Transgenerational Effects of Water-Deficit and Heat Stress on Germination and Seedling Vigour—New Insights from Durum Wheat microRNAs




Abstract
:1. Introduction
2. Results
2.1. Evaluation of Germination and Seedling Establishment
2.2. The microRNAome in the Progenies
2.3. Differentially Expressed miRNAs (DEMs)
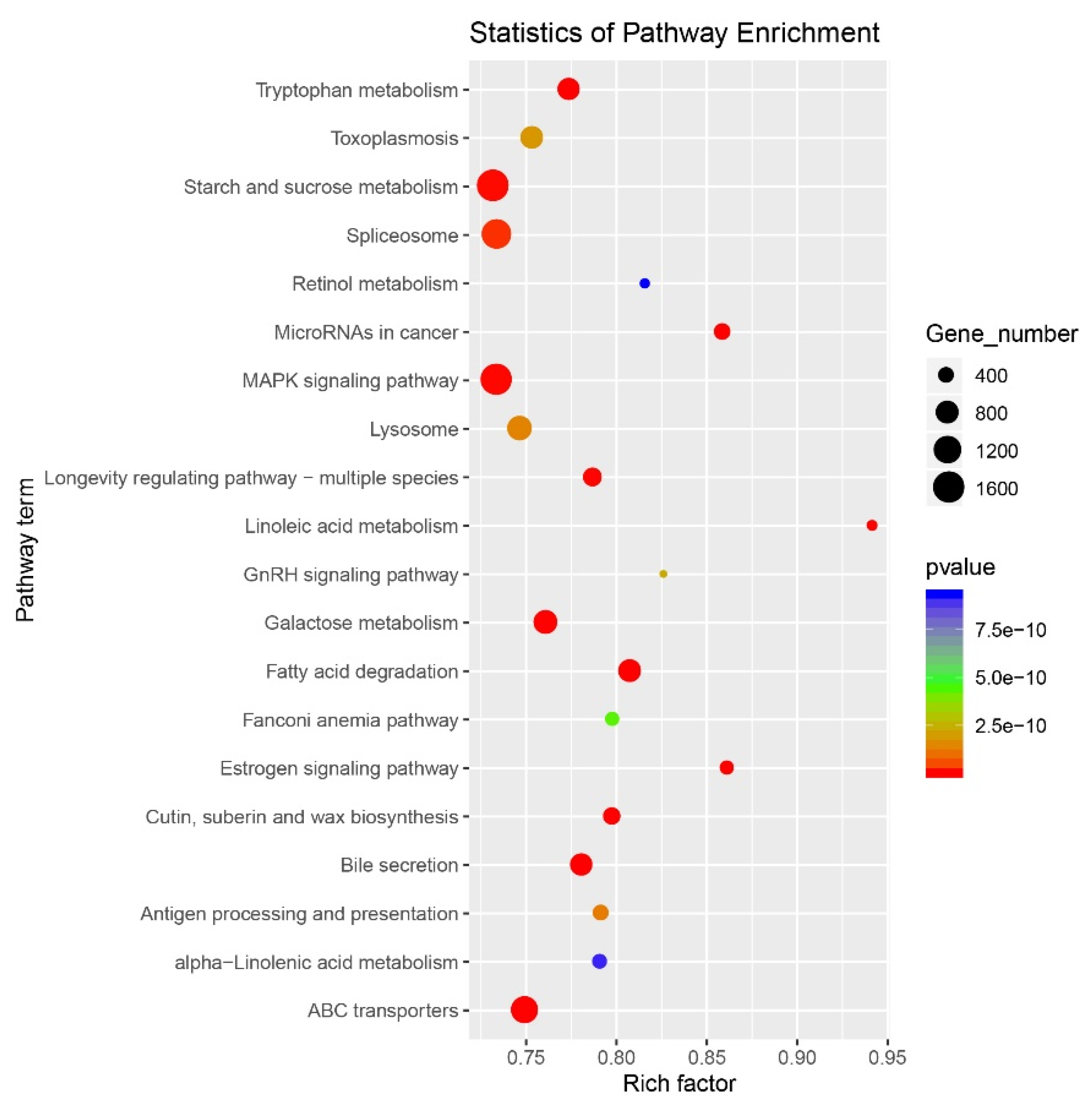
2.4. Target Prediction and Enrichment Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Germination Test
4.3. Measurement of Seedling Establishment
4.4. Small RNA Sequencing
4.5. Analysis of Conserved and Novel miRNAs
4.6. Target Prediction and Functional GO Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Richards, R.A.; Rebetzke, G.J.; Condon, A.G.; van Herwaarden, A.F. Breeding opportunities for increasing the efficiency of water use and crop yield in temperate cereals. Crop Sci. 2002, 42, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Fillery, I.R.; Palta, J.A. Early vigorous growth is a major factor influencing nitrogen uptake in wheat. Funct. Plant Biol. 2004, 31, 121–129. [Google Scholar] [CrossRef]
- Perveen, S.; Nigar, S.; Khalil, S.; Zubair, M. Vigor tests used to rank seed lot quality and predict field emergence in wheat. Pak. J. Bot. 2010, 42, 3147–3155. [Google Scholar]
- Sissons, M. Role of durum wheat composition on the quality of pasta and bread. Food 2008, 2, 75–90. [Google Scholar]
- Li, Y.-F.; Wu, Y.; Hernandez-Espinosa, N.; Peña, R.J. Heat and drought stress on durum wheat: Responses of genotypes, yield, and quality parameters. J. Cereal Sci. 2013, 57, 398–404. [Google Scholar] [CrossRef]
- Liu, H.; Bruce, D.R.; Sissons, M.; Able, A.J.; Able, J.A. Genotype-dependent changes in the phenolic content of durum under water-deficit stress. Cereal Chem. 2018, 95, 59–78. [Google Scholar] [CrossRef]
- Guzmán, C.; Autrique, J.E.; Mondal, S.; Singh, R.P.; Govindan, V.; Morales-Dorantes, A.; Posadas-Romano, G.; Crossa, J.; Ammar, K.; Peña, R.J. Response to drought and heat stress on wheat quality, with special emphasis on bread-making quality, in durum wheat. Field Crop. Res. 2016, 186, 157–165. [Google Scholar] [CrossRef]
- Liu, H.; Able, A.J.; Able, J.A. Genotypic performance of Australian durum under single and combined water-deficit and heat stress during reproduction. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Searle, I.R.; Mather, D.E.; Able, A.J.; Able, J.A. Morphological, physiological and yield responses of durum wheat to pre-anthesis water-deficit stress are genotype-dependent. Crop Pasture Sci. 2015, 66, 1024–1038. [Google Scholar] [CrossRef]
- Hatzig, S.V.; Nuppenau, J.-N.; Snowdon, R.J.; Schießl, S.V. Drought stress has transgenerational effects on seeds and seedlings in winter oilseed rape (Brassica napus L.). BMC Plant Biol. 2018, 18, 297. [Google Scholar] [CrossRef]
