Development of EST-SSR Markers Linked to Flowering Candidate Genes in Elymus sibiricus L. Based on RNA Sequencing

Abstract
:1. Introduction
2. Results
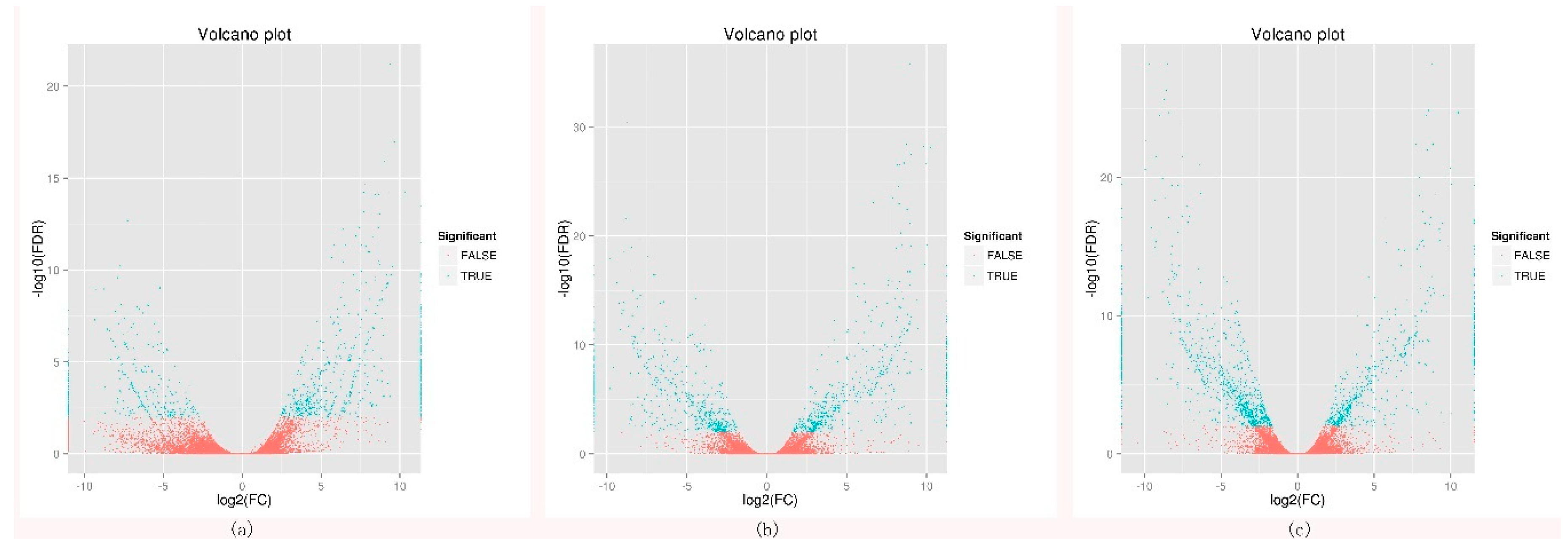
2.1. Transcriptome Sequencing, Unigenes Annotation, and Differentially Expressed Genes Analysis
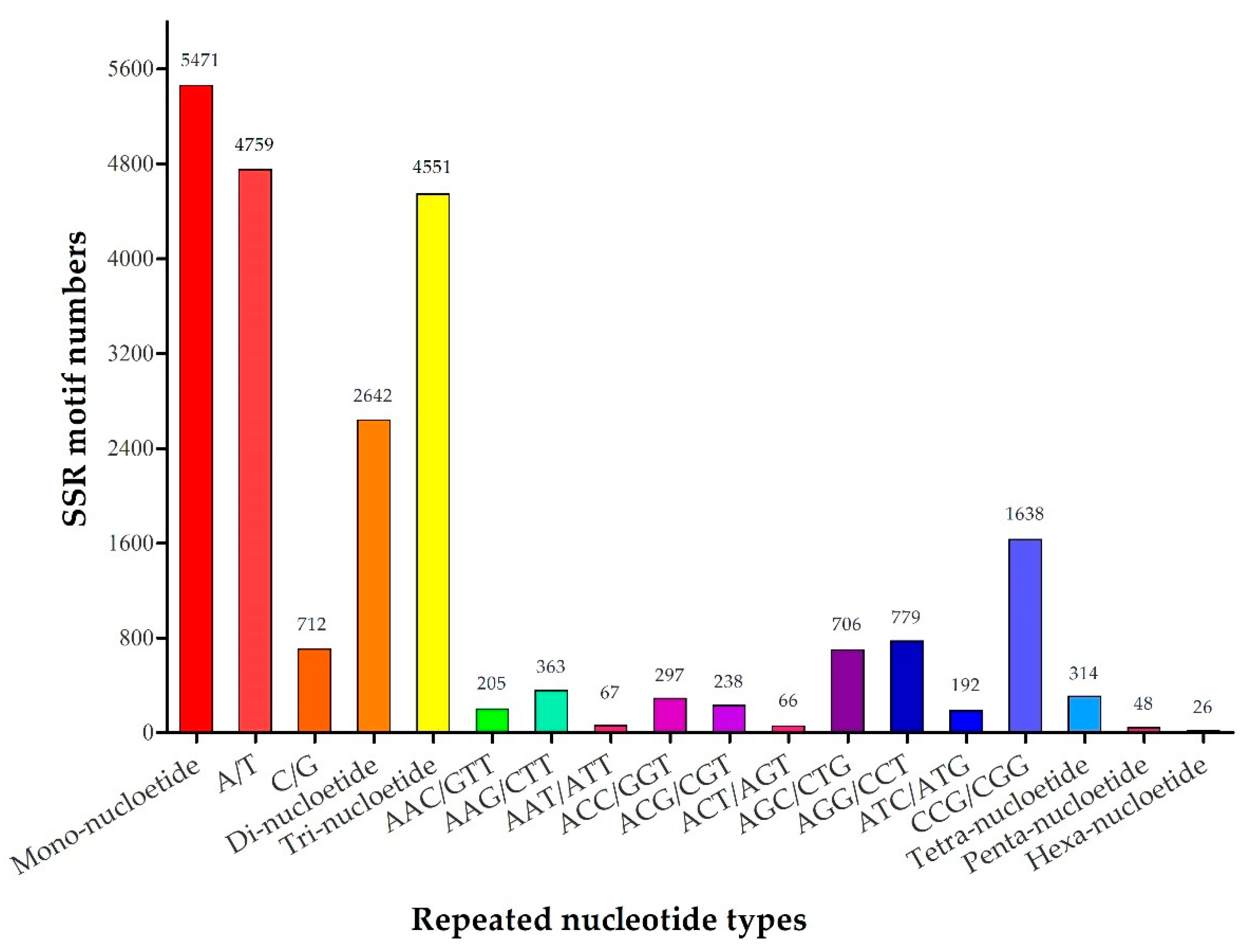
2.2. Frequency and Distribution of EST-SSR Markers
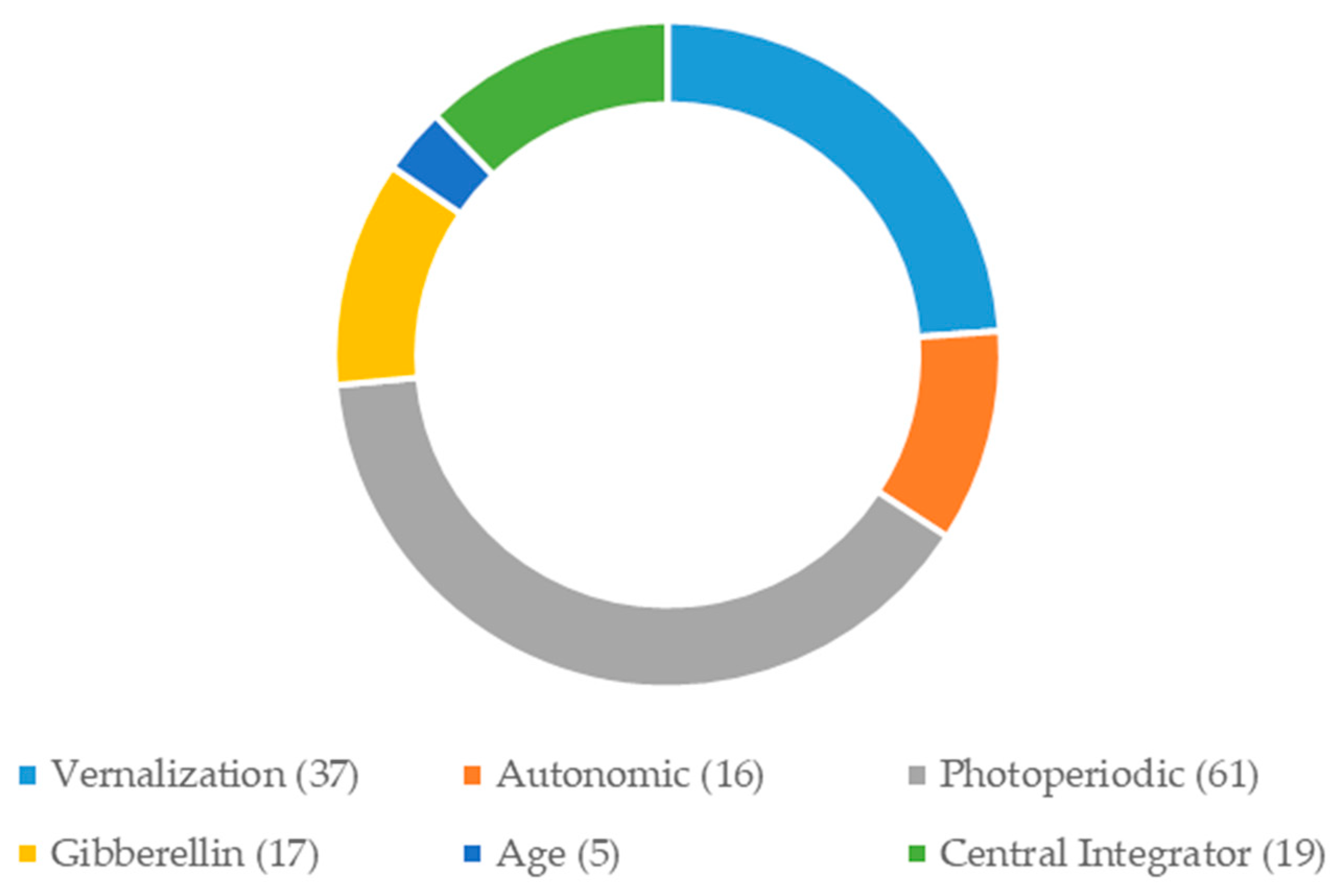
2.3. Development of Candidate Gene-Based EST-SSR Markers
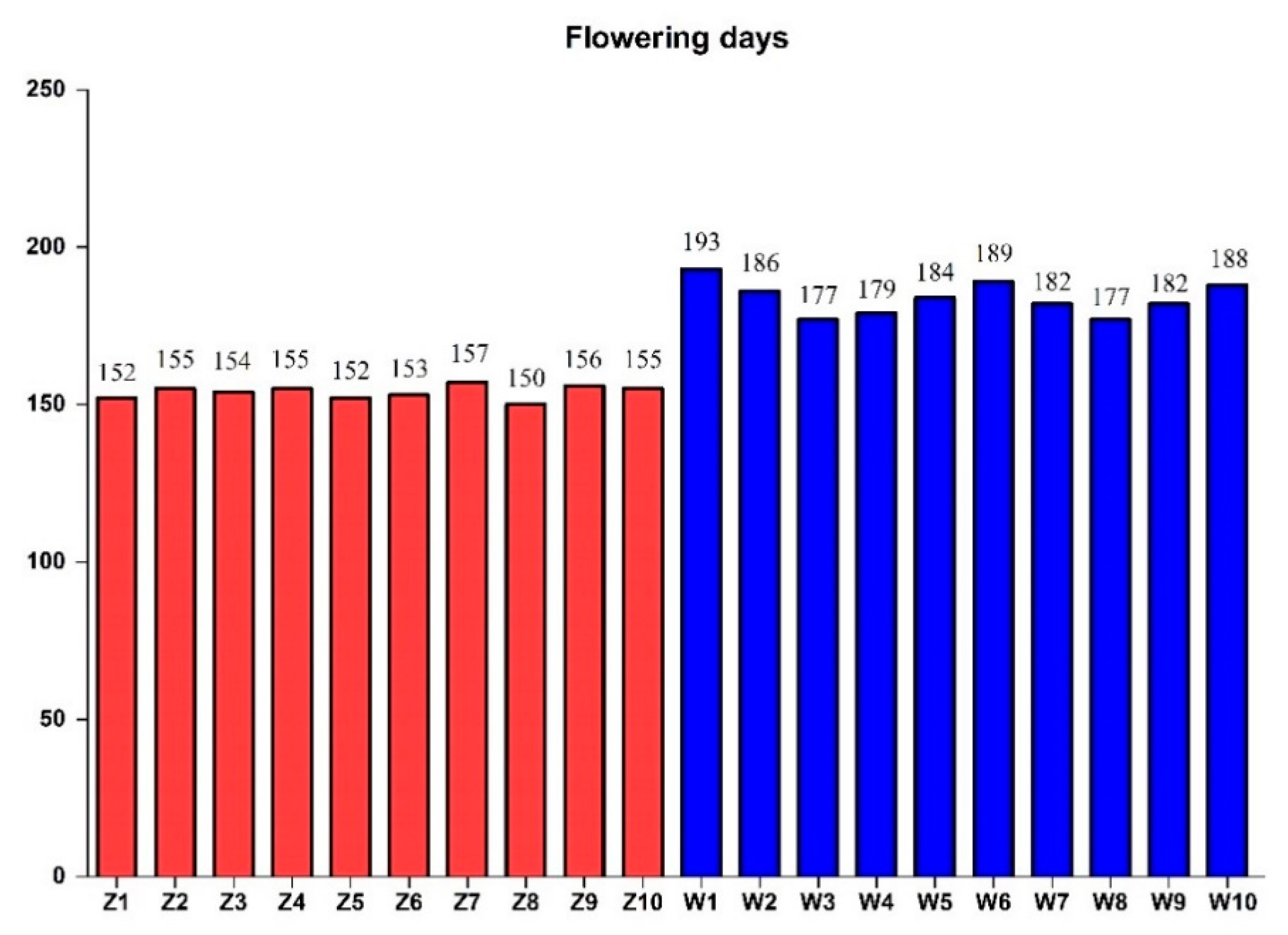
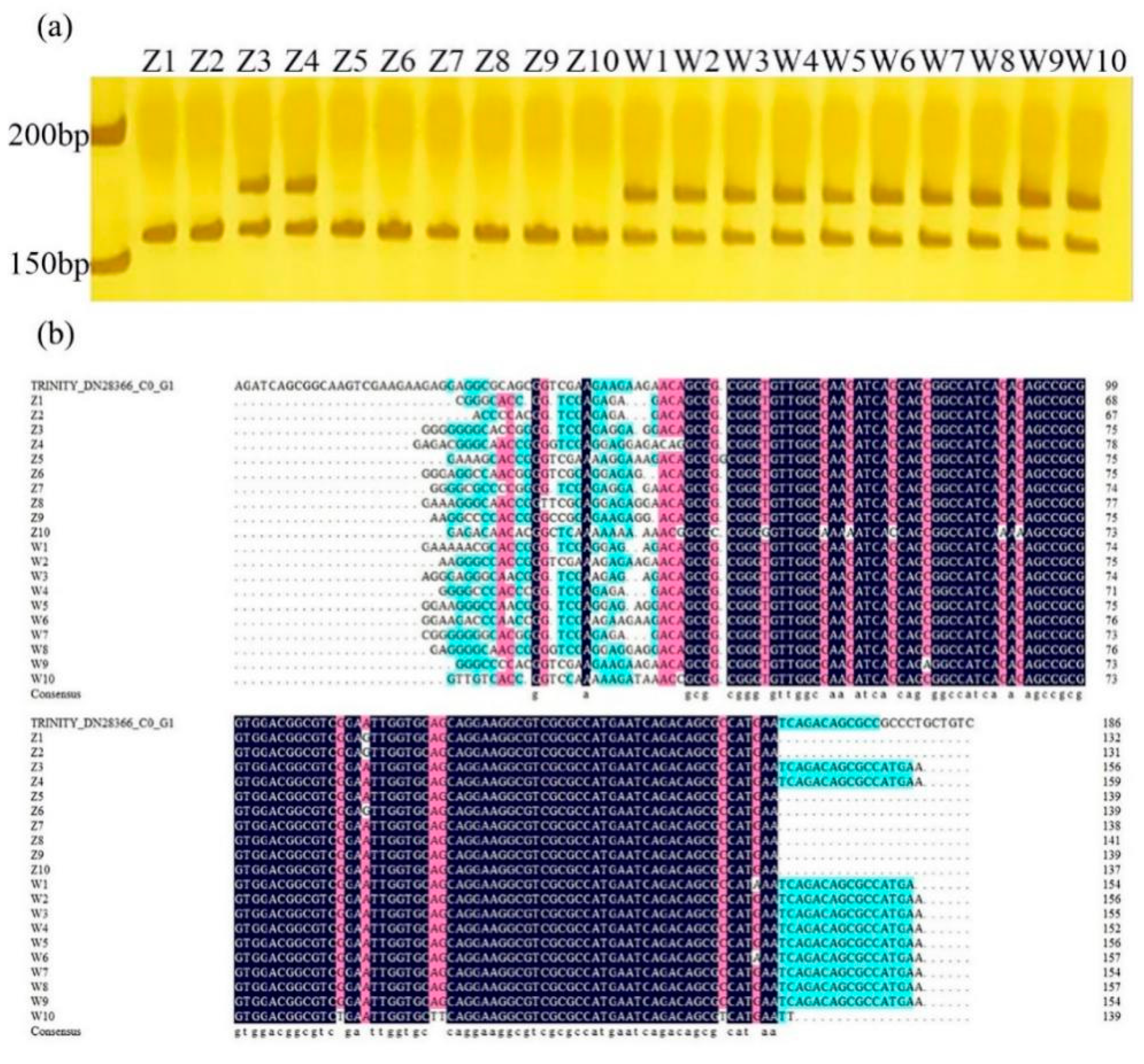
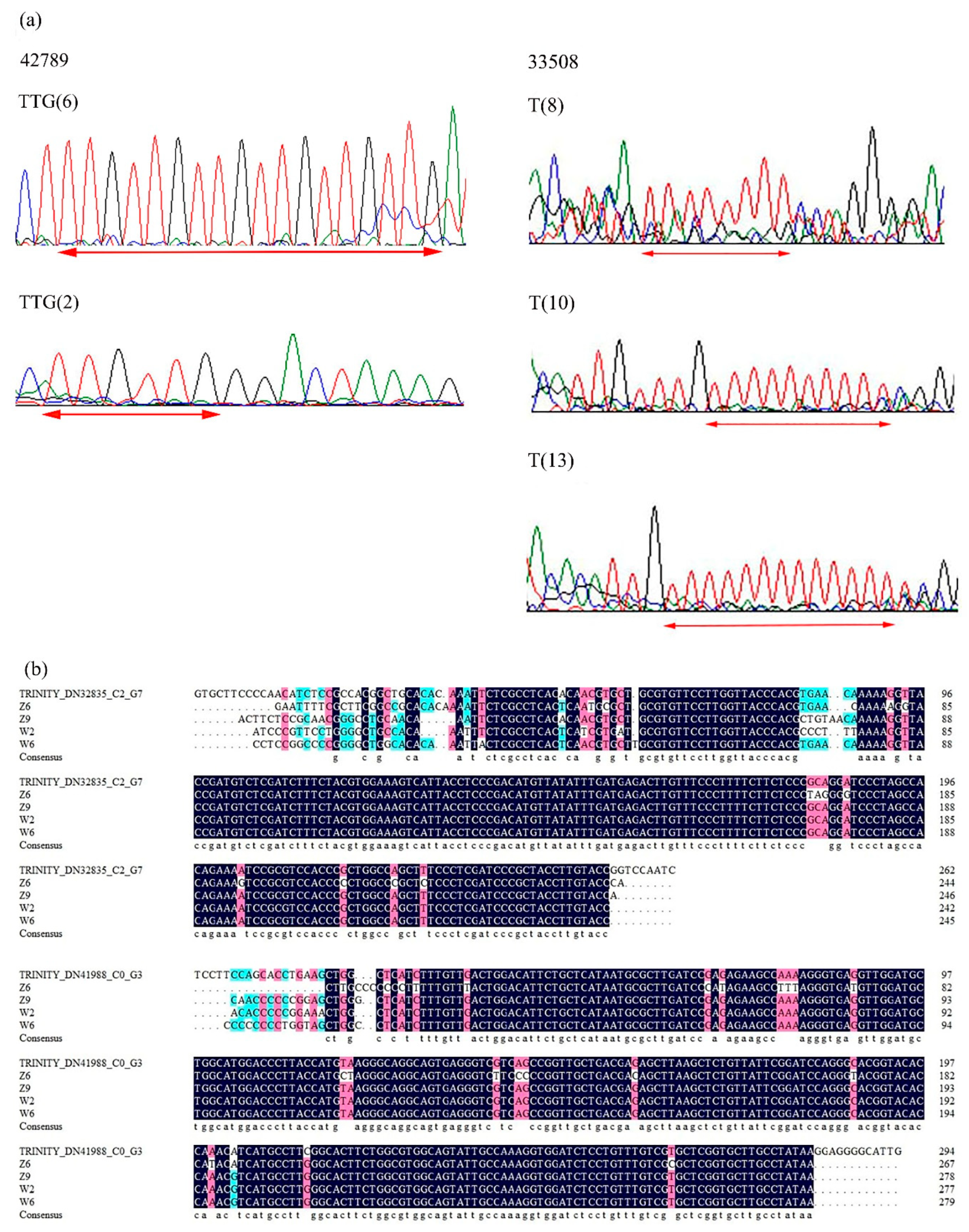
2.4. Validation of Candidate Gene-Based Specific Primers Authenticity
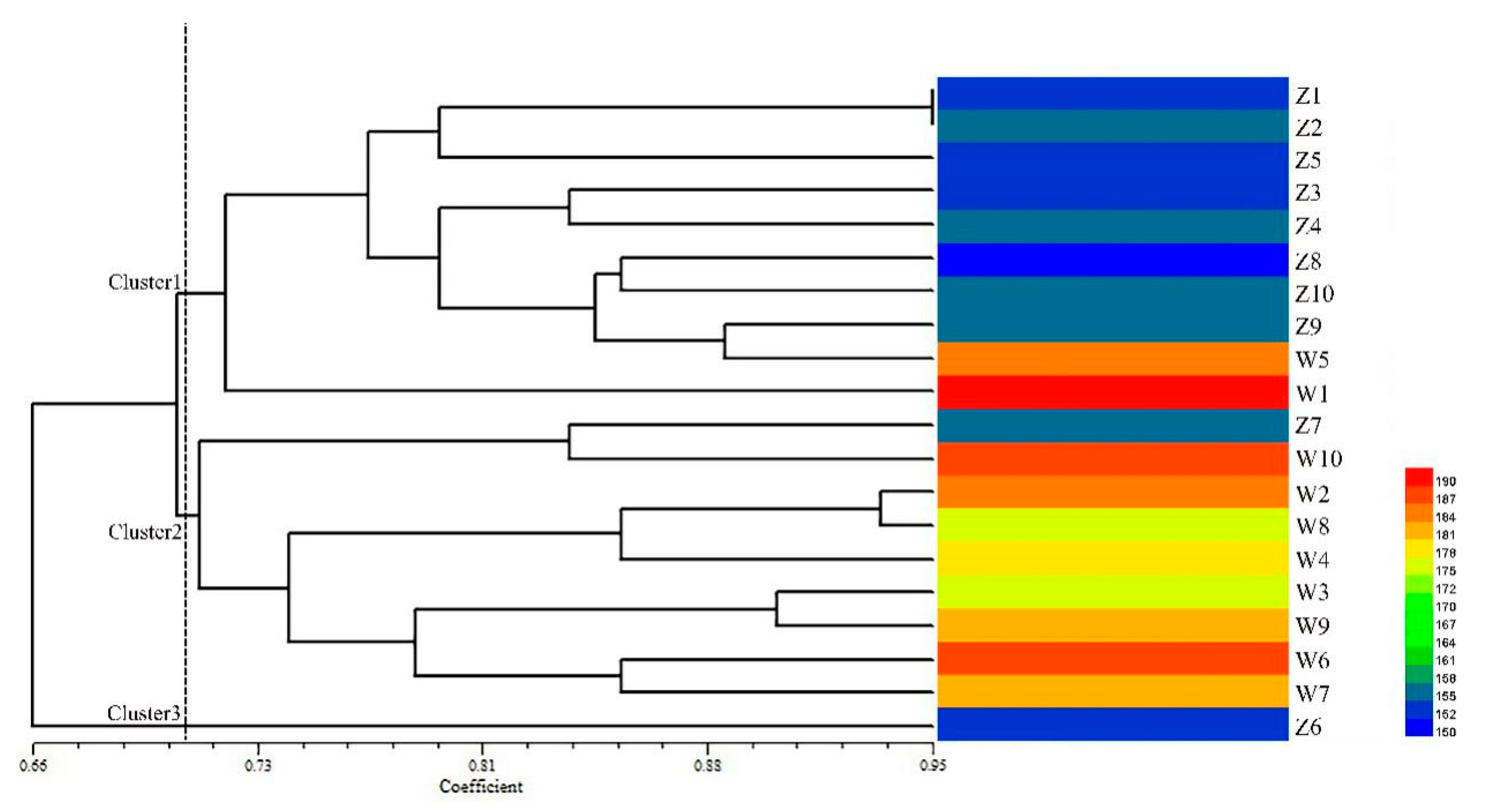
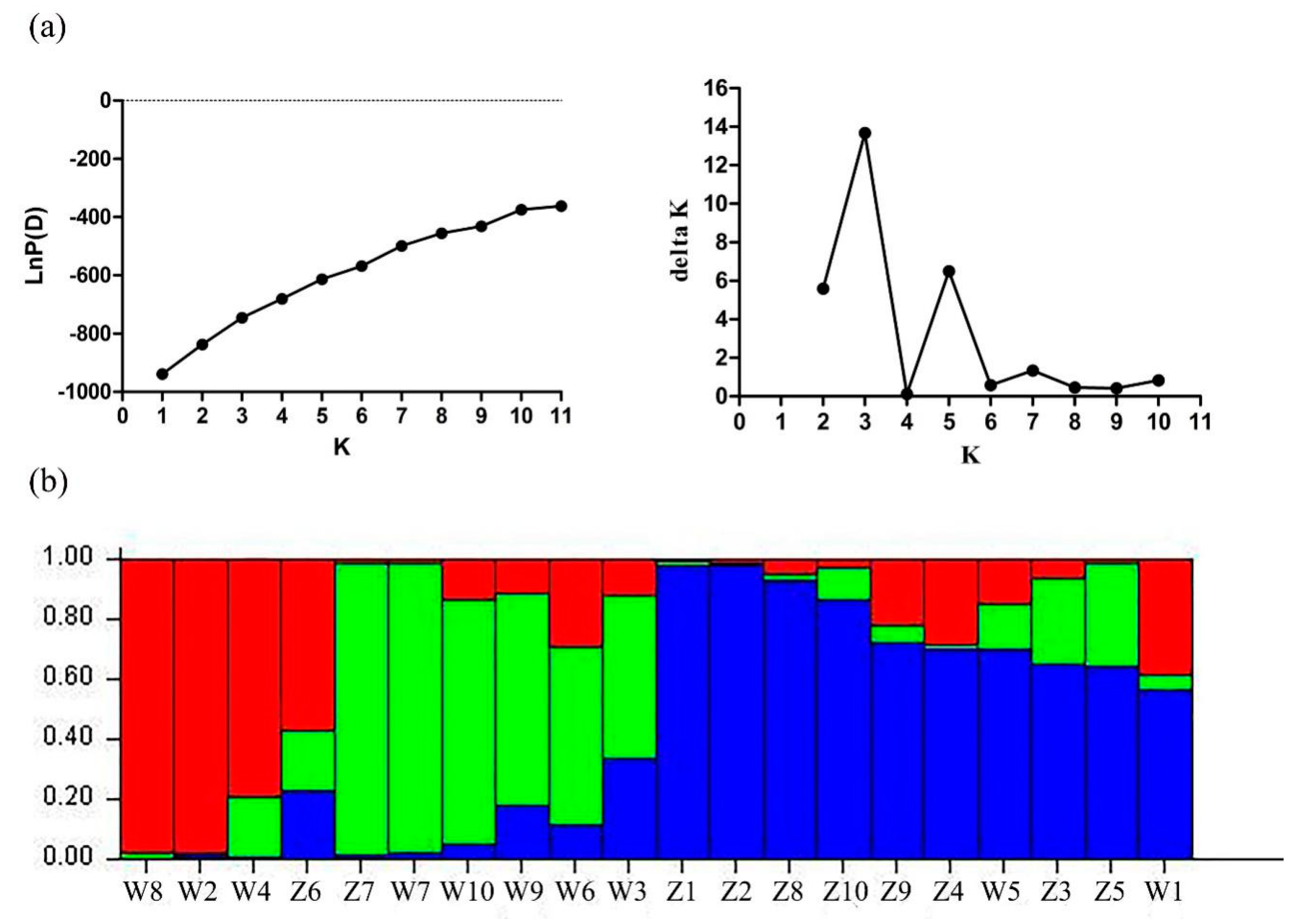
2.5. Genetic Diversity Analysis Using Candidate Gene-Based EST-SSR Markers
3. Discussion
3.1. Candidate Gene-Based EST-SSR Markers with Flowering Time for Marker-Assisted Selection
3.2. Genetic Diversity of Candidate Gene-Based EST-SSR Markers
4. Materials and Methods
4.1. Plant Materials
4.2. Development of Candidate Gene-Based EST-SSR Markers Based on RNA-Seq in E. sibiricus
4.3. DNA Extraction, Genotyping and Primer Validation
4.4. Allele Scoring and Polymorphism Detection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xie, W.; Zhao, X.; Zhang, J.; Wang, Y.; Liu, W. Assessment of genetic diversity of Siberian wild rye (Elymus sibiricus L.) germplasms with variation of seed shattering and implication for future genetic improvement. Biochem. Syst. Ecol. 2015, 58, 211–218. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, X.Q.; Zhou, Y.H.; Bai, S.Q.; Liu, W. Assessing genetic diversity of Elymus sibiricus (Poaceae: Triticeae) populations from Qinghai-Tibet Plateau by ISSR markers. Biochem. Syst. Ecol. 2008, 36, 514–522. [Google Scholar] [CrossRef]
- Xie, W.; Zhang, J.; Zhao, X.; Zhang, J.; Wang, Y. Siberian wild rye (Elymus sibiricus L.): Genetic diversity of germplasm determined using DNA fingerprinting and SCoT markers. Biochem. Syst. Ecol. 2015, 60, 186–192. [Google Scholar] [CrossRef]
- Craufurd, P.Q.; Wheeler, T.R. Climate change and the flowering time of annual crops. J. Exp. Bot. 2009, 60, 2529–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torabi, B.; Adibnya, M.; Rahimi, A.; Azari, A. Modeling flowering response to temperature and photoperiod in safflower. Ind. Crop. Prod. 2020, 151, 10. [Google Scholar] [CrossRef]
