

One-Step Suicide Substrate Inactivation Kinetics of a Ping-Pong Reaction with One Substrate Undergoing Disproportionation: A Theoretical Approach with Approximate Solutions

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
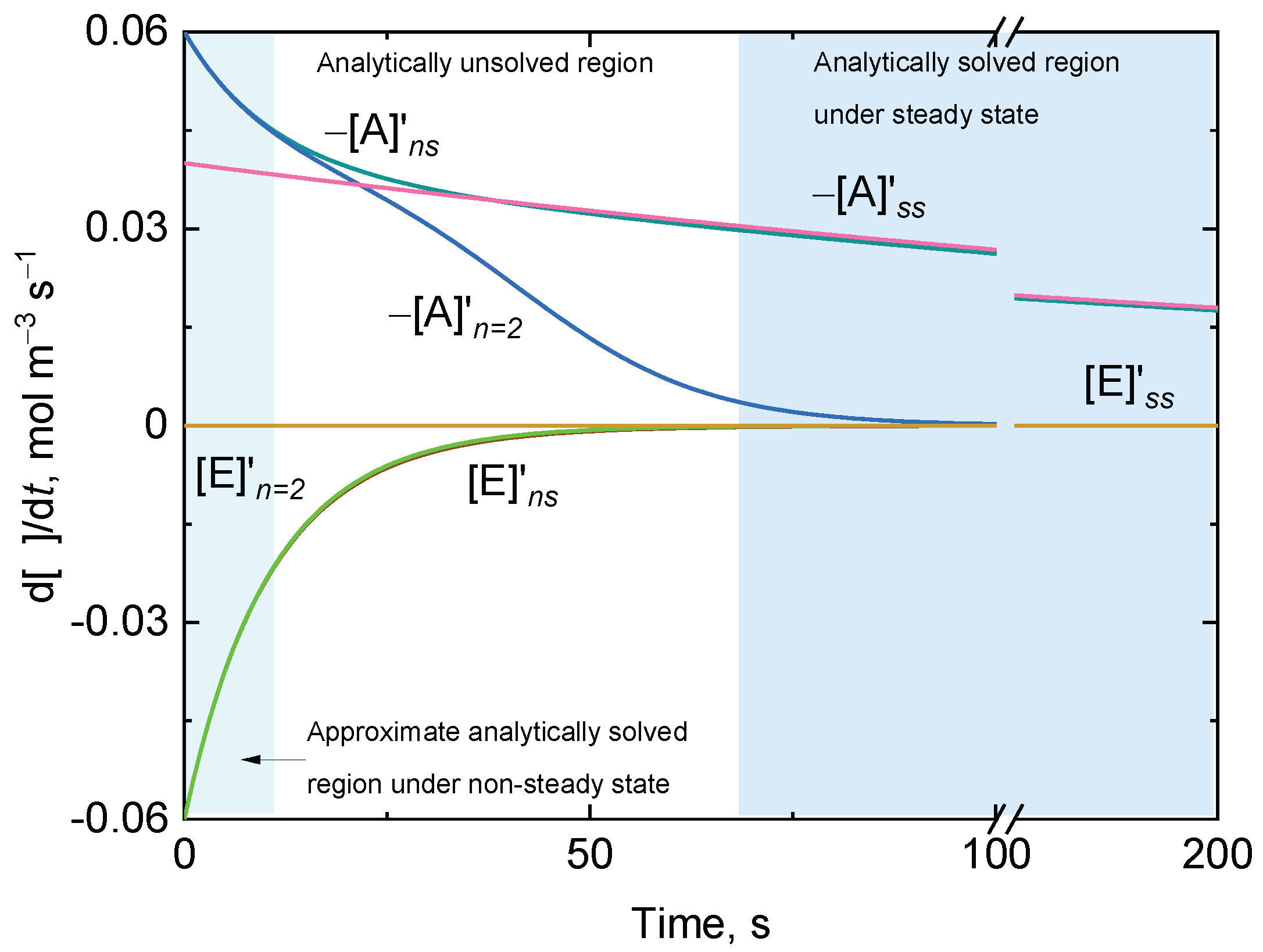
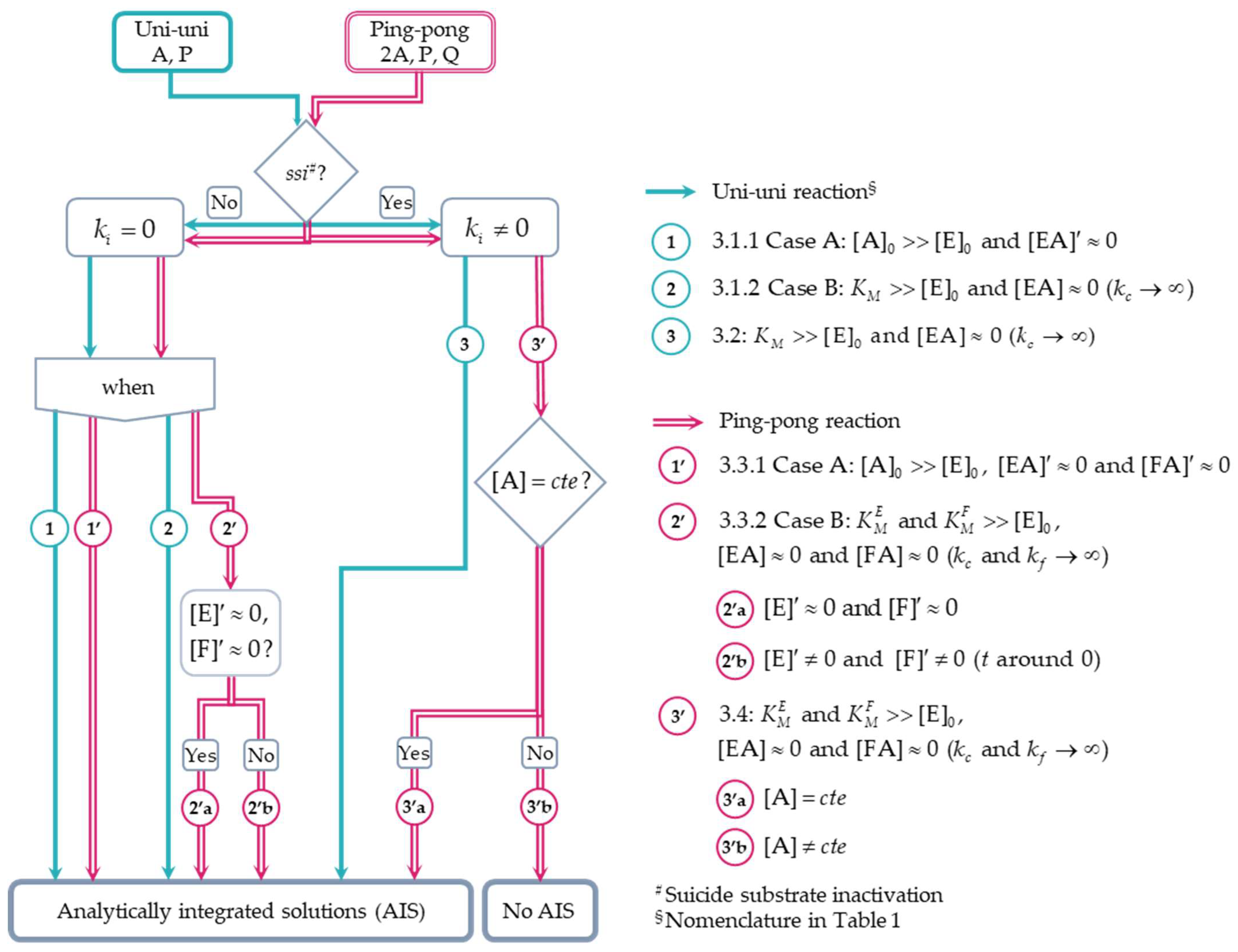
3.1. Approximate Analytical Solutions for the Irreversible Uni–Uni Michaelis–Menten Model in the Absence of Suicide Substrate Inactivation
3.1.1. Case A:
3.1.2. Case B:
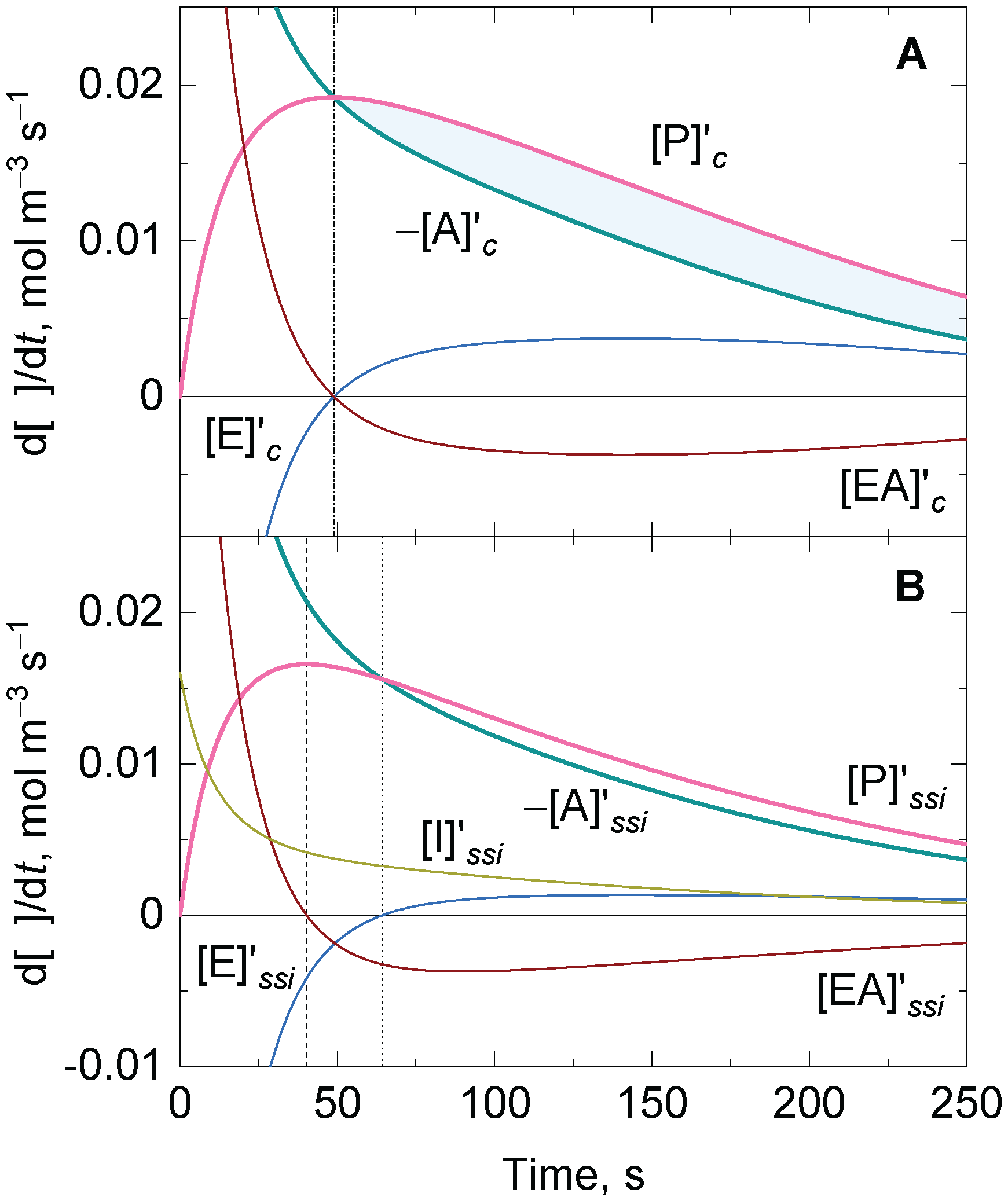
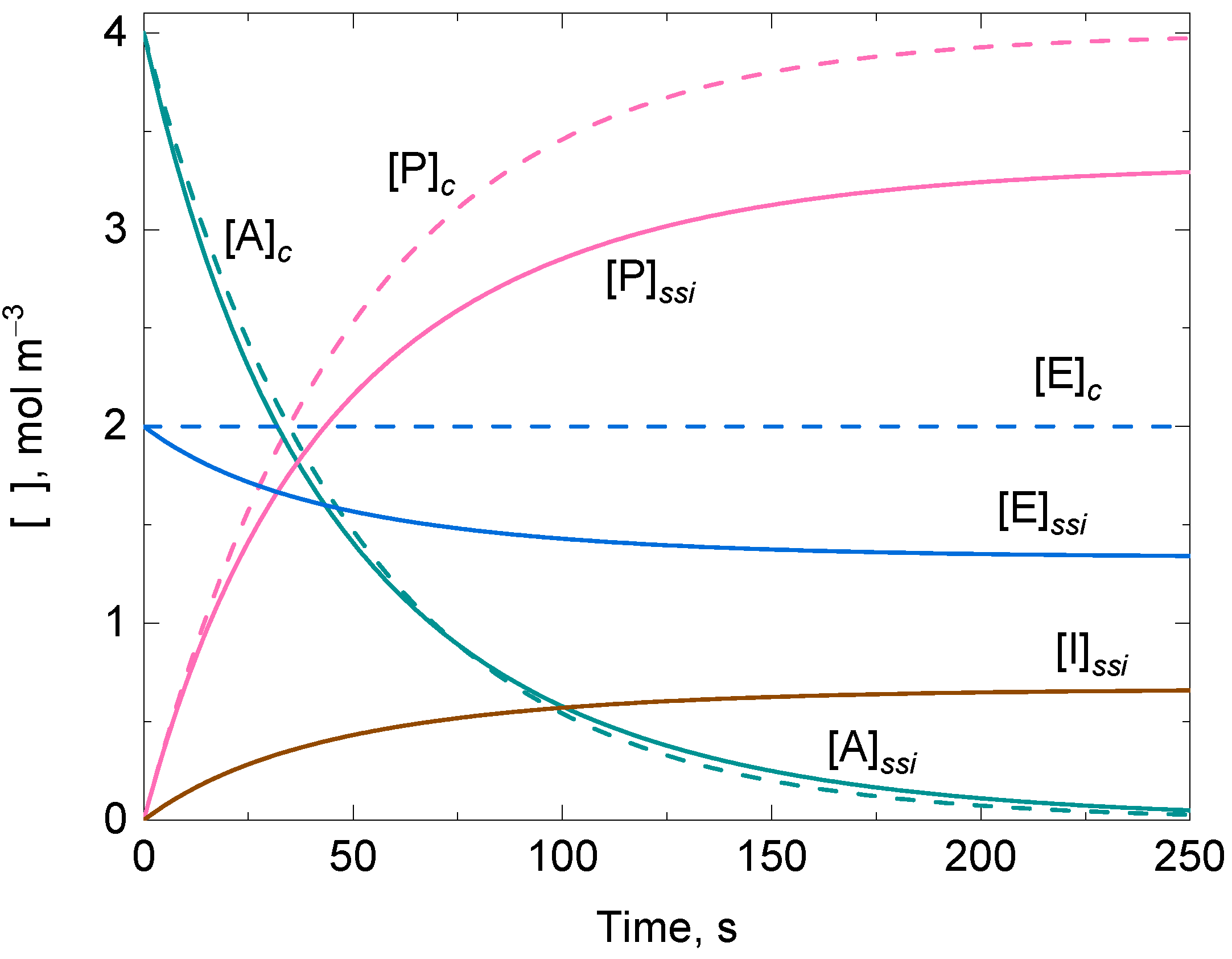
3.2. Approximate Analytical Solution for the Irreversible Uni–Uni Michaelis–Menten Model in the Presence of Suicide Substrate Inactivation
3.3. Analytical Solutions for an Enzyme-Catalyzed Ping-Pong Reaction with One Substrate Undergoing Disproportionation in the Absence of Suicide Substrate Inactivation
3.3.1. Case A:
