Analysis of the Bacterial and Host Proteins along and across the Porcine Gastrointestinal Tract



Abstract
:1. Introduction
2. Material and Methods
2.1. Animal Experiment and Sampling
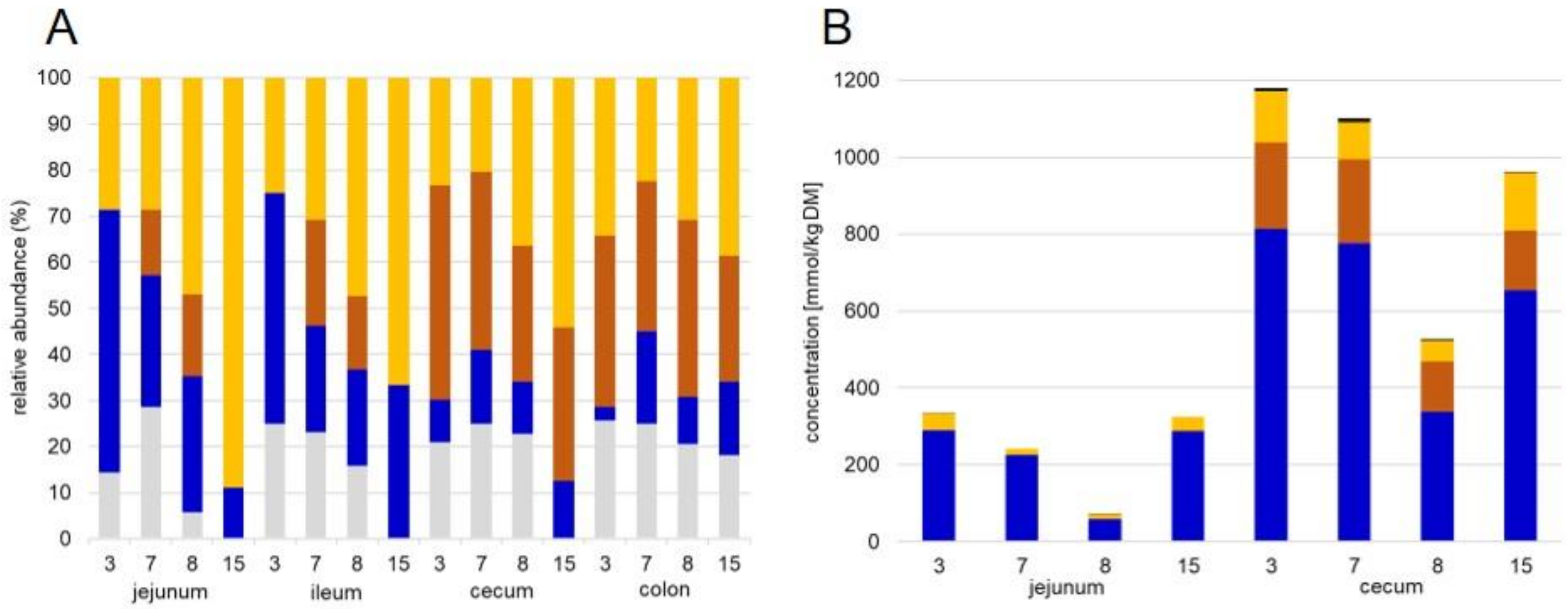
2.2. GC Analysis of Short Chain Fatty Acids
2.3. Sample Preparation
2.4. LC-MS/MS Analysis
2.5. Data Analysis
2.6. PRIDE Accession
3. Results and Discussion
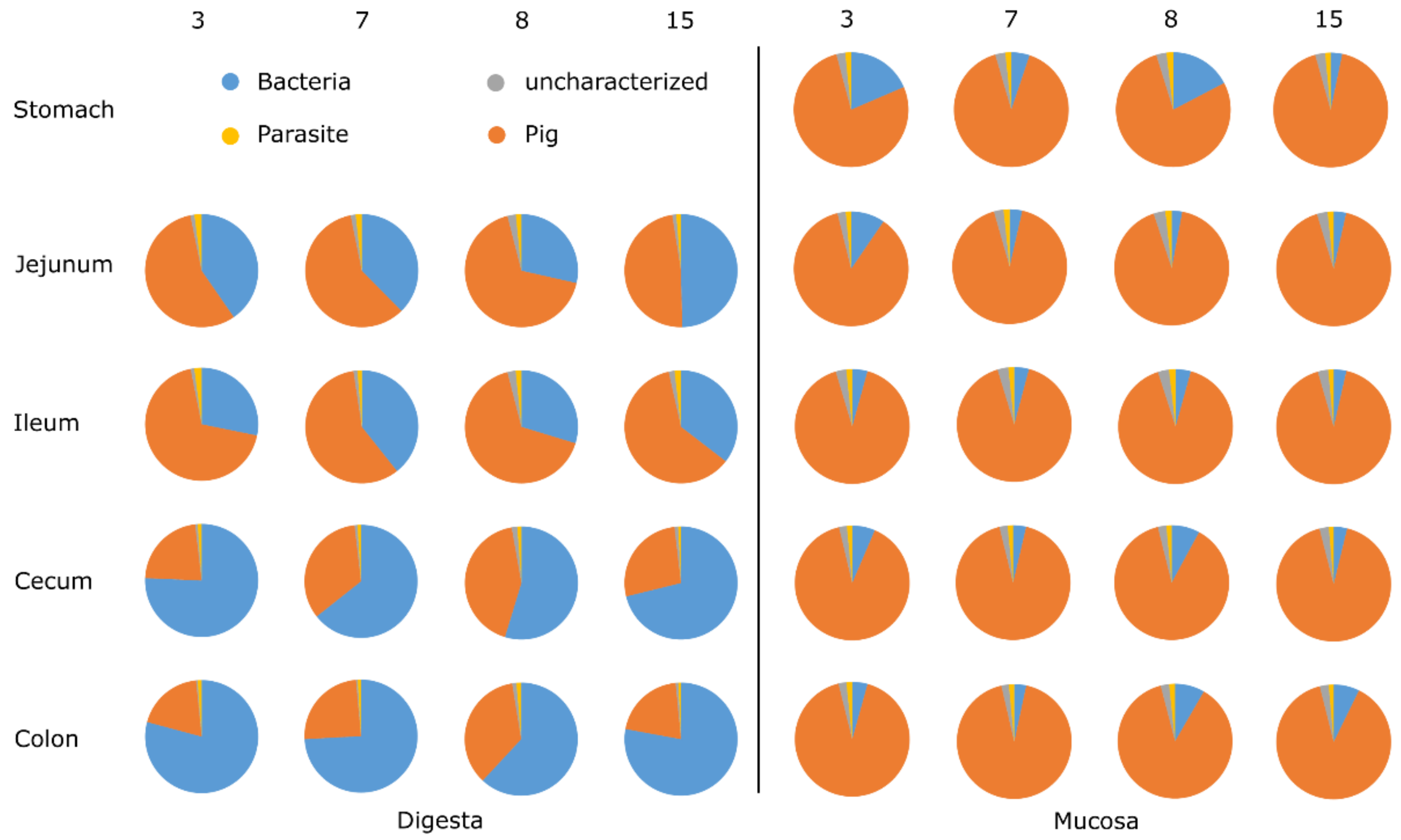
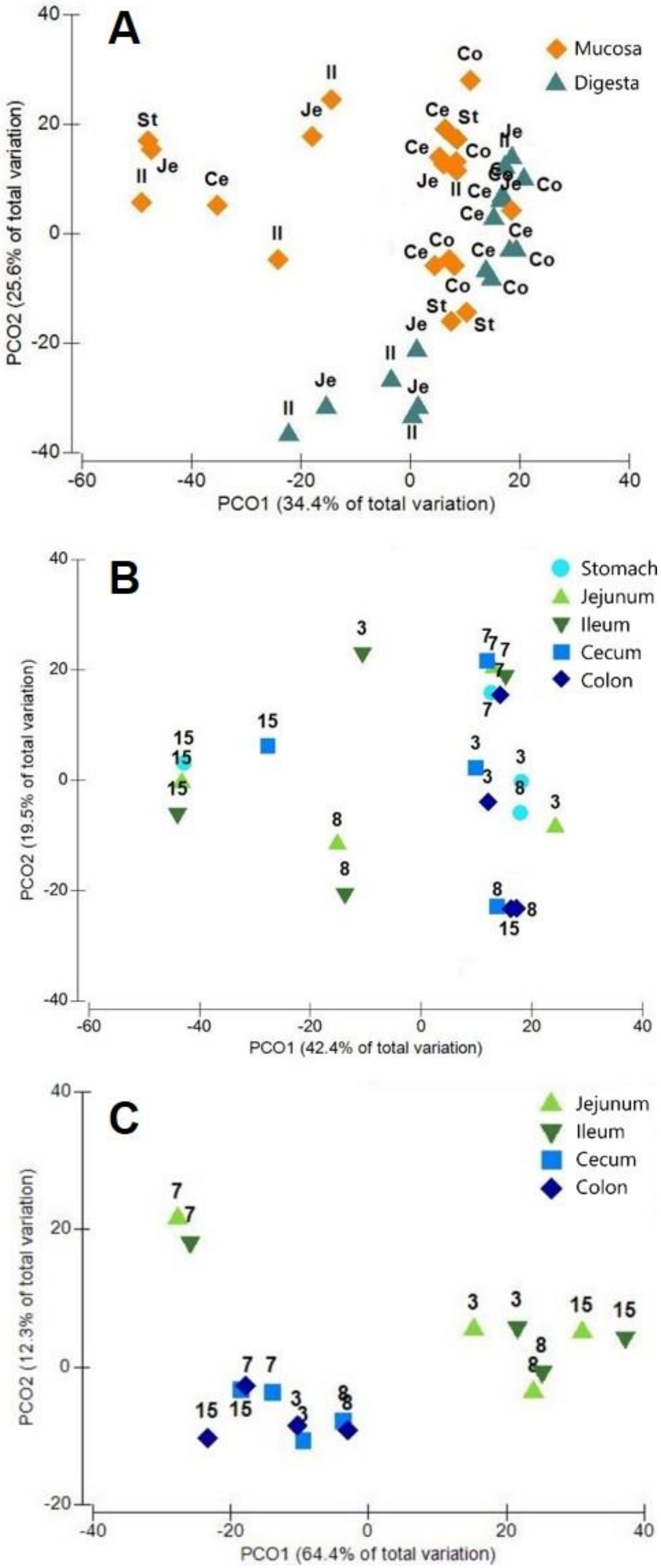
3.1. General Protein Results
3.2. Phylotypes of Bacterial Proteins in Digesta
3.3. Phylotypes of Bacterial Proteins in Mucosa
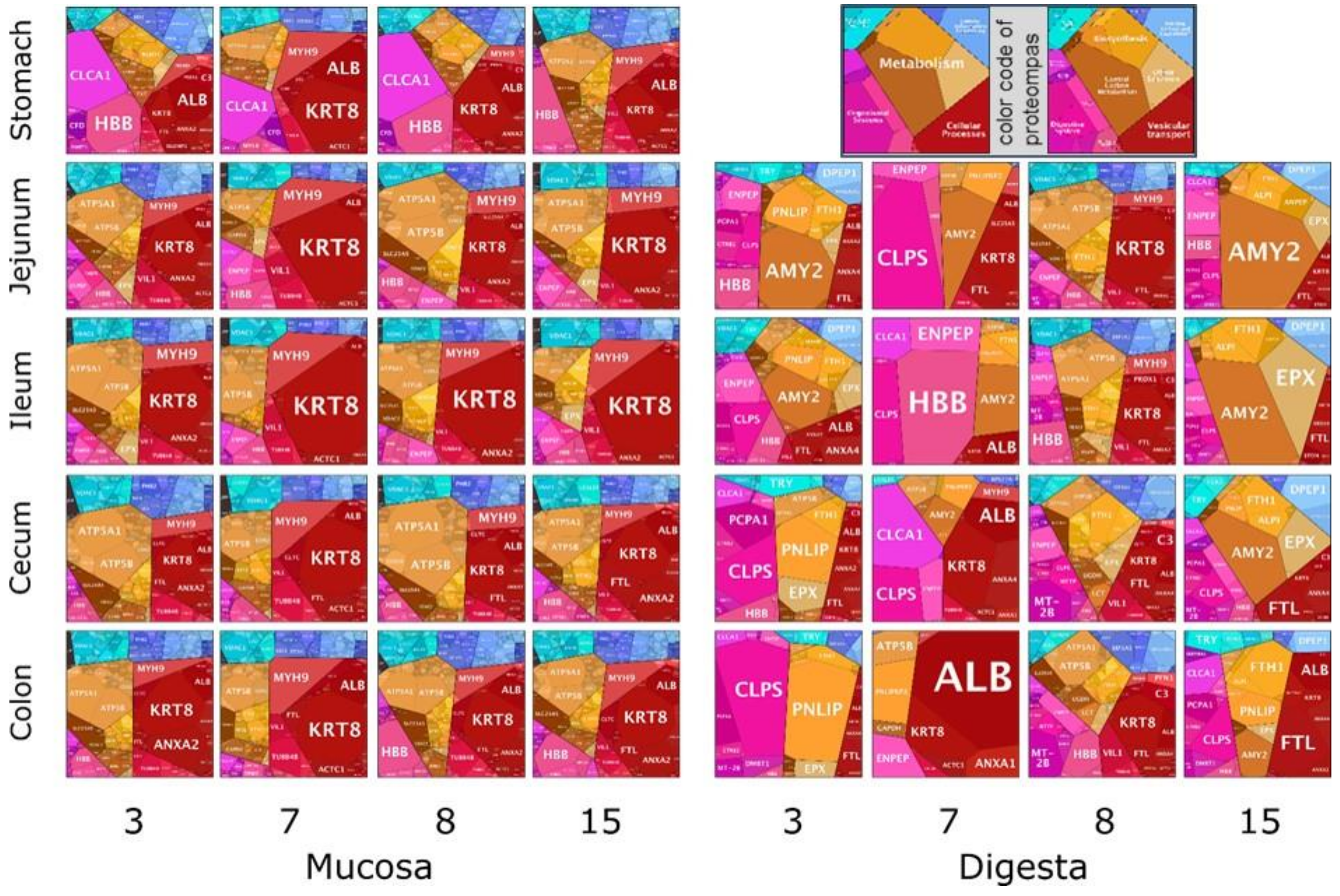
3.4. Functional Annotation and Distribution of Bacterial Proteins in Digesta and Mucosa
3.5. Animal Proteins
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Isaacson, R.; Kim, H.B. The intestinal microbiome of the pig. Anim. Health Res. Rev. 2012, 13, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Pajarillo, E.A.B.; Chae, J.-P.; Balolong, M.P.; Kim, H.B.; Kang, D.-K. Assessment of fecal bacterial diversity among healthy piglets during the weaning transition. J. Gen. Appl. Microbiol. 2014, 60, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.F.; Nyachoti, M. Using probiotics to improve swine gut health and nutrient utilization. Anim. Nutr. 2017, 3, 331–343. [Google Scholar] [CrossRef]
- Hill, J.E.; Hemmingsen, S.M.; Goldade, B.G.; Dumonceaux, T.J.; Klassen, J.; Zijlstra, R.T.; Goh, S.H.; Van Kessel, A.G. Comparison of ileum microflora of pigs fed corn-, wheat-, or barley-based diets by chaperonin-60 sequencing and quantitative PCR. Appl. Environ. Microbiol. 2005, 71, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Richards, J.; Gong, J.; De Lange, C. The gastrointestinal microbiota and its role in monogastric nutrition and health with an emphasis on pigs: Current understanding, possible modulations, and new technologies for ecological studies. Can. J. Anim Sci. 2005, 85, 421–435. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Widmer, G.; Tzipori, S. A pig model of the human gastrointestinal tract. Gut Microbes 2013, 4, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Heinritz, S.N.; Mosenthin, R.; Weiss, E. Use of pigs as a potential model for research into dietary modulation of the human gut microbiota. Nutr. Res. Rev. 2013, 26, 191–209. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Estelle, J.; Kiilerich, P.; Ramayo-Caldas, Y.; Xia, Z.; Feng, Q.; Liang, S.; Pedersen, A.O.; Kjeldsen, N.J.; Liu, C.; et al. A reference gene catalogue of the pig gut microbiome. Nat. Microbiol. 2016, 1. [Google Scholar] [CrossRef]
- Klymiuk, N.; Seeliger, F.; Bohlooly, Y.M.; Blutke, A.; Rudmann, D.G.; Wolf, E. Tailored pig models for preclinical efficacy and safety testing of targeted therapies. Toxicol. Pathol. 2016, 44, 346–357. [Google Scholar] [CrossRef]
- Roura, E.; Koopmans, S.J.; Lalles, J.P.; Le Huerou-Luron, I.; de Jager, N.; Schuurman, T.; Val-Laillet, D. Critical review evaluating the pig as a model for human nutritional physiology. Nutr. Res. Rev. 2016, 29, 60–90. [Google Scholar] [CrossRef]
- Derrien, M.; van Passel, M.W.; van de Bovenkamp, J.H.; Schipper, R.; de Vos, W.; Dekker, J. Mucin-bacterial interactions in the human oral cavity and digestive tract. Gut Microbes 2010, 1, 254–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, M.E.; Sjovall, H.; Hansson, G.C. The gastrointestinal mucus system in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 352–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rodas, B.; Youmans, B.P.; Danzeisen, J.L.; Tran, H.; Johnson, T.J. Microbiome profiling of commercial pigs from farrow to finish. J. Anim. Sci. 2018, 96, 1778–1794. [Google Scholar] [CrossRef] [PubMed]
- Mann, E.; Schmitz-Esser, S.; Zebeli, Q.; Wagner, M.; Ritzmann, M.; Metzler-Zebeli, B.U. Mucosa-associated bacterial microbiome of the gastrointestinal tract of weaned pigs and dynamics linked to dietary calcium-phosphorus. PLoS ONE 2014, 9, e86950. [Google Scholar] [CrossRef] [PubMed]