- Simpson, G.G.; Dean, C. Flowering—Arabidopsis, the rosetta stone of flowering time? Science 2002, 296, 285–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrés, F.; Coupland, G. The genetic basis of flowering responses to seasonal cues. Nat. Rev. Genet. 2012, 13, 627–639. [Google Scholar] [CrossRef]
- Osnato, M.; Castillejo, C.; Matias-Hernandez, L.; Pelaz, S. TEMPRANILLO genes link photoperiod and gibberellin pathways to control flowering in Arabidopsis. Nat. Commun. 2012, 3, 808. [Google Scholar] [CrossRef] [Green Version]
- Michaels, S.D.; Amasino, R.M. Loss of FLOWERING LOCUS C activity eliminates the late-flowering phenotype of FRIGIDA and autonomous pathway mutations but not responsiveness to vernalization. Plant Cell 2001, 13, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Levy, Y.Y.; Mesnage, S.; Mylne, J.S.; Gendall, A.R.; Dean, C. Multiple roles of Arabidopsis VRN1 in vernalization and flowering time control. Science 2002, 297, 243–246. [Google Scholar] [CrossRef]
- Corbesier, L.; Vincent, C.; Jang, S.H.; Fornara, F.; Fan, Q.Z.; Searle, I.; Giakountis, A.; Farrona, S.; Gissot, L.; Turnbull, C.; et al. FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science 2007, 316, 1030–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samach, A.; Onouchi, H.; Gold, S.E.; Ditta, G.S.; Schwarz-Sommer, Z.; Yanofsky, M.F.; Coupland, G. Distinct roles of CONSTANS target genes in reproductive development of Arabidopsis. Science 2000, 288, 1613–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.C.; Qiao, L.F.; Chen, J.C.; Rong, Y.H.; Zhao, Y.H.; Cui, X.K.; Xu, J.P.; Hou, X.M.; Dong, C.H. Arabidopsis REM16 acts as a B3 domain transcription factor to promote flowering time via directly binding to the promoters of SOC1 and FT. Plant J. 2020. [Google Scholar] [CrossRef]
- Li, C.X.; Lin, H.Q.; Chen, A.; Lau, M.; Jernstedt, J.; Dubcovsky, J. Wheat VRN1, FUL2 and FUL3 play critical and redundant roles in spikelet development and spike determinacy. Development 2019, 146, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, M.C.; Zhou, Z.J.; Zho, X.S.; Yang, H.Y.; Li, M.R.; Li, H.Q. Overexpression of OsFTL10 induces early flowering and improves drought tolerance in Oryza sativa L. PeerJ 2019, 7, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Li, T.; Wang, Y.M.; Zheng, J.; Li, H.F.; Hao, C.Y.; Zhang, X.Y. TaZIM-A1 negatively regulates flowering time in common wheat (Triticum aestivum L.). J. Integr. Plant Biol. 2019, 