3.3.2. Case B:
3.4. Analytical Solutions for an Enzyme-Catalyzed Ping-Pong Reaction with One Substrate Undergoing Disproportionation in the Presence of Suicide Substrate Inactivation
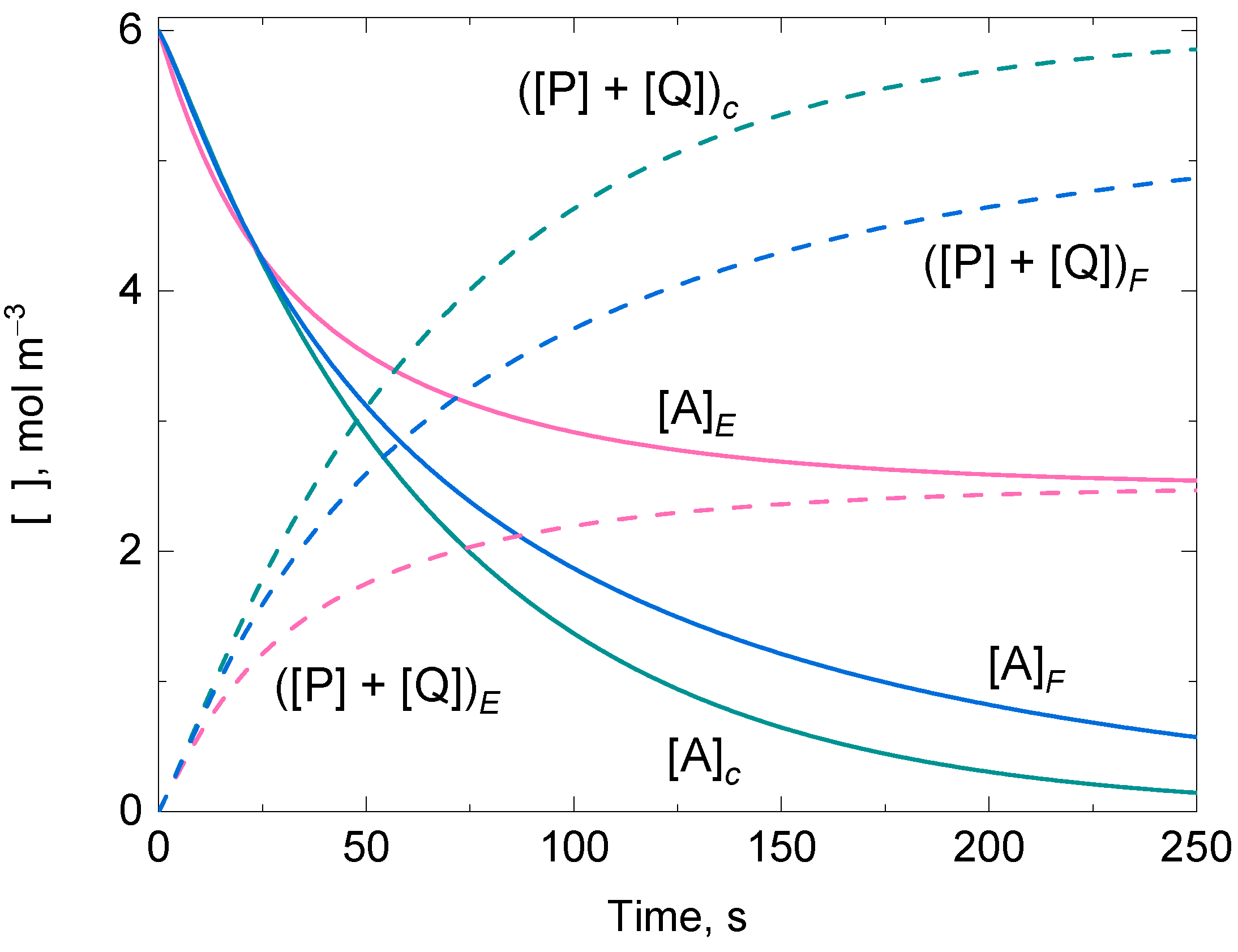
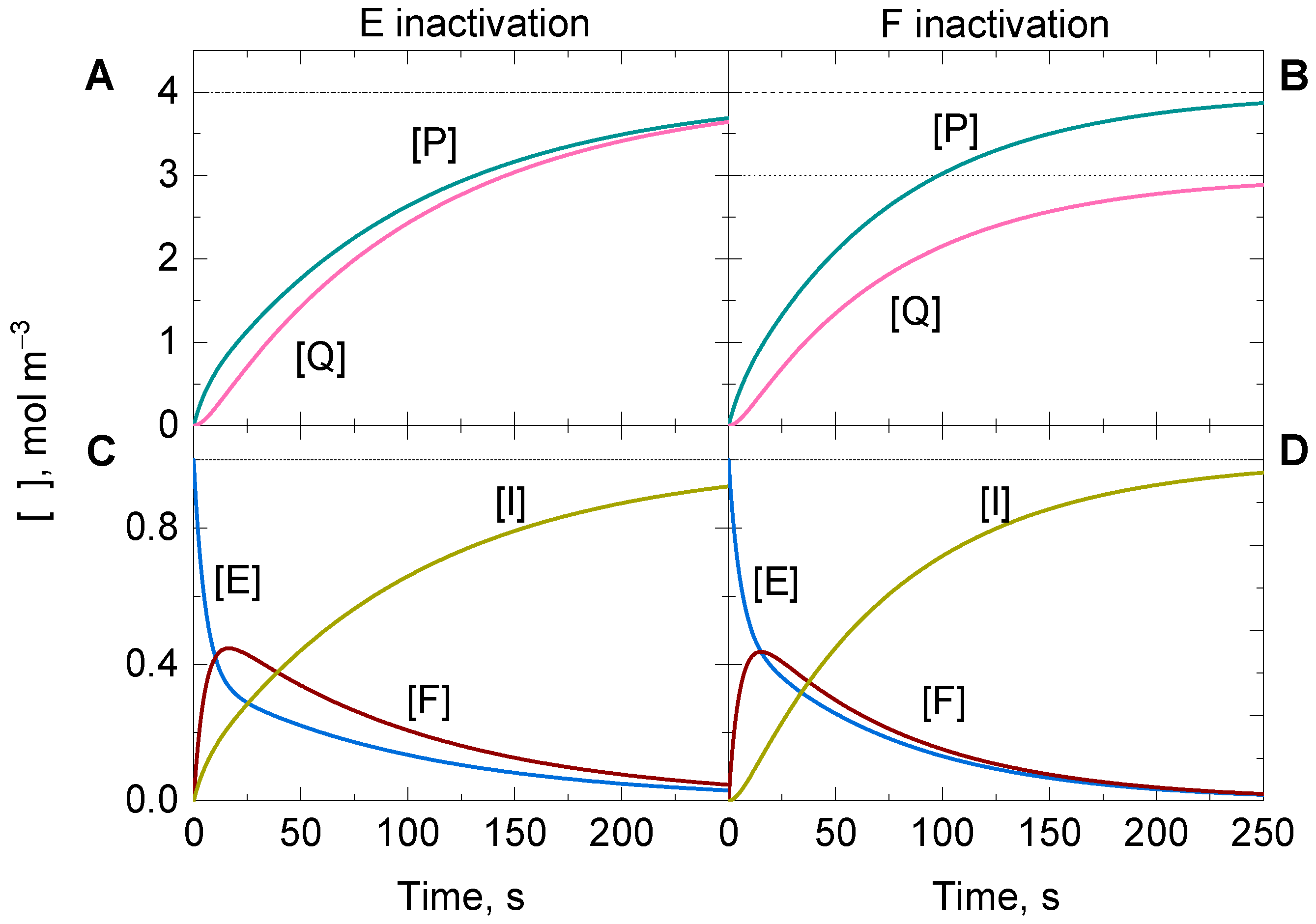
3.4.1. Case A: Suicide Substrate Inactivation on E
3.4.2. Case B: Suicide Substrate Inactivation on F
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mons, E.; Roet, S.; Kim, R.Q.; Mulder, M.P.C. A comprehensive guide for assessing covalent inhibition in enzymatic assays illustrated with kinetic simulations. Curr. Protoc. 2022, 2, e419. [Google Scholar] [CrossRef] [PubMed]
- Purich, D.L. Kinetic behavior of enzyme inhibitors. In Enzyme Kinetics: Catalysis and Control; Elsevier: London, UK, 2010; pp. 485–574. ISBN 978-0-12-380924-7. [Google Scholar]
