The Complete Chloroplast Genome Sequencing and Comparative Analysis of Reed Canary Grass (Phalaris arundinacea) and Hardinggrass (P. aquatica)



Abstract
:1. Introduction
2. Results
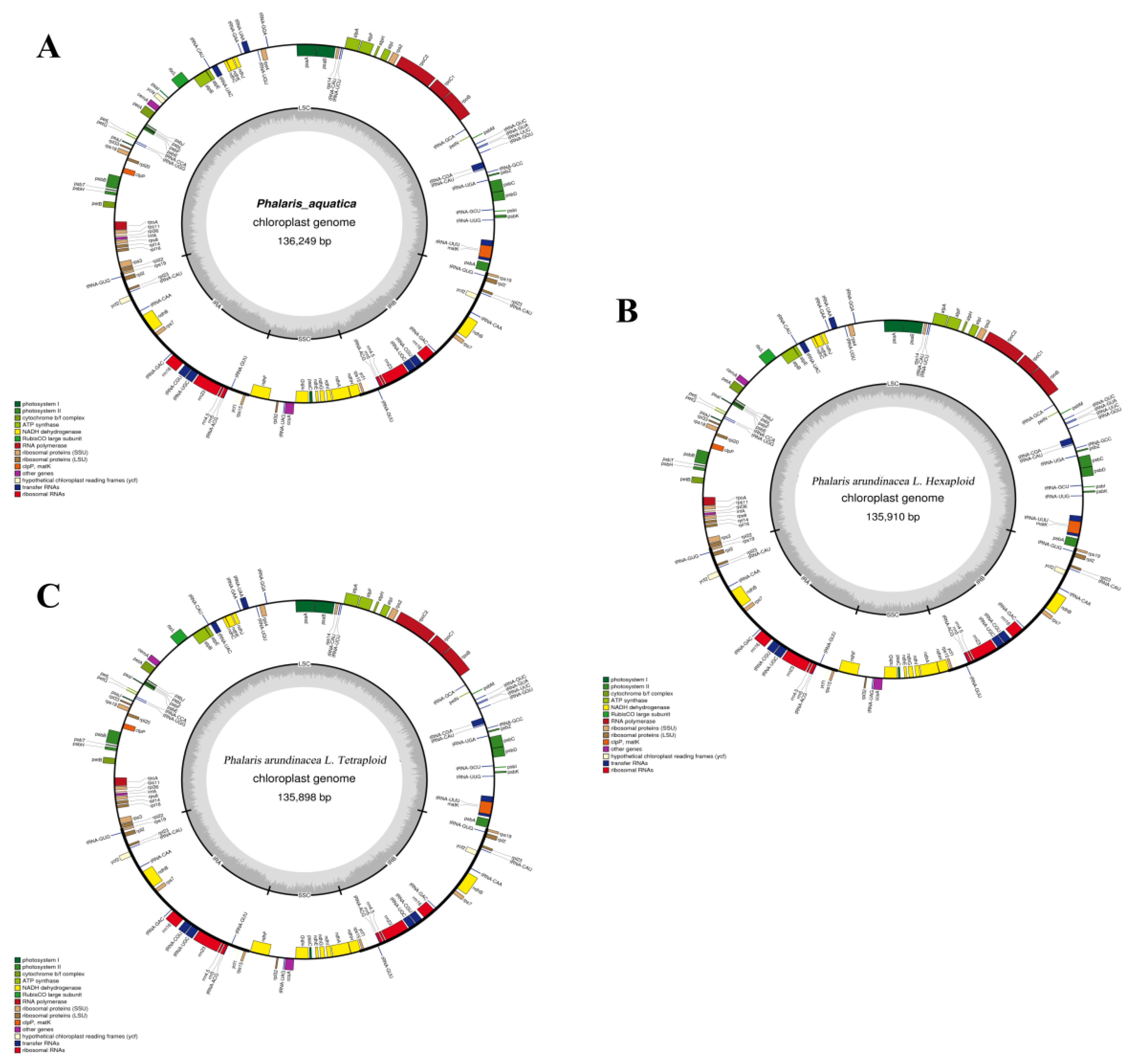
2.1. Genomic Features of Three Phalaris Chloroplast Genomes
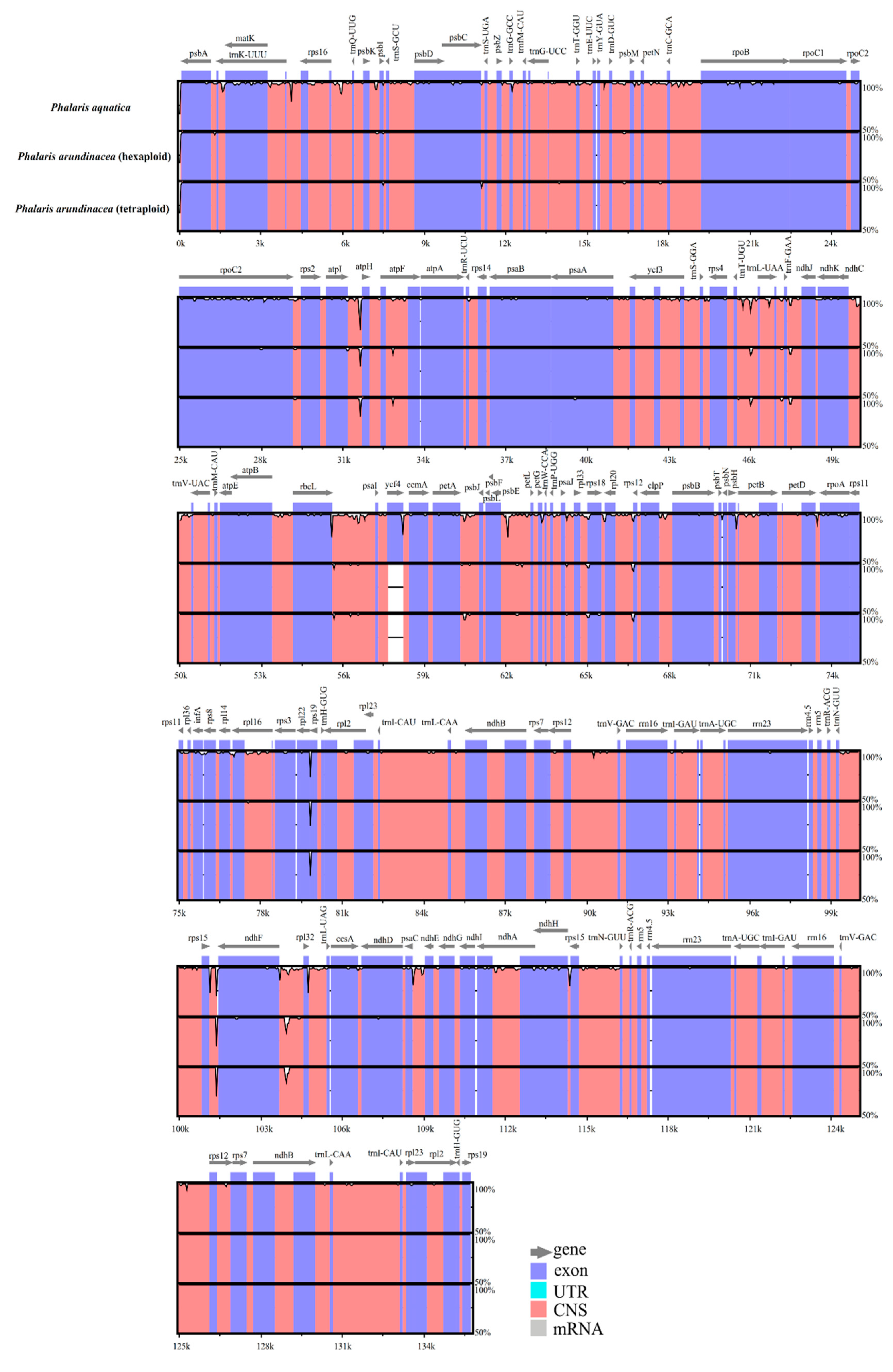
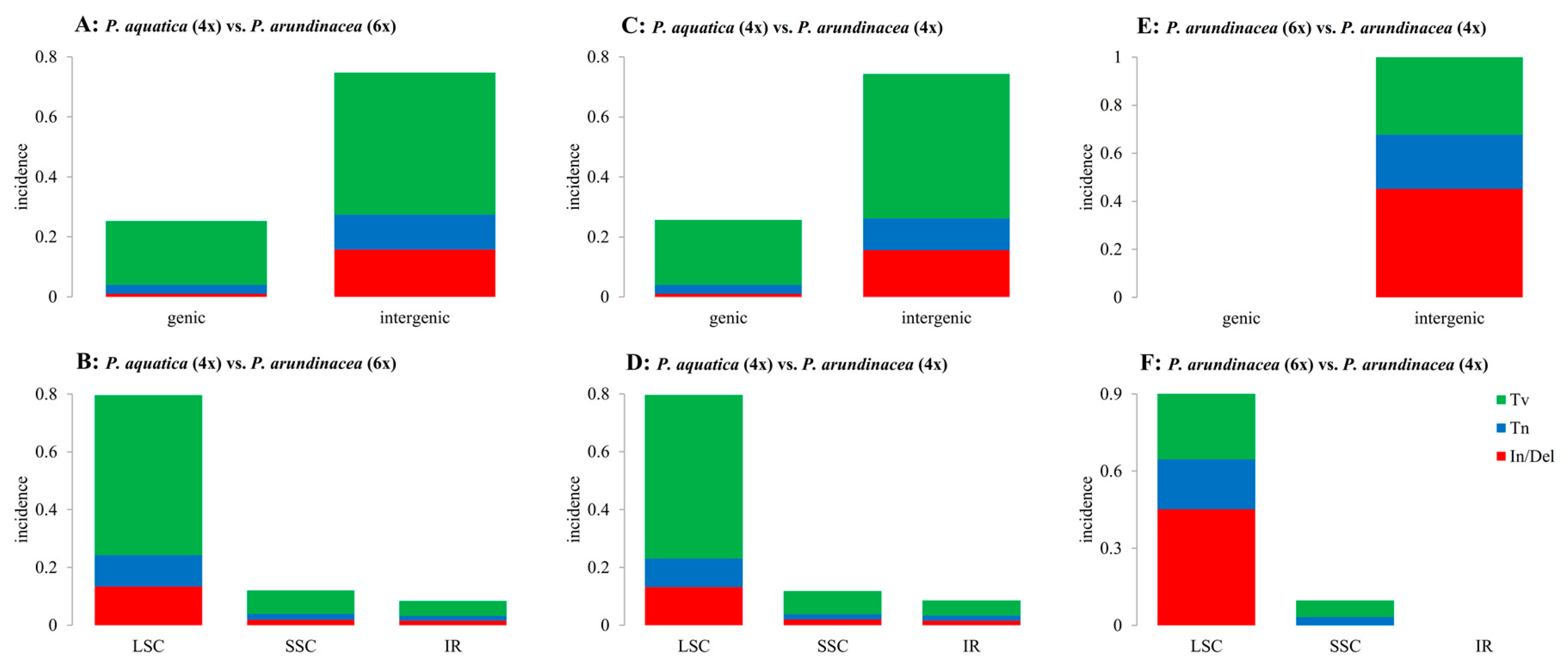
2.2. Variation among Three Chloroplast Genomes
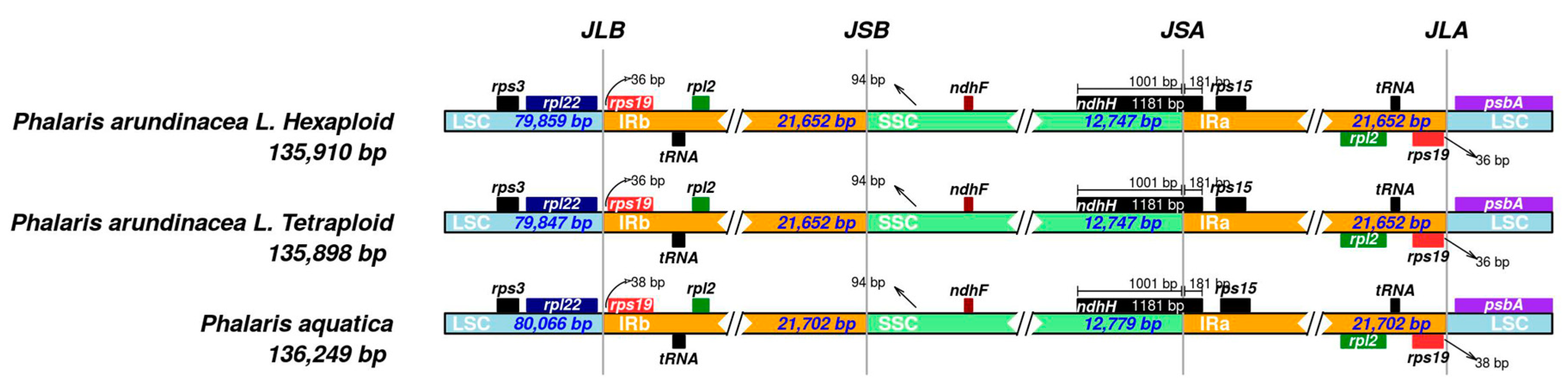
2.3. IR Scope Characteristics
2.4. Nucleotide Diversity
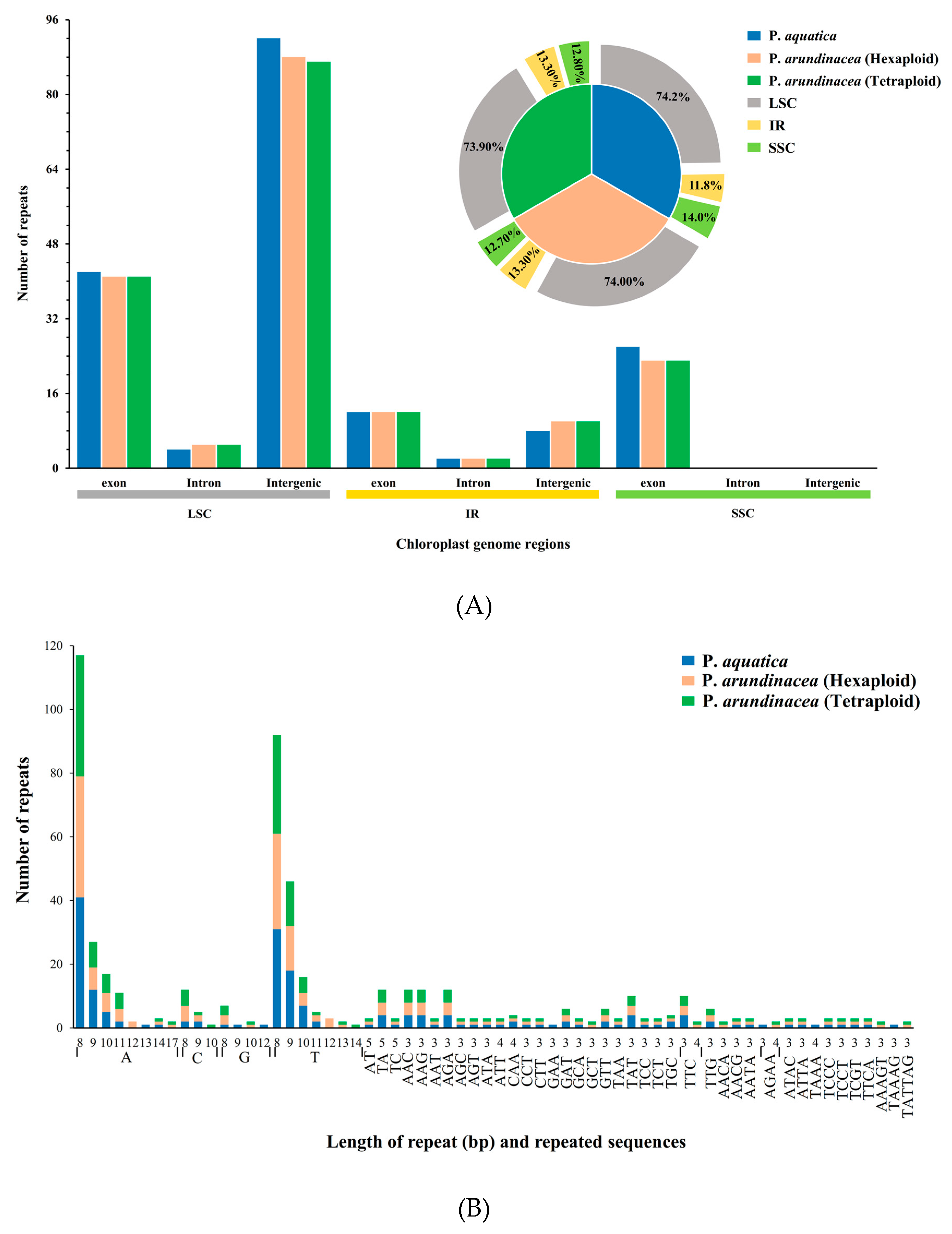
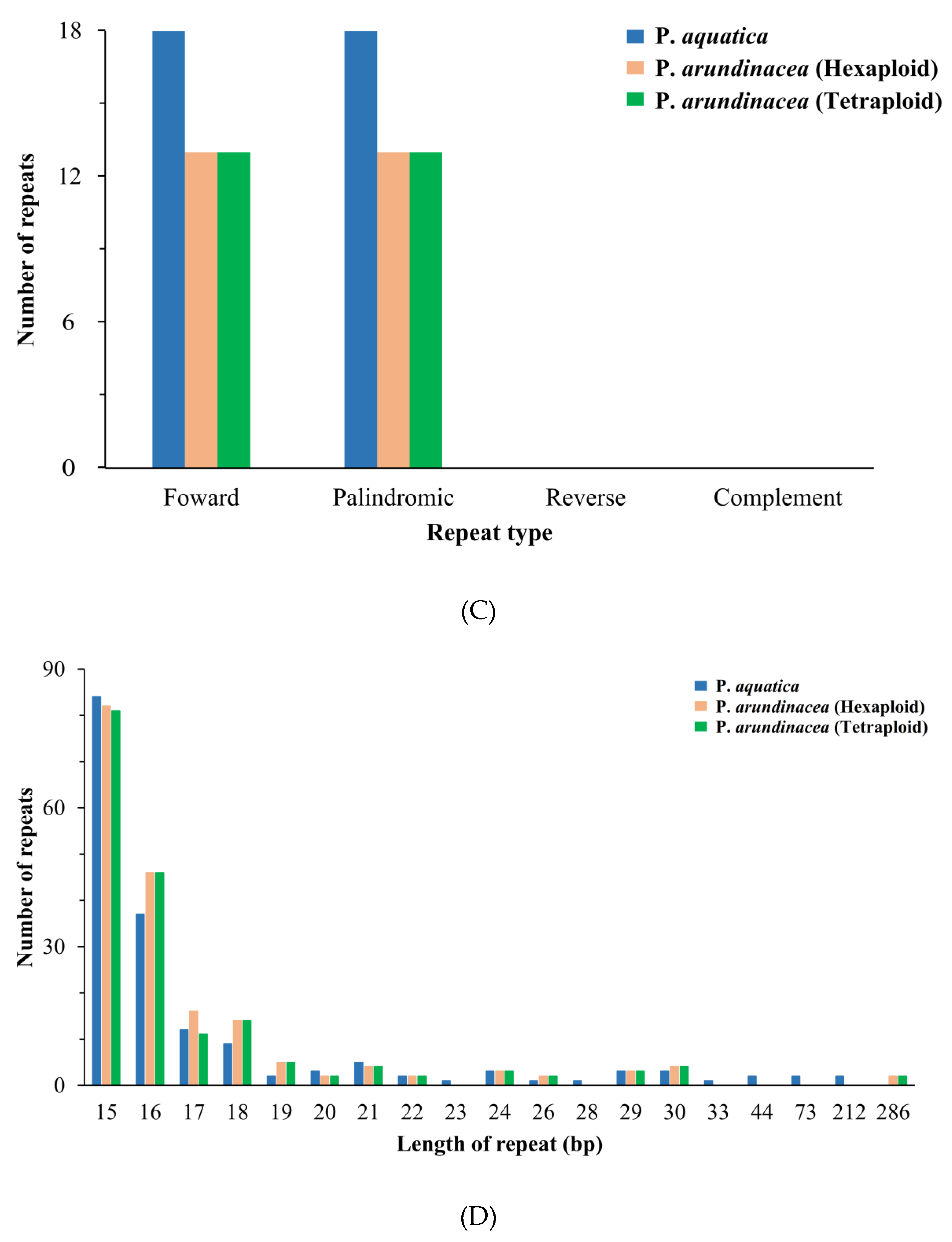
2.5. Repetitive Sequences
2.6. Codon Usage
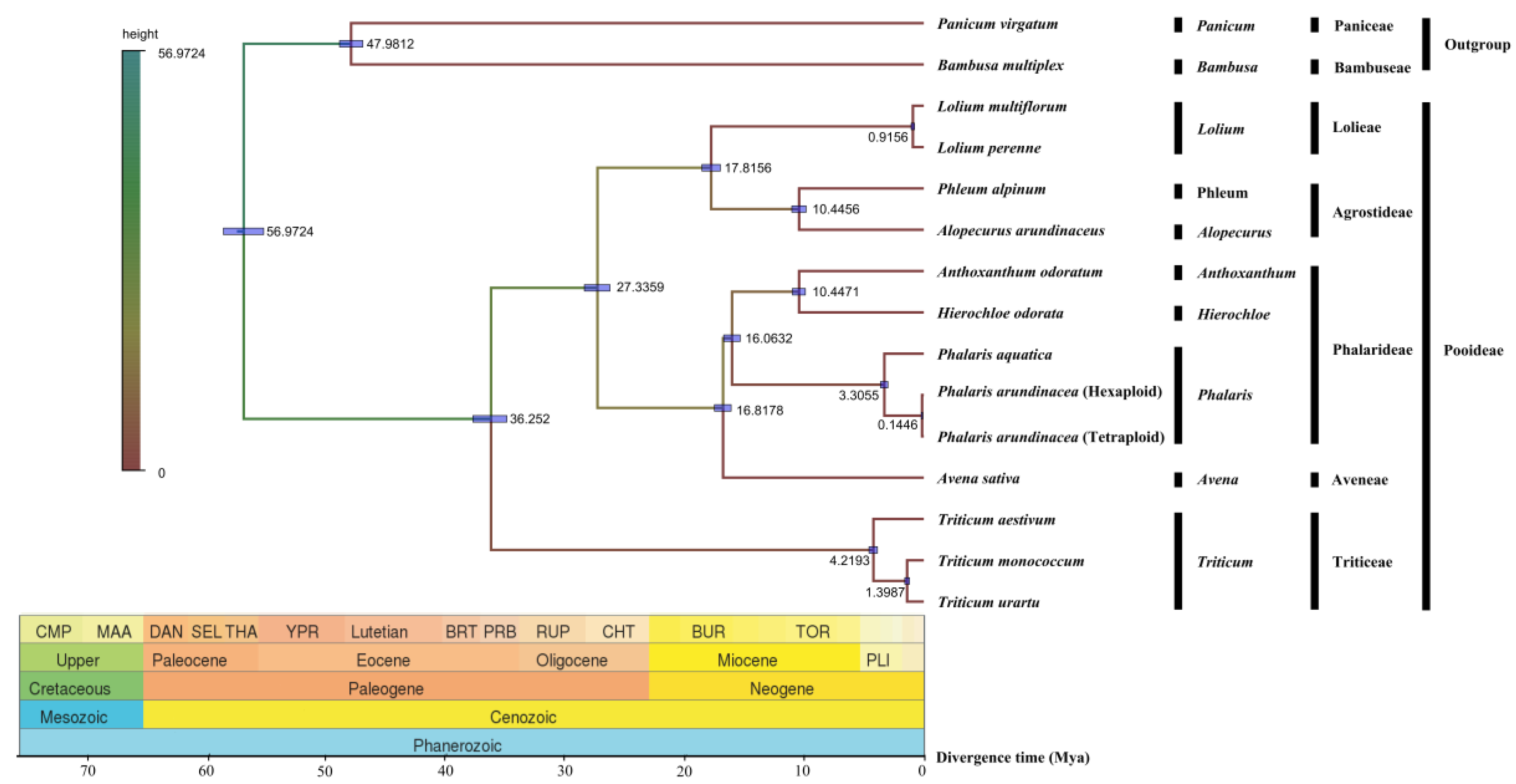
2.7. Phylogenetic Divergence Time
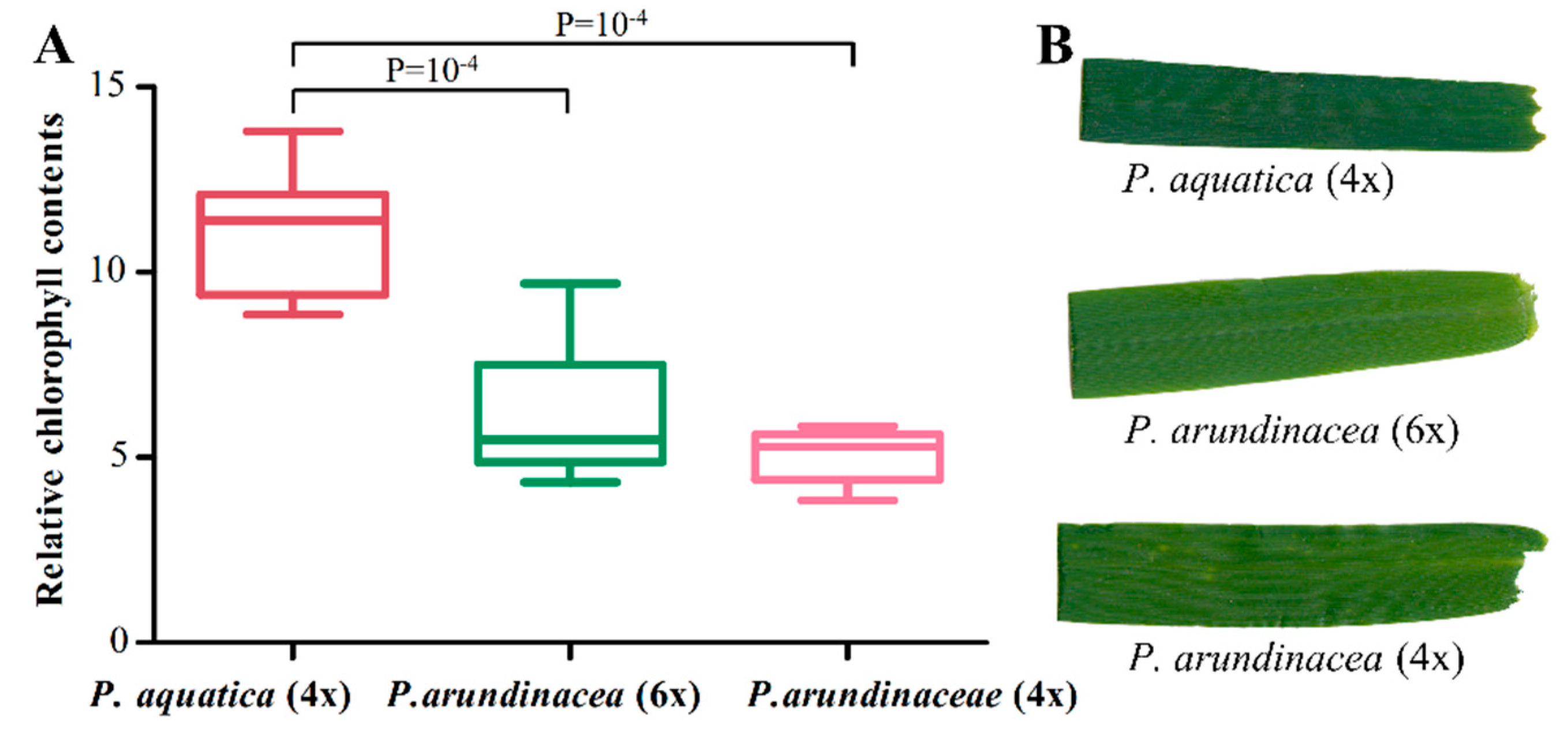
2.8. Diversity and Expression of Chloroplast Genes
3. Discussion
3.1. Chloroplast Genome Characteristics of Three Phalaris Cultivars
3.2. Sequences Variation and Gene Mutation
3.3. CpSSRs and RSCU
3.4. Phylogeny Analysis and Divergence Time
3.5. Gene Expression in Chloroplast Genomes
4. Materials and Methods
4.1. DNA Extraction and Genome Sequencing
4.2. Chloroplast Genome Assembly and Annotation
4.3. Multiple Chloroplast Genome Alignments
4.4. Identification of Repetitive Sequences
4.5. Relative Synonymous Codon Usage Analysis
4.6. Analysis of Non-Synonymous/Synonymous Substitution
4.7. Phylogenetic Analysis
4.8. Chloroplast RNA-Seq and Chlorophyll Measurement
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Availability of Data and Materials
References
- Ravi, V.; Khurana, J.P.; Tyagi, A.K.; Khurana, P. An update on chloroplast genomes. Plant Syst. Evol. 2008, 271, 101–122. [Google Scholar] [CrossRef]
- Allen, F.J. The function of genomes in bioenergetic organelles. Philos. Trans. R. Soc. B 2003, 358, 19–38. [Google Scholar] [CrossRef]
