G.; Zhou, Y.; Pu, X.; Su, W.; Wang, J. Editing Sterol Side Chain Reductase 2 Gene (StSSR2) via CRISPR/Cas9 Reduces the Total Steroidal Glycoalkaloids in Potato. All Life 2021, 14, 401–413. [Google Scholar] [CrossRef]
- Voigt, B. EU Regulation of Gene-Edited Plants—A Reform Proposal. Front. Genome Ed. 2023, 5, 1119442. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azariadis, A.; Andrzejczak, O.A.; Carlsen, F.M.; Westberg, I.; Brinch-Pedersen, H.; Petersen, B.L.; Hebelstrup, K.H. A Walk on the Wild Side: Genome Editing of Tuber-Bearing Solanum bulbocastanum. Plants 2024, 13, 1044. https://doi.org/10.3390/plants13071044
Azariadis A, Andrzejczak OA, Carlsen FM, Westberg I, Brinch-Pedersen H, Petersen BL, Hebelstrup KH. A Walk on the Wild Side: Genome Editing of Tuber-Bearing Solanum bulbocastanum. Plants. 2024; 13(7):1044. https://doi.org/10.3390/plants13071044
Chicago/Turabian StyleAzariadis, Aristotelis, Olga A. Andrzejczak, Frida M. Carlsen, Ida Westberg, Henrik Brinch-Pedersen, Bent L. Petersen, and Kim H. Hebelstrup. 2024. "A Walk on the Wild Side: Genome Editing of Tuber-Bearing Solanum bulbocastanum" Plants 13, no. 7: 1044. https://doi.org/10.3390/plants13071044
APA StyleAzariadis, A., Andrzejczak, O. A., Carlsen, F. M., Westberg, I., Brinch-Pedersen, H., Petersen, B. L., & Hebelstrup, K. H. (2024). A Walk on the Wild Side: Genome Editing of Tuber-Bearing Solanum bulbocastanum. Plants, 13(7), 1044. https://doi.org/10.3390/plants13071044