- Ndou, S.P.; Tun, H.M.; Kiarie, E.; Walsh, M.C.; Khafipour, E.; Nyachoti, C.M. Dietary supplementation with flaxseed meal and oat hulls modulates intestinal histomorphometric characteristics, digesta- and mucosa-associated microbiota in pigs. Sci. Rep. 2018, 8, 5880. [Google Scholar] [CrossRef]
- Kelly, J.; Daly, K.; Moran, A.W.; Ryan, S.; Bravo, D.; Shirazi-Beechey, S.P. Composition and diversity of mucosa-associated microbiota along the entire length of the pig gastrointestinal tract; dietary influences. Environ. Microbiol. 2017, 19, 1425–1438. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wu, W.; Lee, Y.-K.; Xie, J.; Zhang, H. Spatial heterogeneity and co-occurrence of mucosal and luminal microbiome across swine intestinal tract. Front. Microbiol. 2018, 9, 48. [Google Scholar] [CrossRef]
- Burrough, E.R.; Arruda, B.L.; Plummer, P.J. Comparison of the luminal and mucosa-associated microbiota in the colon of pigs with and without swine dysentery. Front. Vet. Sci. 2017, 4, 139. [Google Scholar] [CrossRef]
- Metzler-Zebeli, B.U.; Lawlor, P.G.; Magowan, E.; Zebeli, Q. Interactions between metabolically active bacteria and host gene expression at the cecal mucosa in pigs of diverging feed efficiency. J. Anim. Sci. 2018, 96, 2249–2264. [Google Scholar] [CrossRef]
- Looft, T.; Allen, H.K.; Cantarel, B.L.; Levine, U.Y.; Bayles, D.O.; Alt, D.P.; Henrissat, B.; Stanton, T.B. Bacteria, phages and pigs: The effects of in-feed antibiotics on the microbiome at different gut locations. ISME J. 2014, 8, 1566–1576. [Google Scholar] [CrossRef]
- Holman, D.B.; Brunelle, B.W.; Trachsel, J.; Allen, H.K. Meta-analysis to define a core microbiota in the swine gut. mSystems 2017, 2, e00004–17. [Google Scholar] [CrossRef] [PubMed]