- Liu, L.X.; Wang, Y.W.; He, P.Z.; Li, P.; Fu, C.X. Chloroplast genome analyses and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data. BMC Genom. 2018, 19, 235. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.T.; Bayer, R.J. Molecular phylogenetics of Acacia (Fabaceae: Mimosoideae) based on the chloroplast MATK coding sequence and flanking TRNK intron spacer regions. Am. J. Bot. 2001, 88, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.A.; Scharaschkin, T. Phylogeny and historical biogeography of Anaxagorea (Annonaceae) using morphology and non-coding chloroplast aequence data. Syst. Bot. 2005, 30, 712–735. [Google Scholar]
- Sahramaa, M.; Hömmö, L.; Jauhiainen, L. Variation in seed production traits of reed canarygrass germplasm. Crop Sci. 2004, 44, 988–996. [Google Scholar] [CrossRef]
- Voshell, S.M.; Baldini, R.M.; Hilu, K.W. Infrageneric treatment of Phalaris (Canary grasses, Poaceae) based on molecular phylogenetics and floret structure. Aust. Syst. Bot. 2016, 6, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Perdereau, A.; Klaasm, M.; Barth, S.; Hodkinson, T.R. Plastid genome sequencing reveals biogeographical structure and extensive population genetic variation in wild populations of Phalaris arundinacea L. in north-western Europe. GCB Bioenergy 2017, 1, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.C.; Evans, M. Comparative growth and development of hexaploid and tetraploid reed canarygrass. Crop Sci. 2014, 54, 1062. [Google Scholar] [CrossRef]
- Mcwilliam, J.R.; Nealsmith, C.A. Tetraploid and hexaploid chromosome races of Phalaris arundinacea L. Aust. J. Agric. Res. 1962, 13, 1–9. [Google Scholar] [CrossRef]
- Jansone, B.; Rancane, S.; Berzins, P.; Stesele, V. Reed canary grass (Phalaris arundinacea L.) in natural biocenosis of Latvia, research experiments and production fields. Renew. Energy Energy Effic. Int. Sci. Conf. 2012. [Google Scholar]