- Hiratake, J. Enzyme inhibitors as chemical tools to study enzyme catalysis: Rational design, synthesis, and applications. Chem. Rec. 2005, 5, 209–228. [Google Scholar] [CrossRef] [PubMed]
- Geronikaki, A. Recent trends in enzyme inhibition and activation in drug design. Molecules 2021, 26, 17. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd ed.; Wiley: London, UK, 2013; ISBN 9781118488133. [Google Scholar]
- Punekar, N.S. Enzymes: Catalysis, Kinetics and Mechanisms; Springer: Singapore, 2018; ISBN 978-981-13-0784-3. [Google Scholar]
- Bisswanger, H. Enzyme Kinetics: Principles and Methods; Wiley: Weinheim, Germany, 2009; ISBN 978-3-527-30343-4. [Google Scholar]
- Segel, H.L. Enzyme Kinetics. Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems; John Wiley & Sons, Ltd: New York, NY, USA, 1975; ISBN 978-0-471-30309-1. [Google Scholar]
- Whiteley, C.G. Mechanistic and kinetic studies of inhibition of enzymes. Cell Biochem. Biophys. 2000, 33, 217–225. [Google Scholar] [CrossRef]
- Silverman, R.B. Mechanism-based enzyme inactivators. Methods Enzymol. 1995, 249, 240–283. [Google Scholar] [CrossRef]
- Abeles, R.H.; Maycock, A.L. Suicide enzyme inactivators. Acc. Chem. Res. 1976, 9, 313–319. [Google Scholar] [CrossRef]
- Walsh, C.T. Suicide substrates, mechanism-based enzyme inactivators: Recent developments. Annu. Rev. Biochem. 1984, 53, 493–535. [Google Scholar] [CrossRef]
- Waley, S.G. Kinetics of suicide substrates. Biochem. J. 1980, 185, 771–773. [Google Scholar] [CrossRef]
- Tatsunami, S.; Yago, N.; Hosoe, M. Kinetics of suicide substrates steady-state treatments and computer-aided exact solutions. Biochim. Biophys. Acta. 1981, 662, 226–235. [Google Scholar] [CrossRef]
- Michaelis, L.; Menten, M. The kinetics of the inversion effect. Biochem. Z. 1913, 49, 333–369. [Google Scholar]