- Wijewardana, C.; Reddy, K.R.; Krutz, L.J.; Gao, W.; Bellaloui, N. Drought stress has transgenerational effects on soybean seed germination and seedling vigor. PLoS ONE 2019, 14, e0214977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begcy, K.; Sandhu, J.; Walia, H. Transient heat stress during early seed development primes germination and seedling establishment in rice. Front. Plant Sci. 2018, 9, 1768. [Google Scholar] [CrossRef]
- Carvalho, M.E.A.; Piotto, F.A.; Nogueira, M.L.; Gomes-Junior, F.G.; Chamma, H.M.C.P.; Pizzaia, D.; Azevedo, R.A. Cadmium exposure triggers genotype-dependent changes in seed vigor and germination of tomato offspring. Protoplasma 2018, 255, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Wollmann, H.; Berger, F. Epigenetic reprogramming during plant reproduction and seed development. Curr. Opin. Plant Biol. 2012, 15, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Lämke, J.; Bäurle, I. Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol. 2017, 18, 124. [Google Scholar] [CrossRef]
- Holeski, L.M.; Jander, G.; Agrawal, A.A. Transgenerational defense induction and epigenetic inheritance in plants. Trends Ecol. Evol. 2012, 27, 618–626. [Google Scholar] [CrossRef]
- Liu, H.; Able, A.J.; Able, J.A. SMARTER de-stressed cereal breeding. Trends Plant Sci. 2016, 21, 909–925. [Google Scholar] [CrossRef]
- Budak, H.; Akpinar, B.A. Plant miRNAs: Biogenesis, organization and origins. Funct. Integr. Genom. 2015, 15, 523–531. [Google Scholar] [CrossRef]
- Budak, H.; Kantar, M.; Bulut, R.; Akpinar, B.A. Stress responsive miRNAs and isomiRs in cereals. Plant Sci. 2015, 235, 1–13. [Google Scholar] [CrossRef]
- Giusti, L.; Mica, E.; Bertolini, E.; De Leonardis, A.M.; Faccioli, P.; Cattivelli, L.; Crosatti, C. microRNAs differentially modulated in response to heat and drought stress in durum wheat cultivars with contrasting water use efficiency. Funct. Integr. Genom. 2017, 17, 293–309. [Google Scholar] [CrossRef]
- Zuluaga, D.L.; De Paola, D.; Janni, M.; Curci, P.L.; Sonnante, G. Durum wheat miRNAs in response to nitrogen starvation at the grain filling stage. PLoS ONE 2017, 12, e0183253. [Google Scholar] [CrossRef]
- Liu, H.; Searle, I.R.; Watson-Haigh, N.S.; Baumann, U.; Mather, D.E.; Able, A.J.; Able, J.A. Genome-wide identification of microRNAs in leaves and the developing head of four durum genotypes during water deficit stress. PLoS ONE 2015, 10, e0142799. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Able, A.J.; Able, J.A. Genotypic water-deficit stress responses in durum wheat: Association between physiological traits, microRNA regulatory modules and yield components. Funct. Plant Biol. 2017, 44, 538–551. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Able, A.J.; Able, J.A. Water-deficit stress responsive microRNAs and their targets in four durum wheat genotypes. Funct. Integr. Genom. 2017, 17, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, K.; Chen, L.; Zou, Y.; Liu, H.; Tian, Y.; Li, D.; Wang, R.; Zhao, F.; Ferguson, B.J.; et al. microRNA167-directed regulation of the auxin response factors, GmARF8a and GmARF8b, is required for soybean nodulation and lateral root development. Plant Physiol. 2015, 168, 984–999. [Google Scholar] [CrossRef] [Green Version]
- Zuluaga, D.L.; Sonnante, G. The use of nitrogen and its regulation in cereals: Structural genes, transcription factors, and the role of miRNAs. Plants 2019, 8, 294. [Google Scholar] [CrossRef] [Green Version]
- Ragupathy, R.; Ravichandran, S.; Mahdi, M.S.R.; Huang, D.; Reimer, E.; Domaratzki, M.; Cloutier, S. Deep sequencing of wheat sRNA transcriptome reveals distinct temporal expression pattern of miRNAs in response to heat, light and UV. Sci. Rep. 2016, 6, 39373. [Google Scholar] [CrossRef] [Green Version]
- Ravichandran, S.; Ragupathy, R.; Edwards, T.; Domaratzki, M.; Cloutier, S. MicroRNA-guided regulation of heat stress response in wheat. BMC Genom. 2019, 20, 488. [Google Scholar] [CrossRef] [Green Version]
- Yakovlev, I.A.; Fossdal, C.G. In silico analysis of small RNAs suggest roles for novel and conserved miRNAs in the formation of epigenetic memory in somatic embryos of Norway spruce. Front Physiol. 2017, 8, 674. [Google Scholar] [CrossRef]
- Zuluaga, D.L.; Liuzzi, V.; Curci, P.L.; Sonnante, G. MicroRNAs in durum wheat seedlings under chronic and short-term nitrogen stress. Funct. Integr. Genom. 2018, 18, 645–657. [Google Scholar] [CrossRef]
- Kantar, M.; Lucas, S.J.; Budak, H. miRNA expression patterns of Triticum dicoccoides in response to shock drought stress. Planta 2011, 233, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Akpinar, B.A.; Kantar, M.; Budak, H. Root precursors of microRNAs in wild emmer and modern wheats show major differences in response to drought stress. Funct. Integr. Genom. 2015, 15, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Maccaferri, M.; Harris, N.S.; Twardziok, S.O.; Pasam, R.K.; Gundlach, H.; Spannagl, M.; Ormanbekova, D.; Lux, T.; Prade, V.M.; Milner, S.G. Durum wheat genome highlights past domestication signatures and future improvement targets. Nat. Genet. 2019, 51, 885–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Paola, D.; Zuluaga, D.L.; Sonnante, G. The miRNAome of durum wheat: Isolation and characterisation of conserved and novel microRNAs and their target genes. BMC Genom. 2016, 17, 505. [Google Scholar] [CrossRef] [Green Version]