- Oram, R.N.; Williams, J.D. Variation in concentration and composition of toxic alkaloids among strains of Phalaris tuberosa L. Nature 1967, 213, 946–947. [Google Scholar] [CrossRef]
- Frelich, J.R.; Marten, G.C. Quick test for reed canarygrass (Phalaris arundinacea L.) alkaloid concentration. Crop Sci. 1973, 13, 548–551. [Google Scholar] [CrossRef]
- Ball, D.M.; Hoveland, C.S. Alkaloid levels in Phalaris aquatica L. as affected by environment. Agron. J. 1978, 70, 977–981. [Google Scholar] [CrossRef]
- Das, S.; Ghosh, S.; Pan, A.; Dutta, C. Compositional variation in bacterial genes and proteins with potential expression level. FEBS Lett. 2005, 579, 5210. [Google Scholar] [CrossRef]
- Piskol, R.; Ramaswami, G.; Li, J.B. Reliable identification of genomic variants from RNA-Seq data. Am. J. Hum. Genet. 2013, 93, 641–651. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Wang, S.; Xia, E.H.; Jiang, J.J.; Zeng, F.C. Full transcription of the chloroplast genome in photosynthetic eukaryotes. Sci. Rep. 2016, 6, 30135. [Google Scholar] [CrossRef]
- Silva, S.R.; Pinheiro, D.G.; Penha, H.A.; Płachno, B.J.; Michael, T.P.; Meer, E.J.; Miranda, V.F.O.; Varani, A.M. Intraspecific variation within the Utricularia amethystina species morphotypes based on chloroplast genomes. Int. J. Mplecular Sci. 2019, 24, 6130. [Google Scholar] [CrossRef] [Green Version]
- Frazer, K.A.; Lior, P.; Alexander, P.; Rubin, E.M.; Inna, D. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Darling, A.E.; Craven, M.; Mau, B.; Perna, N.T. Multiple Alignment of Rearranged Genomes. In Proceedings of the 2004 IEEE Computational Systems Bioinformatics Conference, Stanford, CA, USA, 19 August 2004. [Google Scholar]
