Comparative Analyses of the Chloroplast Genomes of Patchouli Plants and Their Relatives in Pogostemon (Lamiaceae)


Abstract
:1. Introduction
2. Results
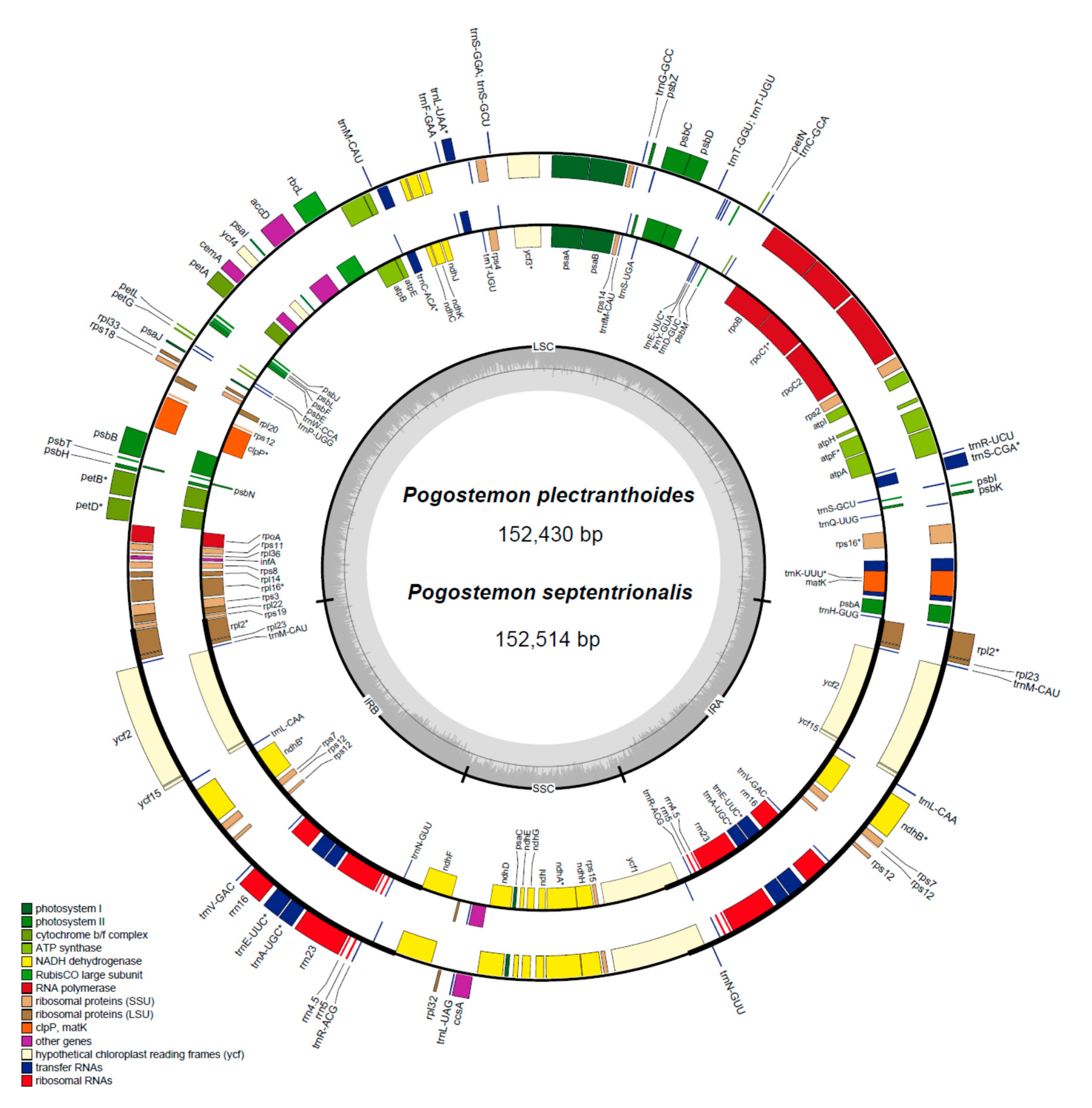
2.1. Characteristics of Chloroplast Genomes
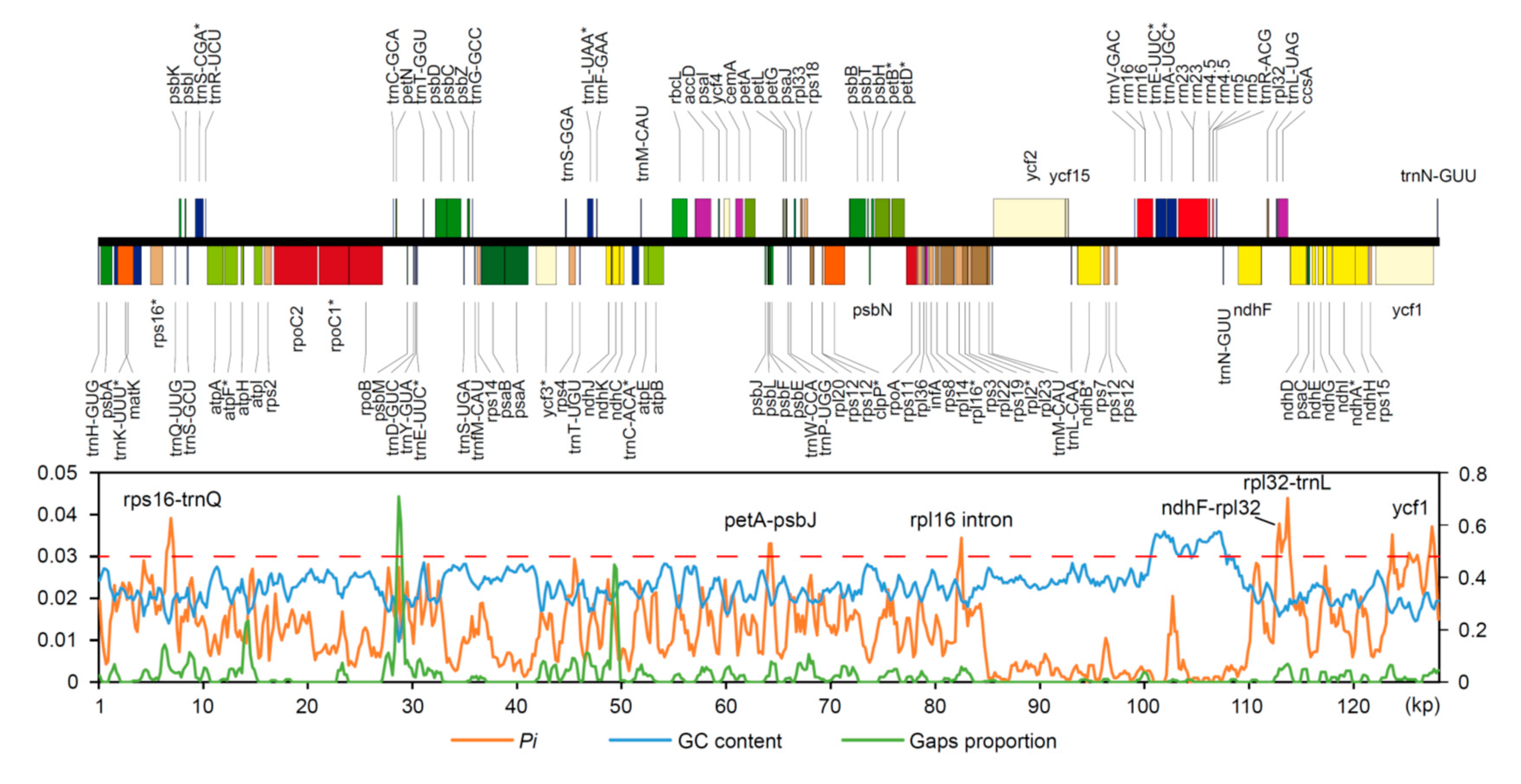
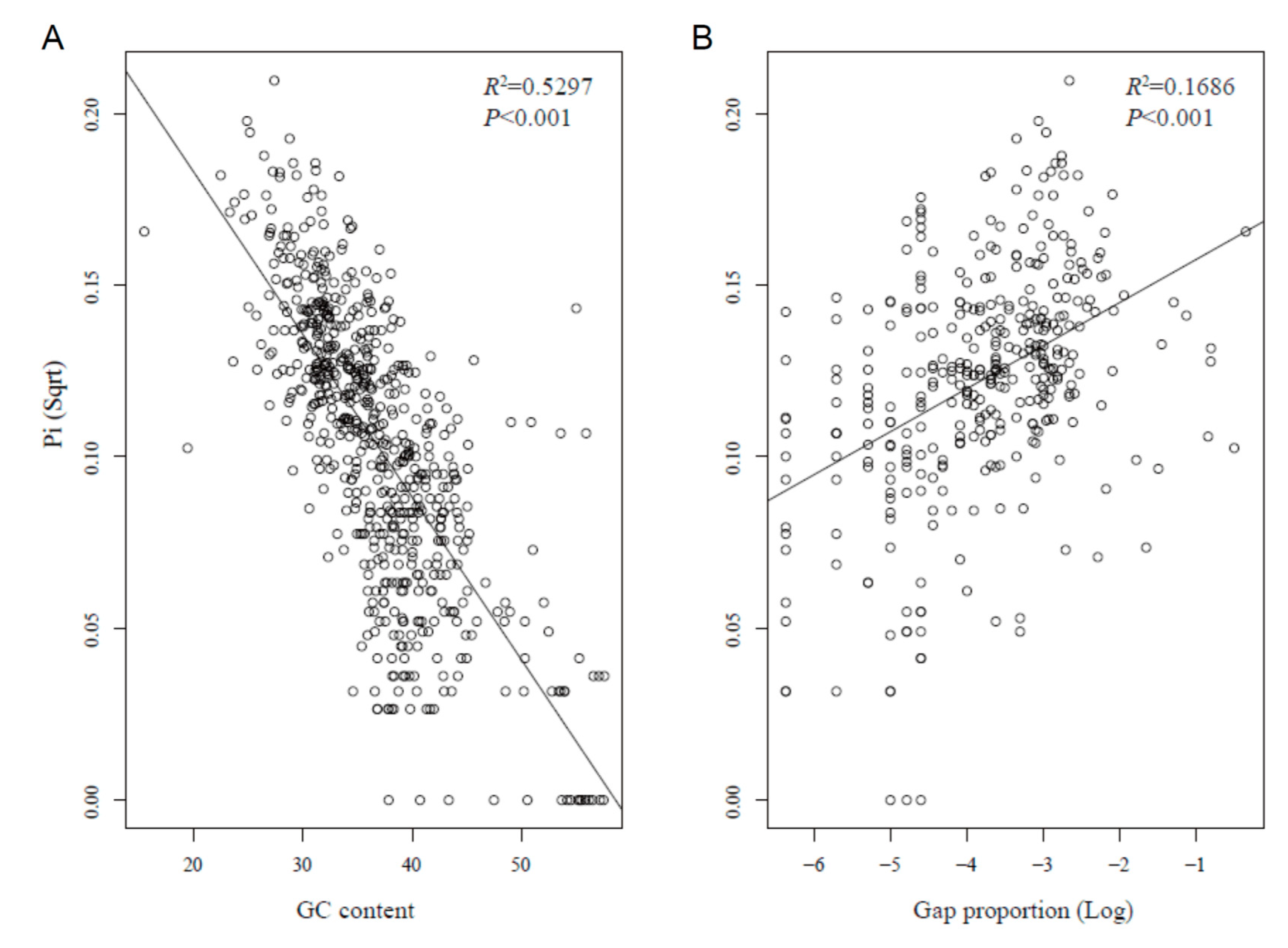
2.2. Divergence Hotspots in Chloroplast Genomes
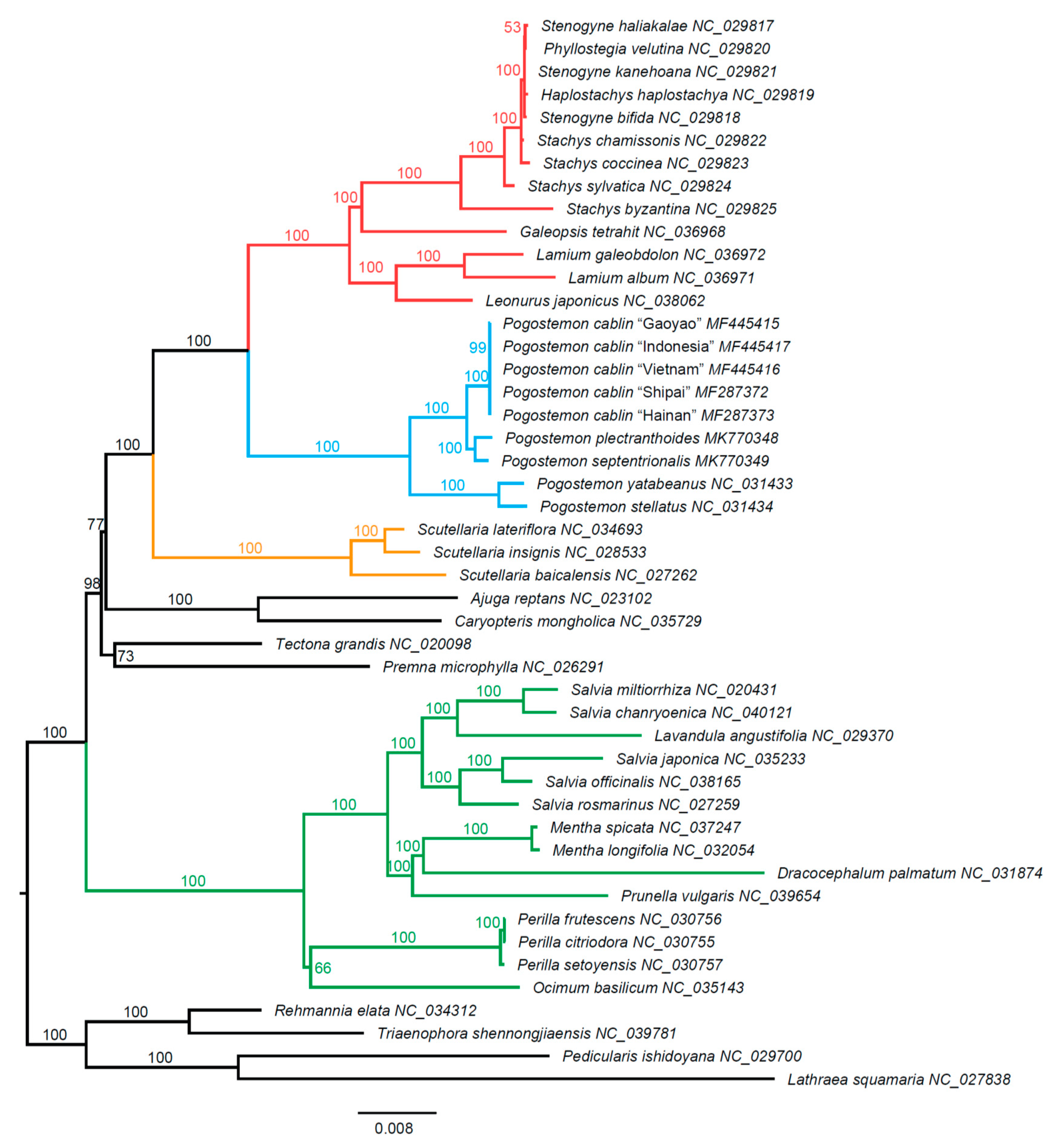
2.3. Phylogenetic Findings
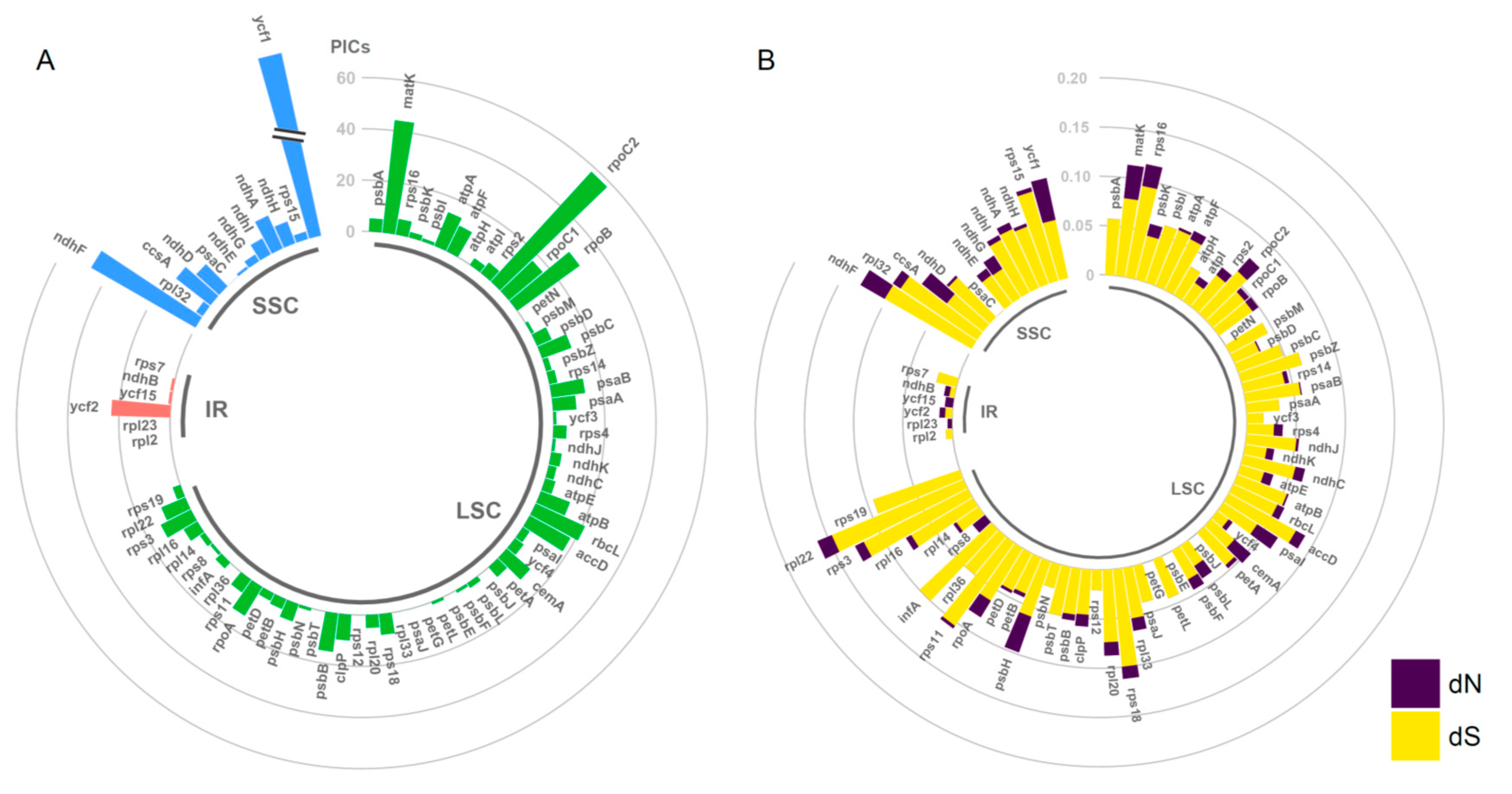
2.4. Estimation of Evolutionary Rates among Protein-Coding Genes
3. Discussion
3.1. Divergence Hotspots in Cp Genomes
3.2. Phylogenetic Positions of Pogostemon and Its Related Taxa
3.3. Substitution Rate Variation in Cp Genomes
4. Materials and Methods
4.1. DNA Extraction and Sequencing
4.2. Assembly and Annotation
4.3. Sequence and Structure Divergence
4.4. Phylogenetic Analyses
4.5. Estimation of Evolutionary Rates
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harley, R.M.; Atkins, S.; Budantsev, A.L.; Cantino, P.D.; Conn, B.J.; Grayer, R.; Harley, M.M.; de Kok, R.; Krestovskaja, T.; Morales, R.; et al. Labiatae. In Flowering Plants Dicotyledons: Lamiales (Except Acanthaceae Including Avicenniaceae); Kadereit, J.W., Ed.; Springer: Berlin/Heidelberg, Germany, 2004; pp. 167–275. [Google Scholar]
- Bhatti, G.R.; Ingrouille, M. Systematics of Pogostemon (Labiatae). Bull. Nat. Hist. Mus. 1997, 27, 77–147. [Google Scholar]