61, 359–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.M.; Zhang, T.J.; Guo, T.; Ding, W.; Long, R.C.; Yang, Q.C.; Wang, Z. Isolation and Functional Characterization of MsFTa, a FLOWERING LOCUS T Homolog from Alfalfa (Medicago sativa). Int. J. Mol. Sci. 2019, 20, 1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.K.; Feng, G.Y.; Yan, H.D.; Zhang, Z.R.; Bushman, B.S.; Wang, J.P.; Bombarely, A.; Li, M.Z.; Yang, Z.F.; Nie, G.; et al. Genome assembly provides insights into the genome evolution and flowering regulation of orchardgrass. Plant Biotechnol. J. 2020, 18, 373–388. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Y.J.; Zhang, Q.; Qi, Y.; Guo, D.J. Global characterization of Artemisia annua glandular trichome transcriptome using 454 pyrosequencing. BMC Genom. 2009, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- Der, J.P.; Barker, M.S.; Wickett, N.J.; de Pamphilis, C.W.; Wolf, P.G. De novo characterization of the gametophyte transcriptome in bracken fern, Pteridium aquilinum. BMC Genom. 2011, 12, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arghavan, A.; Shekoufeh, E.; Sahar, A.; Mahsa, H.; Behrouz, S.; Hassan, J.; Hossein, F.; Sadegh, M.-F.; Fariba, R. Parallel consideration of SSRs and differentially expressed genes under abiotic stress for targeted development of functional markers in almond and related Prunus species. Sci. Hortic. 2015, 198, 462–472. [Google Scholar] [CrossRef]
- Singh, A.K.; Chaurasia, S.; Kumar, S.; Singh, R.; Kumari, J.; Yadav, M.C.; Singh, N.; Gaba, S.; Jacob, S.R. Identification, analysis and development of salt responsive candidate gene based SSR markers in wheat. BMC Plant Biol. 2018, 18, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, B.K.; Agrawal, P.K.; Gupta, H.S.; Kumar, A.; Bhatt, J.C. Identification of candidate gene-based SSR markers for lysine and tryptophan metabolic pathways in maize (Zea mays). Plant Breed. 2012, 131, 20–27. [Google Scholar] [CrossRef]
- Jespersen, D.; Ma, X.Q.; Bonos, S.A.; Belanger, F.C.; Raymer, P.; Huang, B.R. Association of SSR and candidate gene markers with genetic variations in summer heat and drought performance for creeping bentgrass. Crop. Sci. 2018, 58, 2644–2656. [Google Scholar] [CrossRef]