- Kazutaka, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 4, 722–780. [Google Scholar]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-Delbarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 12, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, P.; Gilchrist, M.A. Explaining complex codon usage patterns with selection for translational efficiency, mutation bias, and genetic drift. Proc. Natl. Acad. Sci. USA 2011, 108, 10231–10236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luca, V.D.; Pierre, B.S. The cell and developmental biology of alkaloid biosynthesis. Trends Plant Sci. 2000, 5, 168–173. [Google Scholar] [CrossRef]
- Ozawa, S.I.; Nield, J.; Terao, A.; Stauber, E.J.; Hippler, M.; Koike, H.; Rochaix, J.D.; Takahashi, Y. Biochemical and structural studies of the large ycf4-photosystem I assembly complex of the green alga Chlamydomonas reinhardtii. Plant Cell 2009, 21, 2424–2442. [Google Scholar] [CrossRef] [Green Version]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L.; Palmer, J.D.; Lao, N.T.; Heggie, L.; Kavanagh, T.A.; Hibberd, J.M.; Gray, J.C.; Morden, C.W.; et al. Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 2001, 13, 645–658. [Google Scholar] [CrossRef] [Green Version]
- Young, H.A.; Lanzatella, C.L.; Sarath, G.; Tobias, C.M.; Newbigin, E. Chloroplast genome variation in upland and lowland switchgrass. PLoS ONE 2011, 8, e23980. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.C.; Chen, S.Y.; Zhang, X.Z. Whole-genome comparison reveals divergent IR borders and mutation hotspots in chloroplast genomes of herbaceous Bamboos (Bambusoideae: Olyreae). Molecules 2018, 7, 1537. [Google Scholar] [CrossRef] [Green Version]
- Loewe, L.; Charlesworth, B.; Bartolomé, C.; Nöel, V. Estimating selection on nonsynonymous mutations. Genetics 2006, 2, 1079–1092. [Google Scholar] [CrossRef] [Green Version]