- Yao, G.; Deng, Y.F.; Ge, X.J. A taxonomic revision of Pogostemon (Lamiaceae) from China. Phytotaxa 2015, 200, 1–67. [Google Scholar] [CrossRef] [Green Version]
- Yao, G.; Drew, B.T.; Yi, T.S.; Yan, H.F.; Yuan, Y.M.; Ge, X.J. Phylogenetic relationships, character evolution and biogeographic diversification of Pogostemon s.l. (Lamiaceae). Mol. Phylogen. Evol. 2016, 98, 184–200. [Google Scholar] [CrossRef] [PubMed]
- Swamy, M.K.; Sinniah, U.R. Patchouli (Pogostemon cablin Benth.): Botany, agrotechnology and biotechnological aspects. Ind. Crops Prod. 2016, 87, 161–176. [Google Scholar] [CrossRef]
- Weiss, E.A. Essential Oil Crops; CAB International: Wallingford, UK, 1997. [Google Scholar]
- Kane, N.; Sveinsson, S.; Dempewolf, H.; Yang, J.Y.; Zhang, D.; Engels, J.M.M.; Cronk, Q. Ultra-barcoding in cacao (Theobroma spp.; Malvaceae) using whole chloroplast genomes and nuclear ribosomal DNA. Am. J. Bot. 2012, 99, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Nock, C.J.; Waters, D.L.E.; Edwards, M.A.; Bowen, S.G.; Rice, N.; Cordeiro, G.M.; Henry, R.J. Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol. J. 2011, 9, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Liu, T.J.; Yuan, X.; Huang, H.R.; Yao, G.; Mo, X.L.; Xue, X.; Yan, H.F. The plastid genome and its implications in barcoding specific-chemotypes of the medicinal herb Pogostemon cablin in China. PLoS ONE 2019, 14, e0215512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, D.G.; Kane, N.C.; Ebert, D.P.; Rieseberg, L.H. Genome skimming reveals the origin of the Jerusalem Artichoke tuber crop species: Neither from Jerusalem nor an artichoke. New Phytol. 