- Jeyaraj, A.; Zhang, X.; Hou, Y.; Shangguan, M.; Gajjeraman, P.; Li, Y.; Wei, C. Genome-wide identification of conserved and novel microRNAs in one bud and two tender leaves of tea plant (Camellia sinensis) by small RNA sequencing, microarray-based hybridization and genome survey scaffold sequences. BMC Plant Biol. 2017, 17, 212. [Google Scholar] [CrossRef] [Green Version]
- Changhai, S.; Baodi, D.; Yunzhou, Q.; Yuxin, L.; Lei, S.; Mengyu, L.; Haipei, L. Physiological regulation of high transpiration efficiency in winter wheat under drought conditions. Plant Soil Environ. 2010, 56, 340–347. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Liu, H.; Ji, H.; Wang, Y.; Dong, B.; Qiao, Y.; Liu, M.; Li, X. The wheat GT factor TaGT2L1D negatively regulates drought tolerance and plant development. Sci. Rep. 2016, 6, 27042. [Google Scholar] [CrossRef] [Green Version]
- Dong, B.; Zheng, X.; Liu, H.; Able, J.A.; Yang, H.; Zhao, H.; Zhang, M.; Qiao, Y.; Wang, Y.; Liu, M. Effects of drought stress on pollen sterility, grain yield, abscisic acid and protective enzymes in two winter wheat cultivars. Front. Plant Sci. 2017, 8, 1008. [Google Scholar] [CrossRef]
- Dong, B.; Yang, H.; Liu, H.; Qiao, Y.; Zhang, M.; Wang, Y.; Xie, Z.; Liu, M. Effects of shading stress on grain number, yield, and photosynthesis during early reproductive growth in wheat. Crop Sci. 2019, 59, 363–378. [Google Scholar] [CrossRef]
- Barnabás, B.; Jäger, K.; Fehér, A. The effect of drought and heat stress on reproductive processes in cereals. Plant Cell Environ. 2008, 31, 11–38. [Google Scholar] [CrossRef]
- Grass, L.; Burris, J. Effect of heat stress during seed development and maturation on wheat (Triticum durum) seed quality. I. Seed germination and seedling vigor. Can. J. Plant Sci. 1995, 75, 821–829. [Google Scholar] [CrossRef] [Green Version]
- Alptekin, B.; Langridge, P.; Budak, H. Abiotic stress miRNomes in the Triticeae. Funct. Integr. Genom. 2016, 117, 145–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2018, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Kwok, R.P.; Lundblad, J.R.; Chrivia, J.C.; Richards, J.P.; Bächinger, H.P.; Brennan, R.G.; Roberts, S.G.; Green, M.R.; Goodman, R.H. Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature 1994, 370, 223. [Google Scholar] [CrossRef]
- Gehring, C. Adenyl cyclases and cAMP in plant signaling-past and present. Cell Commum Signal. 2010, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Colcombet, J.; Krysan, P.J. Cellular complexity in MAPK signaling in plants: Questions and emerging tools to answer them. Front. Plant Sci. 2018, 9, 1674. [Google Scholar]
- Smékalová, V.; Doskočilová, A.; Komis, G.; Šamaj, J. Crosstalk between secondary messengers, hormones and MAPK modules during abiotic stress signalling in plants. Biotechnol. Adv. 2014, 32, 2–11. [Google Scholar] [CrossRef]