- Wilmes, P.; Heintz-Buschart, A.; Bond, P.L. A decade of metaproteomics: Where we stand and what the future holds. Proteomics 2015, 15, 3409–3417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deusch, S.; Seifert, J. Catching the tip of the iceberg—Evaluation of sample preparation protocols for metaproteomic studies of the rumen microbiota. Proteomics 2015, 15, 3590–3595. [Google Scholar] [CrossRef] [PubMed]
- Haange, S.B.; Oberbach, A.; Schlichting, N.; Hugenholtz, F.; Smidt, H.; von Bergen, M.; Till, H.; Seifert, J. Metaproteome analysis and molecular genetics of rat intestinal microbiota reveals section and localization resolved species distribution and enzymatic functionalities. J. Proteome Res. 2012, 11, 5406–5417. [Google Scholar] [CrossRef] [PubMed]
- Heyer, C.M.; Schmucker, S.; Aumiller, T.; Föll, A.; Uken, K.; Rodehutscord, M.; Hoelzle, L.E.; Seifert, J.; Stefanski, V.; Mosenthin, R. The impact of dietary phosphorus and calcium on the intestinal microbiota and mitogen-induced proliferation of mesenteric lymph node lymphocytes in pigs. J. Anim. Sci. 2016, 94, 373–376. [Google Scholar] [CrossRef]
- NRC. Nutrient Requirements of Swine, 11th ed.; National Academies Press: Washington, DC, USA, 2012. [Google Scholar]
- Wischer, G.; Boguhn, J.; Steingass, H.; Schollenberger, M.; Hartung, K.; Rodehutscord, M. Effect of monensin on in vitro fermentation of silages and microbial protein synthesis. Arch. Anim. Nutr. 2013, 67, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Apajalahti, J.H.; Särkilahti, L.K.; Mäki, B.R.; Heikkinen, J.P.; Nurminen, P.H.; Holben, W.E. Effective recovery of bacterial DNA and percent-guanine-plus-cytosine-based analysis of community structure in the gastrointestinal tract of broiler chickens. Appl. Environ. Microbiol. 1998, 64, 4084–4088. [Google Scholar]
- Tilocca, B.; Witzig, M.; Rodehutscord, M.; Seifert, J. Variations of phosphorous accessibility causing changes in microbiome functions in the gastrointestinal tract of chickens. PLoS ONE 2016, 11, e0164735. [Google Scholar] [CrossRef]