- Yamane, K.; Kawahara, T. Size homoplasy and mutational behavior of chloroplast simple sequence repeats (cpSSRs) inferred from intra- and interspecific variations in four chloroplast regions of diploid and polyploid Triticum and Aegilops species. Genet. Resour. Crop Evol. 2017, 3, 1–17. [Google Scholar] [CrossRef]
- Ma, Q.P.; Li, C.; Wang, J.; Wang, Y.; Ding, Z.T. Analysis of synonymous codon usage in FAD7 genes from different plant species. Genet. Mol. Res. 2015, 14, 1414–1422. [Google Scholar] [CrossRef] [PubMed]
- Orei, M.; Shalloway, D. Specific correlations between relative synonymous codon usage and protein secondary structure. J. Mol. Biol. 1998, 281, 31–48. [Google Scholar]
- Omura, A.; Maeda, Y.; Kawana, T.; Siringan, F.P.; Berdin, R.D. U-series dates of Pleistocene corals and their implications to the paleo-sea levels and the vertical displacement in the Central Philippines. Quat. Int. 2004, 3, 3–13. [Google Scholar] [CrossRef]
- Lin, C.P.; Ko, C.Y.; Kuo, C.I.; Liu, M.S.; Schafleitner, R.; Chen, L.F.O. Transcriptional slippage and RNA editing increase the diversity of transcripts in chloroplasts: Insight from deep sequencing of Vigna radiata genome and transcriptome. PLoS ONE 2015, 6, e129396. [Google Scholar] [CrossRef] [Green Version]
- Safonova, Y.; Bankevich, A.; Pevzner, P.A. DipSPAdes: Assembler for highly polymorphic diploid genomes. J. Comput. Biol. 2015, 6, 265–279. [Google Scholar] [CrossRef] [Green Version]
- Boetzer, M.; Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol. 2012, 13, R56. [Google Scholar] [CrossRef] [Green Version]
- Forster, J.W.; Jones, E.S.; Kölliker, R.; Drayton, M.C.; Smith, K.F. Development and Implementation of Molecular Markers for Forage Crop Improvement; Springer: Dordrecht, The Netherlands, 2001. [Google Scholar]
- Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Clements, J.; Eddy, S.R.; Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [Green Version]