2014, 201, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Gitzendanner, M.A.; Soltis, P.S.; Yi, T.S.; Li, D.Z.; Soltis, D.E. Plastome phylogenetics: 30 years of inferences into plant evolution. Adv. Bot. Res. 2018, 85, 293–313. [Google Scholar]
- Zhang, M.Y.; Fritsch, P.W.; Ma, P.F.; Wang, H.; Lu, L.; Li, D.Z. Plastid phylogenomics and adaptive evolution of Gaultheria series Trichophyllae (Ericaceae), a clade from sky islands of the Himalaya-Hengduan Mountains. Mol. Phylogen. Evol. 2017, 110, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, E.A.; Wen, J. Using nuclear gene data for plant phylogenetics: Progress and prospects II. Next-gen approaches. J. Syst. Evol. 2015, 53, 371–379. [Google Scholar] [CrossRef]
- McKain, M.R.; Johnson, M.G.; Uribe-Convers, S.; Eaton, D.; Yang, Y. Practical considerations for plant phylogenomics. Appl. Plant Sci. 2018, 6, e1038. [Google Scholar] [CrossRef]
- Shaw, J.; Lickey, E.B.; Beck, J.T.; Farmer, S.B.; Liu, W.; Miller, J.; Siripun, K.C.; Winder, C.T.; Schilling, E.E.; Small, R.L. The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 2005, 92, 142–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef] [Green Version]
- Shaw, J.; Shafer, H.L.; Leonard, O.R.; Kovach, M.J.; Schorr, M.; Morris, A.B. Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: The tortoise and the hare IV. Am. J. Bot. 2014, 101, 1987–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruhlman, T.; Verma, D.; Samson, N.; Daniell, H. The role of heterologous chloroplast sequence elements in transgene integration and expression. Plant Physiol. 2010, 152, 2088–2104. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.F.; Stull, G.W.; Walker-Hale, N.; Vargas, O.M.; Larson, D.A. Characterizing gene tree conflict in plastome-inferred phylogenies. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.P.; Xu, C.; Li, C.H.; Sun, J.H.; Zuo, Y.J.; Shi, S.; Cheng, T.; Guo, J.J.; Zhou, S.L. ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.K.; Ruhlman, T.A. Plastid genomes of seed plants. In Genomics of Chloroplasts and Mitochondria; Bock, R., Knoop, V., Eds.; Springer: Dordrecht, The Netherlands, 2012; pp. 103–126. [Google Scholar]
- Dong, W.P.; Liu, J.; Yu, J.; Wang, L.; Zhou, S.L. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Depamphilis, C.W.; Leebens-Mack, J.; Muller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [Green Version]
- Guisinger, M.M.; Kuehl, J.N.V.; Boore, J.L.; Jansen, R.K. Genome-wide analyses of Geraniaceae plastid DNA reveal unprecedented patterns of increased nucleotide substitutions. Proc. Natl. Acad. Sci. USA 2008, 105, 18424–18429. [Google Scholar] [CrossRef] [Green Version]
- Guisinger, M.M.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: Rearrangements, repeats, and codon usage. Mol. Biol. Evol. 2011, 28, 583–600. [Google Scholar] [CrossRef] [Green Version]