- Olsen, J.V.; Ong, S.-E.; Mann, M. Trypsin cleaves exclusively c-terminal to arginine and lysine residues. Mol. Cell. Proteomics. 2004, 3, 608–614. [Google Scholar] [CrossRef]
- Tilocca, B.; Burbach, K.; Heyer, C.M.E.; Camarinha-Silva, A.; Hölzle, L.E.; Mosenthin, R.; Stefanski, V.; Seifert, J. Dietary changes in nutritional studies shape the structural and functional composition of the pigs fecal microbiome—from days to weeks. Microbiome 2017, 5, 144. [Google Scholar] [CrossRef]
- Mesuere, B.; Debyser, G.; Aerts, M.; Devreese, B.; Vandamme, P.; Dawyndt, P. The unipept metaproteomics analysis pipeline. Proteomics 2015, 15, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhu, Z.; Fu, L.; Niu, B.; Li, W. Webmga: A customizable web server for fast metagenomic sequence analysis. BMC Genomics 2011, 12, 444. [Google Scholar] [CrossRef] [PubMed]
- Liebermeister, W.; Noor, E.; Flamholz, A.; Davidi, D.; Bernhardt, J.; Milo, R. Visual account of protein investment in cellular functions. P. Natl. Acad. Sci. USA 2014, 111, 8488–8493. [Google Scholar] [CrossRef] [PubMed]
- Vizcaino, J.A.; Csordas, A.; Del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the pride database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Wilfart, A.; Montagne, L.; Simmins, H.; Noblet, J.; Van Milgen, J. Effect of fibre content in the diet on the mean retention time in different segments of the digestive tract in growing pigs. Livest. Sci. 2007, 109, 27–29. [Google Scholar] [CrossRef]
- Heyer, C.M.E.; Schmucker, S.; Burbach, K.; Weiss, E.; Eklund, M.; Aumiller, T.; Capezzone, F.; Steuber, J.; Rodehutscord, M.; Hoelzle, L.E.; et al. Phytate degradation, intestinal microbiota, microbial metabolites, and immune values are changed in growing pigs fed diets with varying calcium-phosphorus concentration and fermentable substrates, in preparation.