- Laslett, D. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Marc, L.; Oliver, D.; Sabine, K.; Ralph, B. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 44, W575–W581. [Google Scholar]
- Ali, A.; Jaakko, H.; Peter, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 17, 3030–3031. [Google Scholar]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 16, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.P.; Zhang, Y.B.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinf. 2010, 8, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Dong, X.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.C.; Aaron, O.; Alekseyenko, A.V. MixtureTree annotator: A program for automatic colorization and visual annotation of MixtureTree. PLoS ONE 2015, 10, e118893. [Google Scholar] [CrossRef]
- Cassol, D.; Silva, F.S.P.; Falqueto, A.R.; Bacarin, M.A. An evaluation of non-destructive methods to estimate total chlorophyll content. Photosynthetica 2008, 46, 634–636. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species/Ploidies | Size (bp) | GC Content (%) | tRNA | rRNA | mRNA | Genes | Number of Genes Duplicated in IR | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cp | LSC | SSC | IR | cp | LSC | SSC | IR | ||||||
| P arundinacea (4x) | 135,898 | 79,846 | 12,746 | 21,653 | 38.52 | 36.45 | 32.88 | 44.00 | 28 | 4 | 72 | 104 | 19 |
| P arundinacea (6x) | 135,910 | 79,858 | 12,746 | 21,653 | 38.52 | 36.44 | 32.87 | 44.00 | 28 | 4 | 72 | 104 | 19 |
| P aquatica (4x) | 136,249 | 80,065 | 12,778 | 21,703 | 38.45 | 36.36 | 32.84 | 43.94 | 28 | 4 | 73 | 105 | 19 |
| Category | Function | Name of Gene | |||||
|---|---|---|---|---|---|---|---|
| Self-replication | Ribosomal RNA Genes | rrn4.5 | rrn5 | rrn16 | rrn23 | ||
| Transfer RNA genes | trnA-ACG | trnL-CAA | trnV-GAC | trnH-GUG | trnA-GUU | trnA-UGC * | |
| trnT-CGU * | trnS-CGA * | trnL-UAA * | trnV-UAC * | trnL-UUU * | trnM-CAU # | ||