- Decker-Walters, D.S.; Chung, S.M.; Staub, J.E. Plastid sequence evolution: A new pattern of nucleotide substitutions in the Cucurbitaceae. J. Mol. Evol. 2004, 58, 606–614. [Google Scholar] [CrossRef]
- Li, B.; Cantino, P.D.; Olmstead, R.G.; Bramley, G.L.C.; Xiang, C.L.; Ma, Z.H.; Tan, Y.H.; Zhang, D.X. A large-scale chloroplast phylogeny of the Lamiaceae sheds new light on its subfamilial classification. Sci. Rep. 2016, 6, 34343. [Google Scholar] [CrossRef] [Green Version]
- Bendiksby, M.; Thorbek, L.; Scheen, A.C.; Lindqvist, C.; Ryding, O. An updated phylogeny and classification of Lamiaceae subfamily Lamioideae. Taxon 2011, 60, 471–484. [Google Scholar] [CrossRef]
- Gaut, B.; Yang, L.; Takuno, S.; Eguiarte, L.E. The patterns and causes of variation in plant nucleotide substitution rates. Annu. Rev. Ecol. Evol. Syst. 2011, 42, 245–266. [Google Scholar] [CrossRef] [Green Version]
- Wicke, S.; Schneeweiss, G. Next-generation organellar genomics: Potentials and pitfalls of high-throughput technologies for molecular evolutionary studies and plant systematics. In Next Generation Sequencing in Plant Systematics; Hörandl, E., Appelhans, M., Eds.; Koeltz Scientific Books: Konigstein, Germany, 2015. [Google Scholar]
- Bock, D.G.; Andrew, R.L.; Rieseberg, L.H. On the adaptive value of cytoplasmic genomes in plants. Mol. Ecol. 2014, 23, 4899–4911. [Google Scholar] [CrossRef]
- Drouin, G.; Daoud, H.; Xia, J. Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. Phylogen. Evol. 2008, 49, 827–831. [Google Scholar] [CrossRef]