- Lai, D.Y.; Li, H.Z.; Fan, S.L.; Song, M.Z.; Pang, C.Y.; Wei, H.L.; Liu, J.J.; Wu, D.; Gong, W.F.; Yu, S.X. Generation of ESTs for flowering gene discovery and SSR marker development in upland cotton. PLoS ONE 2011, 6, e28676. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.Q.; Zhang, J.C.; Zhang, Z.Y.; Xie, W.G. Elymus nutans genes for seed shattering and candidate gene-derived EST-SSR markers for germplasm evaluation. BMC Plant Biol. 2019, 19. [Google Scholar] [CrossRef]
- Danilevskaya, O.N.; Meng, X.; McGonigle, B.; Muszynski, M.G. Beyond flowering time Pleiotropic function of the maize flowering hormone florigen. Plant Signal. Behav. 2011, 6, 1267–1270. [Google Scholar] [CrossRef] [Green Version]
- Bushman, B.S.; Robins, J.C.; Jensen, K.B. Dry matter yield, heading date, and plant mortality of Orchardgrass subspecies in a semiarid environment. Crop. Sci. 2012, 52, 745–751. [Google Scholar] [CrossRef]
- Collard, B.C.Y.; Mackill, D.J. Marker-assisted selection: An approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. B-Biol. Sci. 2008, 363, 557–572. [Google Scholar] [CrossRef] [Green Version]
- Unamba, C.I.; Nag, A.; Sharma, R.K. Next Generation Sequencing Technologies: The doorway to the unexplored genomics of non-model plants. Front. Plant Sci. 2015, 6, 1074. [Google Scholar] [CrossRef] [Green Version]
- Powell, W.; Machray, G.C.; Provan, J. Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1996, 1, 215–222. [Google Scholar] [CrossRef]
- Gupta, P.K.; Rustgi, S.; Sharma, S.; Singh, R.; Kumar, N.; Balyan, H.S. Transferable EST-SSR markers for the study of polymorphism and genetic diversity in bread wheat. Mol. Genet. Genom. 2003, 270, 315–323. [Google Scholar] [CrossRef]
- Saha, M.C.; Mian, M.A.R.; Eujayl, I.; Zwonitzer, J.C.; Wang, L.J.; May, G.D. Tall fescue EST-SSR markers with transferability across several grass species. Theor. Appl. Genet. 2004, 109, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Gupta, S.; Biradar, R.S.; Gupta, P.; Dubey, S.; Singh, N.P. Association of functional markers with flowering time in lentil. J. Appl. Genet. 2018, 59, 9–21. [Google Scholar] [CrossRef]
- Li, D.M.; Wu, W.; Zhang, D.; Liu, X.R.; Liu, X.F.; Lin, Y.J. Floral transcriptome analyses of four Paphiopedilum Orchids with distinct flowering behaviors and development of simple sequence repeat markers. Plant Mol. Biol. Rep. 2015, 33, 1928–1952. [Google Scholar] [CrossRef]
- Monna, L.; Lin, H.X.; Kojima, S.; Sasaki, T.; Yano, M. Genetic dissection of a genomic region for a quantitative trait locus, Hd3, into two loci, Hd3a and Hd3b, controlling heading date in rice. Theor. Appl. Genet. 2002, 104, 772–778. [Google Scholar] [CrossRef]