- O’Callaghan, A.; van Sinderen, D. Bifidobacteria and their role as members of the human gut microbiota. Front. Microbiol. 2016, 7, 925. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.; Berrocoso, J. Review: Dietary fiber utilization and its effects on physiological functions and gut health of swine. Animal 2015, 9, 1441–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drissi, F.; Raoult, D.; Merhej, V. Metabolic role of lactobacilli in weight modification in humans and animals. Microb. Pathog. 2017, 106, 182–194. [Google Scholar] [CrossRef]
- Christensen, H.; Bisgaard, M. Taxonomy and biodiversity of members of Pasteurellaceae. In Pasteurellaceae: Biology, Genomics and Molecular Aspects; Caister Academic Press: Poole, UK, 2008; pp. 1–26. [Google Scholar]
- Polansky, O.; Sekelova, Z.; Faldynova, M.; Sebkova, A.; Sisak, F.; Rychlik, I. Important metabolic pathways and biological processes expressed by chicken cecal microbiota. Appl. Environ. Microbiol. 2016, 82, 1569–1576. [Google Scholar] [CrossRef]
- Brestenský, M.; Nitrayová, S.; Bomba, A.; Patráš, P.; Strojný, L.; Szabadošová, V.; Pramuková, B.; Bertková, I. The content of short chain fatty acids in the jejunal digesta, caecal digesta and faeces of growing pigs. Livest. Sci. 2017, 205, 106–110. [Google Scholar] [CrossRef]
- Al-mosauwi, H.; Ryan, E.; McGrane, A.; Riveros-Beltran, S.; Walpole, C.; Dempsey, E.; Courtney, D.; Fearon, N.; Winter, D.; Baird, A.; et al. Differential protein abundance of a basolateral mct1 transporter in the human gastrointestinal tract. Cell Biol. Int. 2016, 40, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Hegele, R.A.; Ramdath, D.D.; Ban, M.R.; Carruthers, M.N.; Carrington, C.V.; Cao, H. Polymorphisms in pnlip, encoding pancreatic lipase, and associations with metabolic traits. J. Hum. Gen. 2001, 46, 320–324. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Jejunum | Ileum | Caecum | Colon | |||||
|---|---|---|---|---|---|---|---|---|
| Ø | SD | Ø | SD | Ø | SD | Ø | SD | |
| Actinobacteria | ||||||||
| Atopobiaceae | 0.3 | 0.5 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Bifidobacteriaceae | 5.7 | 2.9 | 5.6 | 3.5 | 1.1 | 0.7 | 0.3 | 0.6 |
| Streptomycetaceae | 0.3 | 0.5 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Bacteroidetes | ||||||||
| Bacteroidaceae | 1.2 | 1.5 | 1.0 | 1.7 | 5.9 | 2.0 | 5.0 | 1.2 |
| Porphyromonadaceae | 0.6 | 0.6 | 0.4 | 0.6 | 2.5 | 0.5 | 2.6 | 0.4 |
| Prevotellaceae | 20.7 | 19.9 | 17.7 | 18.3 | 26.0 | 3.9 | 27.2 | 3.1 |
| Rikenellaceae | 0.0 | 0.0 | 0.0 | 0.0 | 0.3 | 0.6 | 0.7 | 0.7 |
| Firmicutes | ||||||||
| Acidaminococcaceae | 0.9 | 0.5 | 1.6 | 1.3 | 2.1 | 0.3 | 2.4 | 0.3 |
| Clostridiaceae | 13.0 | 6.0 | 15.7 | 6.4 | 10.4 | 2.2 | 11.6 | 3.3 |
| Erysipelotrichaceae | 1.7 | 1.0 | 1.9 | 1.2 | 1.1 | 0.7 | 1.2 | 0.7 |
| Eubacteriaceae | 1.3 | 0.8 | 1.7 | 1.1 | 2.5 | 0.7 | 2.3 | 0.8 |
| Lachnospiraceae | 4.7 | 0.8 | 3.8 | 1.2 | 7.8 | 1.2 | 8.7 | 0.9 |