- Wang, X.; Xin, C.; Cai, J.; Zhou, Q.; Dai, T.; Cao, W.; Jiang, D. Heat priming induces trans-generational tolerance to high temperature stress in wheat. Front. Plant Sci. 2016, 7, 501. [Google Scholar] [CrossRef] [Green Version]
- Šamaj, J.; Ovecka, M.; Hlavacka, A.; Lecourieux, F.; Meskiene, I.; Lichtscheidl, I.; Lenart, P.; Salaj, J.; Volkmann, D.; Bögre, L. Involvement of the mitogen-activated protein kinase SIMK in regulation of root hair tip growth. EMBO J. 2002, 21, 3296–3306. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Moore, M.J. Spliceosomes. Curr. Biol. 2015, 25, R181–R183. [Google Scholar] [CrossRef] [Green Version]
- Ding, F.; Cui, P.; Wang, Z.; Zhang, S.; Ali, S.; Xiong, L. Genome-wide analysis of alternative splicing of pre-mRNA under salt stress in Arabidopsis. BMC Genom. 2014, 15, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, Y.; Alshareef, S.; Butt, H.; Lozano-Juste, J.; Li, L.; Galal, A.A.; Moustafa, A.; Momin, A.A.; Tashkandi, M.; Richardson, D.N. Pre-mRNA splicing repression triggers abiotic stress signaling in plants. Plant J. 2017, 89, 291–309. [Google Scholar] [CrossRef] [PubMed]
- Laloum, T.; Martín, G.; Duque, P. Alternative splicing control of abiotic stress responses. Trends Plant Sci. 2018, 23, 140–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, K.; Liu, P.; Wu, C.-A.; Yang, G.-D.; Xu, R.; Guo, Q.-H.; Huang, J.-G.; Zheng, C.-C. Stress-induced alternative splicing provides a mechanism for the regulation of microRNA processing in Arabidopsis thaliana. Mol. Cell 2012, 48, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Kruszka, K.; Pacak, A.; Swida-Barteczka, A.; Nuc, P.; Alaba, S.; Wroblewska, Z.; Karlowski, W.; Jarmolowski, A.; Szweykowska-Kulinska, Z. Transcriptionally and post-transcriptionally regulated microRNAs in heat stress response in barley. J. Exp. Bot. 2014, 65, 6123–6135. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; Qu, G.; Wang, T.; Sun, Q.; Liang, D.; Hu, S. Enhancement of germination and seedling growth of wheat seed using dielectric barrier discharge plasma with various gas sources. Plasma Chem. Plasma Process. 2017, 37, 1105–1119. [Google Scholar] [CrossRef]
- Zahoranová, A.; Henselová, M.; Hudecová, D.; Kaliňáková, B.; Kováčik, D.; Medvecká, V.; Černák, M. Effect of cold atmospheric pressure plasma on the wheat seedlings vigor and on the inactivation of microorganisms on the seeds surface. Plasma Chem. Plasma Process. 2016, 36, 397–414. [Google Scholar] [CrossRef]