- Park, S.; Ruhlman, T.A.; Weng, M.L.; Hajrah, N.H.; Sabir, J.S.M.; Jansen, R.K. Contrasting patterns of nucleotide substitution rates provide insight into dynamic evolution of plastid and mitochondrial genomes of Geranium. Genome Biol. Evol. 2017, 9, 1766–1780. [Google Scholar] [CrossRef]
- Sloan, D.B.; Triant, D.A.; Forrester, N.J.; Bergner, L.M.; Wu, M.; Taylor, D.R. A recurring syndrome of accelerated plastid genome evolution in the angiosperm tribe Sileneae (Caryophyllaceae). Mol. Phylogen. Evol. 2014, 72, 82–89. [Google Scholar] [CrossRef]
- Wicke, S.; Naumann, J. Molecular evolution of plastid genomes in parasitic flowering plants. In Advances in Botanical Research; Chaw, S.-M., Jansen, R.K., Eds.; Academic Press: London, UK, 2018; Volume 85, pp. 315–347. [Google Scholar]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Andrews, S.; FASTQC. A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 20 September 2019).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [Green Version]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brudno, M.; Malde, S.; Poliakov, A.; Do, C.B.; Couronne, O.; Dubchak, I.; Batzoglou, S. Glocal alignment: Finding rearrangements during alignment. Bioinformatics 2003, 19, i54–i62. [Google Scholar] [CrossRef] [Green Version]
- Sukumaran, J.; Holder, M.T. DendroPy: A python library for phylogenetic computing. Bioinformatics 2010, 26, 1569–1571. [Google Scholar] [CrossRef] [Green Version]