| Lactobacillaceae | 15.0 | 7.8 | 17.7 | 10.7 | 8.9 | 2.0 | 6.4 | 1.4 |
| Oscillospiraceae | 0.5 | 0.9 | 0.4 | 0.6 | 1.0 | 1.1 | 1.1 | 1.1 |
| Paenibacillaceae | 0.3 | 0.6 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Peptostreptococcaceae | 1.5 | 1.5 | 1.8 | 1.9 | 0.7 | 0.7 | 0.9 | 0.9 |
| Ruminococcaceae | 3.0 | 1.1 | 2.7 | 1.8 | 7.6 | 2.5 | 6.9 | 3.3 |
| Selenomonadaceae | 8.9 | 3.3 | 8.9 | 3.2 | 5.8 | 2.1 | 5.6 | 2.0 |
| Streptococcaceae | 1.7 | 1.1 | 2.1 | 1.7 | 1.7 | 1.6 | 2.0 | 1.7 |
| Veillonellaceae | 10.5 | 6.3 | 8.8 | 5.7 | 5.6 | 2.5 | 4.8 | 2.0 |
| Proteobacteria | ||||||||
| Pasteurellaceae | 0.3 | 0.5 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Spirochaetes | ||||||||
| Brachyspiraceae | 0.8 | 0.8 | 0.6 | 1.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Leptospiraceae | 0.3 | 0.5 | 0.0 | 0.0 | 0.0 | 0.0 | 0.3 | 0.4 |
| other | 6.9 | 2.1 | 7.8 | 1.0 | 9.0 | 1.7 | 10.1 | 0.3 |
| no. of peptides | 192 | 46.2 | 176 | 60.1 | 509 | 75 | 486 | 95 |
| Stomach | Jejunum | Ileum | Caecum | Colon | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ø | SD | Ø | SD | Ø | SD | Ø | SD | Ø | SD | |
| Actinobacteria | ||||||||||
| Atopobiaceae | 0.4 | 0.7 | 0.5 | 0.9 | 0.5 | 0.9 | 0.8 | 0.8 | 0.4 | 0.7 |
| Bifidobacteriaceae | 1.2 | 1.3 | 0.0 | 0.0 | 1.6 | 2.7 | 0.0 | 0.0 | 0.4 | 0.7 |
| Coriobacteriaceae | 0.0 | 0.0 | 0.0 | 0.0 | 1.6 | 2.7 | 0.0 | 0.0 | 0.0 | 0.0 |
| Eggerthellaceae | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.3 | 0.6 | 0.7 | 0.7 |
| Streptomycetaceae | 1.9 | 2.6 | 4.6 | 3.7 | 4.1 | 1.5 | 2.3 | 1.1 | 1.9 | 0.6 |
| Bacteroidetes | ||||||||||
| Bacteroidaceae | 2.4 | 2.2 | 3.5 | 4.1 | 3.8 | 3.8 | 5.4 | 4.4 | 5.0 | 2.2 |
| Flavobacteriaceae | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.7 | 1.8 | 1.5 | 1.5 |
| Porphyromonadaceae | 0.6 | 0.6 | 0.7 | 1.3 | 0.0 | 0.0 | 1.5 | 1.7 | 2.2 | 1.3 |
| Prevotellaceae | 20.3 | 7.8 | 19.8 | 4.5 | 14.7 | 4.3 | 22.3 | 8.5 | 26.5 | 6.3 |
| Rhodothermaceae | 1.6 | 2.7 | 4.1 | 4.1 | 3.6 | 2.3 | 1.9 | 1.5 | 2.2 | 0.6 |
| Rikenellaceae | 0.8 | 1.3 | 0.0 | 0.0 | 0.0 | 0.0 | 0.5 | 0.8 | 0.0 | 0.0 |
| Firmicutes | ||||||||||
| Acidaminococcaceae | 0.7 | 0.7 | 1.2 | 1.3 | 0.5 | 0.9 | 0.8 | 0.8 | 1.1 | 0.7 |
| Anaerolineaceae | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.3 | 0.6 | 0.7 | 0.7 |
| Clostridiaceae | 4.3 | 3.6 | 5.7 | 4.9 | 6.8 | 10.6 | 5.8 | 4.5 | 10.5 | 7.1 |
| Erysipelotrichaceae | 0.8 | 0.8 | 0.5 | 0.9 | 0.5 | 0.9 | 0.7 | 1.2 | 3.3 | 1.5 |
| Eubacteriaceae | 1.1 | 1.1 | 1.2 | 2.1 | 0.0 | 0.0 | 1.5 | 1.7 | 3.2 | 2.1 |
| Lachnospiraceae | 7.7 | 3.4 | 9.8 | 1.8 | 8.4 | 3.8 | 8.9 | 3.1 | 7.5 | 2.0 |
| Lactobacillaceae | 19.0 | 14.4 | 18.0 | 18.7 | 19.9 | 15.0 | 17.9 | 8.9 | 8.0 | 3.1 |
| Oscillospiraceae | 0.3 | 0.5 | 0.0 | 0.0 | 1.1 | 2.0 | 1.3 | 1.5 | 0.0 | 0.0 |
| Paenibacillaceae | 0.0 | 0.0 | 1.0 | 1.0 | 0.9 | 1.5 | 0.3 | 0.6 | 0.7 | 0.7 |
| Peptoniphilaceae | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.3 | 0.6 | 0.7 | 0.7 |