| trnT-CCA | trnP-GAA | trnC-GCA | trnG-GCC | trnS-GCU | trnS-GGA | ||
| trnT-GGU | trnT-GUA | trnA-GUC | trnL-UAG | trnA-UCU | trnS-UGA | ||
| trnP-UGG | trnT-UGU | trnG-UUC | trnG-UUG | ||||
| Ribosomal proteins (translation) | Small subunit of ribosome (SSU) | rps2 | rps3 | rps4 | rps7 | rps8 | rps11 |
| rps14 | rps15 | rps18 | rps19 | ||||
| Transcription | Large subunit of ribosome (LSU) | rpl2 | rpl14 | rpl16 | rpl20 | rpl22 | rpl23 |
| rpl32 | rpl33 | rpl36 | |||||
| RNA polymerase subunits | rpoA | rpoB | rpoC1 | rpoC2 | |||
| Translation initiation factor | infA | ||||||
| Photosynthesis related genes | Large subunit of Rubisco | rbcL | |||||
| Subunits of Photosystem I | psaA | psaB | psaC | psaI | psaJ | ycf4aq | |
| Subunits of Photosystem II | psbA | psbB | psbC | psbD | psbE | psbF | |
| psbH | psbI | psbJ | psbK | psbL | psbM | ||
| psbT | psbZ | ||||||
| Subunits of ATP synthase | atpA | atpB | atpE | atpF * | atpH | atpI | |
| Cytochrome b/f complex | petA | petB | petG | petL | petN | ||
| C-type cytochrome synthesis gene | ccsA | ||||||
| Subunits of NADH dehydrogenase | ndhA * | ndhB * | ndhC | ndhD | ndhE | ndhF | |
| ndhG | ndhH | ndhI | ndhJ | ndhK | |||
| Other genes | Maturase | matK | |||||
| Protease | clpP | ||||||
| Chloroplast envelope membrane protein | cemA | ||||||
| Hypothetical protein | ycf1 | ||||||
| Hypothetical open reading frames | ycf2 | ||||||
| Gene | P. aquatica | P. arundinacea (4x & 6x) | ||||||
|---|---|---|---|---|---|---|---|---|
| Location | Exon (bp) | Intron I (bp) | Exon II (bp) | Location | Exon I (bp) | Intron I (bp) | Exon II (bp) | |
| atpF | LSC | 160 | 818 | 407 | LSC | 160 | 826 | 407 |
| ndhA | SSC | 550 | 1020 | 539 | SSC | 550 | 1023 | 539 |
| ndhB | IRA | 775 | 712 | 758 | IRA | 775 | 712 | 758 |