- Feng, H.; Zhang, Q.; Wang, B.; Fu, Y.; Huang, L.; Wang, X.; Kang, Z. Exploration of microRNAs and their targets engaging in the resistance interaction between wheat and stripe rust. Frontiers in Plant Science 2015, 6, 469. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Shahid, M.Q.; Wu, J.; Wang, L.; Liu, X.; Lu, Y. Comparative small RNA analysis of pollen development in autotetraploid and diploid rice. Int. J. Mol. 2016, 17, 499. [Google Scholar] [CrossRef]
- Li, W.; Jia, Y.; Liu, F.; Wang, F.; Fan, F.; Wang, J.; Zhu, J.; Xu, Y.; Zhong, W.; Yang, J. Integration analysis of small RNA and degradome sequencing reveals microRNAs responsive to Dickeya zeae in resistant rice. Int. J. Mol. 2019, 20, 222. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Seed Source | Gp, % | Gr, % | Gi | Coleoptile Length (mm) | Young Root Length (mm) |
|---|---|---|---|---|---|
| AuCG | 32.2% ± 1.1% | 98.9% ± 1.1% | 35.1 ± 0.8 | 31.88 ± 0.52 | 48.56 ± 1.06 |
| AuWH | 26.7% ± 1.9% | 97.8% ± 1.1% | 32.2 ± 0.9 | 31.06 ± 0.67 | 50.44 ± 1.19 |
| L6CG | 17.8% ± 1.1% | 93.3% ± 1.9% | 27.6 ± 0.2 | 30.38 ± 0.75 | 38.88 ± 1.28 |
| L6WH | 14.4% ± 1.1% | 92.2% ± 1.1% | 24.4 ± 0.6 | 30.50 ± 0.75 | 33.75 ± 1.23 |
| F pr. Genotype | <0.001 | 0.004 | <0.001 | 0.134 | <0.001 |
| F pr. Parent treatment | 0.011 | 0.438 | 0.002 | 0.614 | 0.178 |
| F pr. Genotype × Parent treatment | 0.438 | 1.000 | 0.842 | 0.493 | 0.005 |
| l.s.d Genotype | 3.1% | 3.1% | 1.6 | n.a | 2.39 |
| l.s.d Parent treatment | 3.1% | n.a | 1.6 | n.a | n.a |
| l.s.d Genotype × Parent treatment | n.a | n.a | n.a | n.a | 3.38 |
| Seed Source | Root Fresh Weight (g) | Root Dry Weight (g) | Root Length (cm) | Shoot Fresh Weight (g) | Shoot Dry Weight (g) | Seedling Height (cm) | Seedling Vigour Index I | Seedling Vigour Index II |
|---|---|---|---|---|---|---|---|---|
| AuCG | 0.718 ± 0.016 | 0.0807 ± 0.0016 | 25.86 ± 0.18 | 0.878 ± 0.025 | 0.1244 ± 0.0030 | 27.33 ± 0.26 | 1577.28 ± 30.48 | 202.80 ± 2.42 |
| AuWH | 0.736 ± 0.014 | 0.0812 ± 0.0022 | 28.46 ± 0.22 | 0.836 ± 0.022 | 0.1189 ± 0.0023 | 26.36 ± 0.23 | 1537.56 ± 28.01 | 195.63 ± 3.70 |
| L6CG | 0.710 ± 0.013 | 0.0721 ± 0.0017 | 25.10 ± 0.19 | 0.844 ± 0.020 | 0.1141 ± 0.0025 | 26.98 ± 0.24 | 1450.17 ± 21.74 | 173.78 ± 2.92 |
| L6WH | 0.658 ± 0.014 | 0.0645 ± 0.0014 | 26.56 ± 0.21 | 0.778 ± 0.016 | 0.1116 ± 0.0033 | 25.39 ± 0.31 | 1323.39 ± 16.81 | 162.32 ± 3.79 |
| F pr. Genotype | 0.005 | <0.001 | <0.001 | 0.034 | 0.004 | 0.018 | <0.001 | <0.001 |
| F pr. Parent treatment | 0.242 | 0.055 | <0.001 | 0.015 | 0.160 | <0.001 | 0.002 | 0.008 |
| F pr. Genotype × Parent treatment | 0.018 | 0.030 | 0.008 | 0.551 | 0.608 | 0.245 | 0.091 | 0.516 |
| l.s.d Genotype | 0.029 | 0.0036 | 0.41 | 0.043 | 0.0057 | 0.54 | 50.90 | 6.67 |
| l.s.d Parent treatment | n.a | n.a | 0.41 | 0.043 | n.a | 0.54 | 50.90 | 6.67 |
| l.s.d Genotype × Parent treatment | 0.041 | 0.0051 | 0.58 | n.a | n.a | 0.76 | n.a | n.a |