- Kojima, S.; Takahashi, Y.; Kobayashi, Y.; Monna, L.; Sasaki, T.; Araki, T.; Yano, M. Hd3a, a rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions. Plant Cell Physiol. 2002, 43, 1096–1105. [Google Scholar] [CrossRef] [Green Version]
- Armstead, I.P.; Turner, L.B.; Farrell, M.; Skot, L.; Gomez, P.; Montoya, T.; Donnison, I.S.; King, I.P.; Humphreys, M.O. Synteny between a major heading-date QTL in perennial ryegrass (Lolium perenne L.) and the Hd3 heading-date locus in rice. Theor. Appl. Genet. 2004, 108, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Anwer, M.U.; Davis, A.; Davis, S.J.; Quint, M. Photoperiod sensing of the circadian clock is controlled by EARLY FLOWERING 3 and GIGANTEA. Plant J. 2020, 101, 1397–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Hao, X.Y.; Lu, Q.H.; Zhang, W.F.; Zhang, H.J.; Wang, L.; Yang, Y.J.; Xiao, B.; Wang, X.C. Genome-wide identification and expression analysis of flowering-related genes reveal putative floral induction and differentiation mechanisms in tea plant (Camellia sinensis). Genomics 2020, 112, 2318–2326. [Google Scholar] [CrossRef]
- Li, C.; Zheng, L.L.; Wang, X.N.; Hu, Z.B.; Zheng, Y.; Chen, Q.H.; Hao, X.C.; Xiao, X.; Wang, X.B.; Wang, G.D.; et al. Comprehensive expression analysis of Arabidopsis GA2-oxidase genes and their functional insights. Plant Sci. 2019, 285, 1–13. [Google Scholar] [CrossRef]
- Martin-Tryon, E.L.; Harmer, S.L. XAP5 CIRCADIAN TIMEKEEPER coordinates light signals for proper timing of photomorphogenesis and the circadian clock in Arabidopsis. Plant Cell 2008, 20, 1244–1259. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Mo, Y.L.; Chen, J.J.; Li, C.M.; Zhao, L.; Liu, Y.H. Expression and interaction proteins analysis of BjuFKF1 in stem mustard. Sci. Hortic. 2020, 269, 6. [Google Scholar] [CrossRef]
- Asp, T.; Frei, U.K.; Didion, T.; Nielsen, K.K.; Lubberstedt, T. Frequency, type, and distribution of EST-SSRs from three genotypes of Lolium perenne, and their conservation across orthologous sequences of Festuca arundinacea, Brachypodium distachyon, and Oryza sativa. BMC Plant Biol. 2007, 7. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Wu, F.; Luo, K.; Zhao, Y.; Yan, Q.; Zhang, Y.; Wang, Y.; Zhang, J. Cross-species transferability of EST-SSR markers developed from the transcriptome of Melilotus and their application to population genetics research. Sci. Rep. 2017, 7, 17959. [Google Scholar] [CrossRef]