| ndhB | IRB | 775 | 712 | 758 | IRB | 775 | 712 | 758 |
| trnS-CGA | LSC | 32 | 655 | 63 | LSC | 32 | 655 | 63 |
| trnT-CGU | IRA | 32 | 787 | 59 | IRA | 32 | 786 | 59 |
| trnT-CGU | IRB | 33 | 785 | 60 | IRB | 33 | 784 | 60 |
| trnL-UAA | LSC | 36 | 543 | 51 | LSC | 36 | 549 | 51 |
| trnV-UAC | LSC | 39 | 579 | 54 | LSC | 39 | 579 | 54 |
| trnA-UGC | IRA | 37 | 811 | 36 | IRA | 37 | 811 | 36 |
| trnA-UGC | IRB | 38 | 809 | 37 | IRB | 38 | 809 | 37 |
| trnK-UUU | LSC | 39 | 2465 | 37 | LSC | 39 | 2463 | 37 |
| Species | NPGS ID | Improvement Status | GenBank Accession |
|---|---|---|---|
| Phalaris arundinacea L. (tetraploid) | PI 272122 | Cultivar | MT274594 |
| Phalaris arundinacea L. (hexaploid) | PI 422031 | Cultivar | MT274595 |
| Phalaris aquatica L. (tetraploid) | PI 434985 | Cultivar | MT274596 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, Y.; Xiong, Y.; Jia, S.; Ma, X. The Complete Chloroplast Genome Sequencing and Comparative Analysis of Reed Canary Grass (Phalaris arundinacea) and Hardinggrass (P. aquatica). Plants 2020, 9, 748. https://doi.org/10.3390/plants9060748
Xiong Y, Xiong Y, Jia S, Ma X. The Complete Chloroplast Genome Sequencing and Comparative Analysis of Reed Canary Grass (Phalaris arundinacea) and Hardinggrass (P. aquatica). Plants. 2020; 9(6):748. https://doi.org/10.3390/plants9060748
Chicago/Turabian StyleXiong, Yi, Yanli Xiong, Shangang Jia, and Xiao Ma. 2020. "The Complete Chloroplast Genome Sequencing and Comparative Analysis of Reed Canary Grass (Phalaris arundinacea) and Hardinggrass (P. aquatica)" Plants 9, no. 6: 748. https://doi.org/10.3390/plants9060748
APA StyleXiong, Y., Xiong, Y., Jia, S., & Ma, X. (2020). The Complete Chloroplast Genome Sequencing and Comparative Analysis of Reed Canary Grass (Phalaris arundinacea) and Hardinggrass (P. aquatica). Plants, 9(6), 748. https://doi.org/10.3390/plants9060748