| Peptostreptococcaceae | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.9 | 1.5 | 0.7 | 0.7 |
| Ruminococcaceae | 2.5 | 2.3 | 3.0 | 3.3 | 4.3 | 4.3 | 3.7 | 3.7 | 3.8 | 1.7 |
| Selenomonadaceae | 9.3 | 7.9 | 2.0 | 2.4 | 1.5 | 2.6 | 1.6 | 1.7 | 2.3 | 1.8 |
| Streptococcaceae | 2.3 | 2.4 | 4.6 | 3.7 | 4.1 | 1.5 | 2.7 | 0.9 | 1.5 | 1.0 |
| Symbiobacteriaceae | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.3 | 0.6 | 0.7 | 0.7 |
| Veillonellaceae | 10.2 | 6.9 | 3.7 | 4.1 | 4.8 | 4.8 | 5.4 | 3.8 | 2.4 | 3.5 |
| Nitrospirae | ||||||||||
| Nitrospiraceae | 0.4 | 0.7 | 0.5 | 0.9 | 0.5 | 0.9 | 0.5 | 0.8 | 0.4 | 0.7 |
| Proteobacteria | ||||||||||
| Alcaligenaceae | 0.0 | 0.0 | 2.3 | 3.9 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Anaplasmataceae | 0.0 | 0.0 | 2.8 | 3.7 | 0.5 | 0.9 | 0.5 | 0.8 | 0.8 | 0.8 |
| Burkholderiaceae | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.4 | 0.7 |
| Comamonadaceae | 0.4 | 0.7 | 0.0 | 0.0 | 0.5 | 0.9 | 0.5 | 0.8 | 0.4 | 0.7 |
| Enterobacteriaceae | 0.0 | 0.0 | 0.0 | 0.0 | 1.1 | 2.0 | 1.9 | 1.5 | 0.7 | 0.7 |
| Helicobacteraceae | 0.8 | 1.3 | 2.3 | 3.9 | 0.9 | 1.5 | 0.5 | 0.8 | 1.1 | 1.2 |
| Pasteurellaceae | 1.4 | 1.4 | 1.5 | 1.7 | 8.0 | 6.1 | 3.3 | 2.0 | 3.4 | 0.5 |
| Pelagibacteraceae | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.3 | 0.6 | 0.7 | 0.7 |
| Vibrionaceae | 0.0 | 0.0 | 0.0 | 0.0 | 1.1 | 2.0 | 0.0 | 0.0 | 0.4 | 0.7 |
| Spirochaetes | ||||||||||
| Brachyspiraceae | 0.0 | 0.0 | 0.0 | 0.0 | 1.1 | 2.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Leptospiraceae | 2.4 | 2.3 | 2.3 | 2.9 | 1.6 | 1.9 | 1.5 | 1.7 | 1.1 | 1.2 |
| Synergistetes | ||||||||||
| Synergistaceae | 0.4 | 0.7 | 0.5 | 0.9 | 0.5 | 0.9 | 0.5 | 0.8 | 0.4 | 0.7 |
| Tenericutes | ||||||||||
| Mycoplasmataceae | 1.9 | 2.6 | 0.5 | 0.9 | 1.6 | 2.7 | 0.3 | 0.6 | 1.9 | 0.6 |
| other | 5.0 | 5.0 | 3.5 | 6.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| no. of peptides | 100.8 | 62.3 | 44.0 | 36.3 | 29.0 | 12.8 | 50.8 | 17.8 | 58.0 | 14.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tröscher-Mußotter, J.; Tilocca, B.; Stefanski, V.; Seifert, J. Analysis of the Bacterial and Host Proteins along and across the Porcine Gastrointestinal Tract. Proteomes 2019, 7, 4. https://doi.org/10.3390/proteomes7010004
Tröscher-Mußotter J, Tilocca B, Stefanski V, Seifert J. Analysis of the Bacterial and Host Proteins along and across the Porcine Gastrointestinal Tract. Proteomes. 2019; 7(1):4. https://doi.org/10.3390/proteomes7010004
Chicago/Turabian StyleTröscher-Mußotter, Johanna, Bruno Tilocca, Volker Stefanski, and Jana Seifert. 2019. "Analysis of the Bacterial and Host Proteins along and across the Porcine Gastrointestinal Tract" Proteomes 7, no. 1: 4. https://doi.org/10.3390/proteomes7010004
APA StyleTröscher-Mußotter, J., Tilocca, B., Stefanski, V., & Seifert, J. (2019). Analysis of the Bacterial and Host Proteins along and across the Porcine Gastrointestinal Tract. Proteomes, 7(1), 4. https://doi.org/10.3390/proteomes7010004