- Šmarda, P.; Bureš, P. The Variation of Base Composition in Plant Genomes. In Plant Genomes, Their Residents, and Their Evolutionary Dynamics; Wendel, J.F., Greilhuber, J., Dolezel, J., Leitch, I.J., Eds.; Springer: Vienna, Austria, 2012; pp. 209–235. [Google Scholar] [CrossRef]
- Team, R.D.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Goldman, N.; Yang, Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 1994, 11, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.H. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Feature | P. plectranthoides | P. septentrionalis | P. yatabeanus | P. stellatus | P. cablin “Gaoyao” |
|---|---|---|---|---|---|
| Genome size | 152,430 | 152,514 | 152,707 | 151,824 | 152,461 |
| LSC length | 83,514 | 83,614 | 83,791 | 83,012 | 83,553 |
| IR length | 25,666 | 25,665 | 25,674 | 25,644 | 25,662 |
| SSC length | 17,584 | 17,570 | 17,568 | 17,524 | 17,584 |
| Total coding length | 91,424 | 91,445 | 91,132 | 91,135 | 91,442 |
| Protein-coding length | 79,500 | 79,521 | 79,275 | 79,278 | 79,518 |
| rRNA-coding length | 9064 | 9064 | 9064 | 9064 | 9064 |
| tRNA-coding length | 2860 | 2860 | 2793 | 2793 | 2860 |
| Total GC content (%) | 38.3 | 38.2 | 38.2 | 38.2 | 38.2 |
| LSC GC content (%) | 36.4 | 36.4 | 36.2 | 36.3 | 36.4 |
| IR GC content (%) | 43.4 | 43.4 | 43.4 | 43.4 | 43.4 |
| SSC GC content (%) | 32.1 | 32.1 | 32 | 32.1 | 32.1 |
| Total number of genes (total/different) | 132/114 | 132/114 | 132/114 | 132/114 | 132/114 |
| Number of duplicated genes in IR | 18 | 18 | 18 | 18 | 18 |