- Pan, L.; Huang, T.; Yang, Z.; Tang, L.; Cheng, Y.; Wang, J.; Ma, X.; Zhang, X. EST-SSR marker characterization based on RNA-sequencing of Lolium multiflorum and cross transferability to related species. Mol. Breed. 2018, 38. [Google Scholar] [CrossRef]
- Zhang, Z.; Xie, W.; Zhao, Y.; Zhang, J.; Wang, N.; Ntakirutimana, F.; Yan, J.; Wang, Y. EST-SSR marker development based on RNA-sequencing of E. sibiricus and its application for phylogenetic relationships analysis of seventeen Elymus species. BMC Plant Biol. 2019, 19, 235. [Google Scholar] [CrossRef]
- Wang, Z.; Yu, G.; Shi, B.; Wang, X.; Qiang, H.; Gao, H. Development and characterization of simple sequence repeat (SSR) markers based on RNA-sequencing of Medicago sativa and in silico mapping onto the M. truncatula genome. PLoS ONE 2014, 9, e92029. [Google Scholar] [CrossRef]
- Zhang, M.A.; Mao, W.H.; Zhang, G.P.; Wu, F.B. Development and characterization of polymorphic EST-SSR and genomic SSR markers for Tibetan annual wild barley. PLoS ONE 2014, 9, e94881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.P.; Liu, P.; Luo, D.; Liu, W.X.; Wang, Y.R. Exploiting illumina sequencing for the development of 95 novel polymorphic EST-SSR markers in common vetch (Vicia sativa subsp sativa). Molecules 2014, 19, 5777–5789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajkova, L.; Koznarova, V.; Mozny, M.; Bartosova, L. Influence of climate change on flowering season of birch in the Czech Republic. Int. J. Biometeorol. 2020, 64, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Grenier, C.; Bramel-Cox, P.J.; Hamon, P. Core collection of sorghum: I. Stratification based on eco-geographical data. Crop. Sci. 2001, 41, 234–240. [Google Scholar] [CrossRef]
- Stuerz, S.; Shrestha, S.P.; Schmierer, M.; Vu, D.H.; Hartmann, J.; Sow, A.; Razafindrazaka, A.; Abera, B.B.; Chuma, B.A.; Asch, F. Climatic determinants of lowland rice development. J. Agron. Crop. Sci. 2020, 206, 466–477. [Google Scholar] [CrossRef]
- Burgarella, C.; Chantret, N.; Gay, L.; Prosperi, J.M.; Bonhomme, M.; Tiffin, P.; Young, N.D.; Ronfort, J. Adaptation to climate through flowering phenology: A case study in Medicago truncatula. Mol. Ecol. 2016, 25, 3397–3415. [Google Scholar] [CrossRef]
- Wang, Q.B.; Wang, Y.P.; Zhang, L. Inheritance and molecular marker for flowering time in radish (Raphanus sativus L.). Plant Mol. Biol. Rep. 2018, 36, 878–887. [Google Scholar] [CrossRef]