| Read Type | TCG | TWH | SCG | SWH | ||||
|---|---|---|---|---|---|---|---|---|
| Total Reads | Unique Reads | Total Reads | Unique Reads | Total Reads | Unique Reads | Total Reads | Unique Reads | |
| Raw reads | 20,042,833 | 8,293,425 | 20,900,186 | 8,593,357 | 24,262,343 | 8,865,724 | 18,786,468 | 7,221,909 |
| 3ADT & Length filter | 6,000,364 | 1,418,893 | 5,651,835 | 1,475,235 | 7,669,010 | 1,832,961 | 6,589,933 | 1,676,502 |
| Junk reads | 92,538 | 68,473 | 101,066 | 73,192 | 111,339 | 79,088 | 76,371 | 56,322 |
| Rfam - rRNA | 215,716 | 5440 | 228,715 | 5378 | 308,312 | 6809 | 266,089 | 7444 |
| Rfam - tRNA | 230,895 | 2119 | 215,340 | 2207 | 248,091 | 2853 | 310,937 | 3964 |
| Rfam - snoRNA | 9600 | 393 | 9813 | 396 | 10,886 | 363 | 8009 | 304 |
| Rfam - snRNA | 2364 | 181 | 2581 | 194 | 3250 | 264 | 3580 | 215 |
| Other Rfam RNA | 13,680 | 733 | 14,276 | 706 | 19,445 | 886 | 17,165 | 893 |
| mRNA | 878,039 | 23,905 | 915,468 | 25,170 | 1,090,076 | 26,584 | 798,629 | 20,880 |
| Repeats | 6104 | 186 | 6073 | 193 | 9312 | 265 | 7704 | 243 |
| Clean reads | 12,631,656 | 6,773,722 | 13,795,248 | 7,011,288 | 14,830,872 | 6,916,493 | 10,781,672 | 5,456,127 |
| Group | Total | TCG | TWH | SCG | SWH | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pre-miRNA | Unique miRNA | Pre-miRNA | Unique miRNA | Pre-miRNA | Unique miRNA | Pre-miRNA | Unique miRNA | Pre-miRNA | Unique miRNA | |
| G1 | 161 | 270 | 158 | 252 | 158 | 256 | 158 | 259 | 158 | 257 |
| G2 | 446 | 483 | 368 | 392 | 372 | 405 | 382 | 410 | 300 | 322 |
| G3 | 838 | 890 | 663 | 694 | 701 | 736 | 722 | 763 | 611 | 644 |
| G4 | 48 | 49 | 34 | 34 | 31 | 33 | 42 | 43 | 34 | 34 |
| G5 | 437 | 485 | 426 | 446 | 429 | 445 | 429 | 455 | 421 | 440 |
| miR_name | miR_seq | up/down | log2(FC) | TCG | TWH |
| osa-miR168a-3p_L-3 | CCCGCCTTGCACCAAGTGAAT | up | 2.04 | 3571 | 14,660 |
| ata-miR169a-3p | TGGGCAAGTCACCCTGGCTACC | up | 1.11 | 533 | 1154 |
| miR_name | miR_seq | up/down | log2(FC) | SCG | SWH |
| tae-miR9661-5p_1ss14GA | TGAAGTAGAGCAGAGACCTCA | down | −1.06 | 3513 | 1689 |
| tae-miR9774_L+2 | AACAAGATATTGGGTATTTCTGTC | up | 1.27 | 1589 | 3841 |
| miR_name | miR_seq | up/down | log2(FC) | TCG | SCG |
| ata-miR167b-5p | TGAAGCTGCCAGCATGATCTGA | down | −1.21 | 20,089 | 8687 |
| tae-MIR7757-p3_1ss19AG | TGGATAGTTTGAGGTTTTGTTT | down | −1.04 | 6524 | 3162 |
| ata-miR167b-3p | AGGTCATGCTGGAGTTTCATC | down | −1.11 | 5746 | 2653 |
| ttu-miR160 | TGCCTGGCTCCCTGTATGCCA | down | −1.04 | 4739 | 2304 |
| mtr-miR398a-5p_1ss7AC | GGAGTGCCACTGAGAACACAAG | down | −1.36 | 2354 | 917 |
| csi-miR166c-3p_R+1 | TCGGACCAGGCTTCATTCCCA | down | −1.52 | 1062 | 370 |
| tae-miR9654b-3p_1ss19GA | TTCCGAAAGGCTTGAAGCAAAT | down | −1.10 | 1542 | 721 |