| Number of genes with introns (with 3 exons) | 18(2) | 18(2) | 18(2) | 18(2) | 18(2) |
| Number of protein-coding genes (total/in IR) | 80/7 | 80/7 | 80/7 | 80/7 | 80/7 |
| Number of tRNA genes (total/in IR) | 30/7 | 30/7 | 30/7 | 30/7 | 30/7 |
| Number of rRNA genes (total/in IR) | 4/4 | 4/4 | 4/4 | 4/4 | 4/4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.-Y.; Liu, T.-J.; Mo, X.-L.; Huang, H.-R.; Yao, G.; Li, J.-R.; Ge, X.-J.; Yan, H.-F. Comparative Analyses of the Chloroplast Genomes of Patchouli Plants and Their Relatives in Pogostemon (Lamiaceae). Plants 2020, 9, 1497. https://doi.org/10.3390/plants9111497
Zhang C-Y, Liu T-J, Mo X-L, Huang H-R, Yao G, Li J-R, Ge X-J, Yan H-F. Comparative Analyses of the Chloroplast Genomes of Patchouli Plants and Their Relatives in Pogostemon (Lamiaceae). Plants. 2020; 9(11):1497. https://doi.org/10.3390/plants9111497
Chicago/Turabian StyleZhang, Cai-Yun, Tong-Jian Liu, Xiao-Lu Mo, Hui-Run Huang, Gang Yao, Jian-Rong Li, Xue-Jun Ge, and Hai-Fei Yan. 2020. "Comparative Analyses of the Chloroplast Genomes of Patchouli Plants and Their Relatives in Pogostemon (Lamiaceae)" Plants 9, no. 11: 1497. https://doi.org/10.3390/plants9111497
APA StyleZhang, C. -Y., Liu, T. -J., Mo, X. -L., Huang, H. -R., Yao, G., Li, J. -R., Ge, X. -J., & Yan, H. -F. (2020). Comparative Analyses of the Chloroplast Genomes of Patchouli Plants and Their Relatives in Pogostemon (Lamiaceae). Plants, 9(11), 1497. https://doi.org/10.3390/plants9111497