- Xie, W.G.; Robins, J.G.; Bushman, B.S. A genetic linkage map of tetraploid orchardgrass (Dactylis glomerata L.) and quantitative trait loci for heading date. Genome 2012, 55, 360–369. [Google Scholar] [CrossRef]
- Wang, N. Evaluation of Flowering Time and Mining of Candidate Genes in Elymus Sibiricus. Master’s Thesis, Lanzhou University, Lanzhou, China, 1 May 2020. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef] [Green Version]
- Guerra, V.; Beule, L.; Lehtsaar, E.; Liao, H.L.; Karlovsky, P. Improved protocol for DNA extraction from subsoils using phosphate lysis buffer. Microorganisms 2020, 8, 532. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Paul, J. Nouvelles recherches sur la distribution florale. Bull. Soc. Vaud. Sci. Nat. 1908, 44, 223–270. [Google Scholar] [CrossRef]
- Hudson, R.R. Gennetic data-analysis-methods for discrete population genetic data-Weir, BS. Science 1990, 250, 575. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Searching Item | Number |
|---|---|
| Total number of sequences examined | 40,639 |
| Total size of examined sequences (bp) | 78,157,064 |
| Total number of identified SSRs | 13,052 |
| Number of SSR containing sequences | 10,286 |
| Number of sequences containing more than 1 SSR | 2176 |
| Number of SSRs present in compound formation | 742 |
| Mono-nucleotide | 5471 |
| Di-nucleotide | 2642 |
| Tri-nucleotide | 4551 |
| Tetra-nucleotide | 314 |
| Penta-nucleotide | 48 |
| Hexa-nucleotide | 26 |
| Primer Code | Flowering Genes | Flowering Pathways/Functions | He | Ho | PIC |
|---|---|---|---|---|---|
| Primer 46865 | FPF1 | Flowering | 0.6592 | 0.9231 | 0.4767 |
| Primer 33680 | FPF1 | Flowering | 0.2268 | 0.1500 | 0.1275 |
| Primer 43088 | ELF3 | Flowering | 0.7966 | 0.9412 | 0.4210 |
| Primer 41496 | GID1 | Gibberellin | 0.4911 | 0.5000 | 0.2367 |
| Primer 40505 | GA2OX6 | Gibberellin | 0.6655 | 0.7000 | 0.2513 |
| Primer 37852 | CIGR | Gibberellin | 0.1975 | 0.0588 | 0.2500 |
| Primer 34975 | XCT | Circadian Clock | 0.5170 | 0.8000 | 0.1567 |
| Primer 48198 | GIGANTEA | Circadian Clock | 0.6584 | 1.0000 | 0.1400 |
| Primer 34500 | MBD9 | CONSTANS-Like | 0.8731 | 1.0000 | 0.2606 |
| Primer 31670 | NFYC4 | CONSTANS-Like | 0.3200 | 0.5946 | 0.1875 |
| Primer 43287 | COL4 | CONSTANS-Like | 0.7138 | 0.9730 | 0.3325 |
| Primer 36067 | COL13 | CONSTANS-Like | 0.8117 | 0.9730 | 0.3025 |
| Primer 36927 | FLC | Central Integrator | 0.8147 | 1.0000 | 0.1500 |