| ata-miR396e-3p | GTTCAATAAAGCTGTGGGAAA | down | −1.10 | 737 | 345 |
| sbi-miR396c_R+1 | TTCCACAGCTTTCTTGAACTTT | down | −1.07 | 758 | 361 |
| PC-5p-1206_1154 | TTTGCGCAGAAGGGAGAAATC | up | inf | 0 | 2228 |
| tae-MIR5048-p3_1ss18TG | AATATATTTGCAGGTTTGAGG | up | 1.08 | 4677 | 9898 |
| ata-miR169f-3p_R-1 | GGCAAGTCCGTCCTTGGCTAC | up | 1.22 | 4609 | 10,772 |
| osa-miR168a-3p_L-3 | CCCGCCTTGCACCAAGTGAAT | up | 1.26 | 3571 | 8570 |
| ata-MIR169d-p3 | TTGTCCTTGGCTACACCTAGT | up | 1.50 | 1742 | 4936 |
| tae-miR9661-5p_1ss14GA | TGAAGTAGAGCAGAGACCTCA | up | 3.49 | 312 | 3513 |
| ata-MIR396d-p3 | AAGCCCATGGAAACCATGCCC | up | 4.66 | 29 | 735 |
| ata-miR169i-5p_1ss15TC | TAGCCAAGGATGACCTGCCTG | up | 3.39 | 53 | 553 |
| ata-miR169a-3p | TGGGCAAGTCACCCTGGCTACC | up | 1.49 | 533 | 1494 |
| PC-5p-726_2034 | TACGGCAAAGCCGTCGGCATA | up | 2.03 | 194 | 793 |
| ata-miR169h-3p_L-1 | CAAGTTGTTCTTGGCTAGC | up | 1.25 | 484 | 1154 |
| tae-MIR164-p3 | CATGTGCCTTTCTTCTCCACC | up | 1.02 | 419 | 848 |
| miR_name | miR_seq | up/down | log2(FC) | TWH | SWH |
| mtr-miR398a-5p_1ss7AC | GGAGTGCCACTGAGAACACAAG | down | −1.69 | 1516 | 471 |
| tae-miR9659-3p | TCCAATGGTTGTTCACGGCATC | down | −1.17 | 910 | 404 |
| osa-miR160f-5p_L-2R+2 | CCTGGCTCCCTGAATGCCATC | down | −1.14 | 817 | 370 |
| sbi-miR396c_R+1 | TTCCACAGCTTTCTTGAACTTT | down | −1.12 | 609 | 280 |
| PC-5p-1206_1154 | TTTGCGCAGAAGGGAGAAATC | up | inf | 0 | 2473 |
| ata-miR169f-3p_R-1 | GGCAAGTCCGTCCTTGGCTAC | up | 1.14 | 4103 | 9018 |
| tae-miR9774_L+2 | AACAAGATATTGGGTATTTCTGTC | up | 1.57 | 1296 | 3841 |
| tae-miR9661-5p_1ss14GA | TGAAGTAGAGCAGAGACCTCA | up | 2.55 | 288 | 1689 |
| ata-MIR396d-p3 | AAGCCCATGGAAACCATGCCC | up | 4.97 | 23 | 710 |
| ata-miR169i-5p_1ss15TC | TAGCCAAGGATGACCTGCCTG | up | 2.52 | 82 | 469 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Able, A.J.; Able, J.A. Transgenerational Effects of Water-Deficit and Heat Stress on Germination and Seedling Vigour—New Insights from Durum Wheat microRNAs. Plants 2020, 9, 189. https://doi.org/10.3390/plants9020189
Liu H, Able AJ, Able JA. Transgenerational Effects of Water-Deficit and Heat Stress on Germination and Seedling Vigour—New Insights from Durum Wheat microRNAs. Plants. 2020; 9(2):189. https://doi.org/10.3390/plants9020189
Chicago/Turabian StyleLiu, Haipei, Amanda J. Able, and Jason A. Able. 2020. "Transgenerational Effects of Water-Deficit and Heat Stress on Germination and Seedling Vigour—New Insights from Durum Wheat microRNAs" Plants 9, no. 2: 189. https://doi.org/10.3390/plants9020189
APA StyleLiu, H., Able, A. J., & Able, J. A. (2020). Transgenerational Effects of Water-Deficit and Heat Stress on Germination and Seedling Vigour—New Insights from Durum Wheat microRNAs. Plants, 9(2), 189. https://doi.org/10.3390/plants9020189