| Primer 34261 | HD3 | Central Integrator | 0.7794 | 1.0000 | 0.2590 |
| Primer 28366 | HD3a | Central Integrator | 0.6509 | 1.0000 | 0.1600 |
| Mean | 0.6117 | 0.7742 | 0.2475 |
| Code | Accession Number | Sample ID | Origins | Longitude/Latitude | Altitude (m) |
|---|---|---|---|---|---|
| 1 | PI598781 | Z1 | Baikal, Buryat, Russia | - | - |
| 2 | PI610876 | Z2 | - | - | - |
| 3 | PI610994 | Z3 | Siberia, Russia | - | 1250 |
| 4 | PI636676 | Z4 | Xiahe, Gansu, China | 35.19 N, 102.65 E | 2720 |
| 5 | PI655199 | Z5 | Hongyuan, Sichuan, China | 32.08 N, 102.57 E | 3280 |
| 6 | W610305 | Z6 | Siberia, Russia | - | - |
| 7 | W630476 | Z7 | Kyrgyzstan | - | - |
| 8 | HZ03 | Z8 | Hezuo, Gansu, China | - | - |
| 9 | LT05 | Z9 | Lintan, Gansu, China | - | - |
| 10 | LQ02 | Z10 | Luqu, Gansu, China | - | - |
| 11 | PI435088 | W1 | Ninshan, Xinjiang, China | - | - |
| 12 | PI504462 | W2 | Xining, Qinghai, China | - | - |
| 13 | PI531665 | W3 | Beijing, China | - | - |
| 14 | PI531669 | W4 | Xining, Qinghai, China | - | 2400 |
| 15 | PI595149 | W5 | Xinjiang, China | 44.17 N, 84.57 E | 1500 |
| 16 | PI595156 | W6 | Xinjiang, China | 45.03 N, 81.12 E | 1300 |
| 17 | PI595162 | W7 | Xinjiang, China | 44.12 N, 87.97 E | 1680 |
| 18 | PI595169 | W8 | Xinjiang, China | 43.77 N, 89.45 E | 1300 |
| 19 | PI595174 | W9 | Xinjiang, China | 43.68 N, 89.30 E | 1870 |
| 20 | PI655092 | W10 | Mongolia | 49.80 N, 94.96 E | 1370 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Y.; Zhang, Z.; Wan, Y.; Tian, J.; Xie, W. Development of EST-SSR Markers Linked to Flowering Candidate Genes in Elymus sibiricus L. Based on RNA Sequencing. Plants 2020, 9, 1371. https://doi.org/10.3390/plants9101371
Zheng Y, Zhang Z, Wan Y, Tian J, Xie W. Development of EST-SSR Markers Linked to Flowering Candidate Genes in Elymus sibiricus L. Based on RNA Sequencing. Plants. 2020; 9(10):1371. https://doi.org/10.3390/plants9101371
Chicago/Turabian StyleZheng, Yuying, Zongyu Zhang, Yiyang Wan, Jiaoyang Tian, and Wengang Xie. 2020. "Development of EST-SSR Markers Linked to Flowering Candidate Genes in Elymus sibiricus L. Based on RNA Sequencing" Plants 9, no. 10: 1371. https://doi.org/10.3390/plants9101371
APA StyleZheng, Y., Zhang, Z., Wan, Y., Tian, J., & Xie, W. (2020). Development of EST-SSR Markers Linked to Flowering Candidate Genes in Elymus sibiricus L. Based on RNA Sequencing. Plants, 9(10), 1371. https://doi.org/10.3390/plants9101371