- Henri, V. Théorie générale de l’action de quelques diastases par Victor Henri. C.R. Acad. Sci. Paris 1902, 135, 916–919. [Google Scholar] [CrossRef]
- Borghans, J.A.M.; De Boer, R.J.; Segel, L.A. Extending the quasi-steady state approximation by changing variables. Bull. Math. Biol. 1996, 58, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Bersani, A.M.; Bersani, E.; Dell’Acqua, G.; Pedersen, M.G. New trends and perspectives in nonlinear intracellular dynamics: One century from Michaelis–Menten paper. Contin. Mech. Thermodyn. 2015, 27, 659–684. [Google Scholar] [CrossRef]
- Briggs, G.E.; Haldane, J.B.S. A note on the kinetics of enzyme action. Biochem. J. 1925, 19, 338. [Google Scholar] [CrossRef] [Green Version]
- Swoboda, P.A. The kinetics of enzyme action. Biochim. Biophys. Acta 1957, 23, 70–80. [Google Scholar] [CrossRef]
- Hommes, F.A. Integrated Michaelis-Menten equation. Arch. Biochem. Biophys. 1962, 96, 28–31. [Google Scholar] [CrossRef]
- Laidler, K.J. Theory of the transient phase in kinetics, with special reference to enzyme systems. Can. J. Chem. 1955, 33, 1614–1624. [Google Scholar] [CrossRef]
- Segel, L.A. On the validity of the steady state assumption of enzyme kinetics. Bull. Math. Biol. 1988, 50, 579–593. [Google Scholar] [CrossRef]
- Schnell, S. Validity of the Michaelis-Menten equation—Steady-state or reactant stationary assumption: That is the question. FEBS J. 2014, 281, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Waley, S.G. Kinetics of suicide substrates. Practical procedures for determining parameters. Biochem. J. 1985, 227, 843–849. [Google Scholar] [CrossRef] [Green Version]
- Goeke, A.; Schilli, C.; Walcher, S.; Zerz, E. A note on the kinetics of suicide substrates. J. Math. Chem. 2012, 50, 1373–1377. [Google Scholar] [CrossRef]
- Wang, Z.X. Kinetics of suicide substrates. J. Theor. Biol. 1990, 147, 497–508. [Google Scholar] [CrossRef]
- Dhatt, S. Indicators for suicide substrate inactivation: A kinetic investigation. J. Chem. Sci. 2017, 129, 1921–1928. [Google Scholar] [CrossRef] [Green Version]
- Galvez, J.; Varon, R.; Garcia-Carmona, F., III. Kinetics of enzyme reactions with inactivation steps. J. Theor. Biol. 1981, 89, 37–44. [Google Scholar] [CrossRef]
- Duggleby, R.G. Progress curves of reactions catalyzed by unstable enzymes. A theoretical approach. J. Theor. Biol. 1986, 123, 67–80. [Google Scholar] [CrossRef]
- Tudela, J.; Garcia-Canovas, F.; Varon, R.; Garcia-Carmona, F.; Galvez, J.; Lozano, J.A. Transient-phase kinetics of enzyme inactivation induced by suicide substrates. Biochim. Biophys. Acta 1987, 912, 408–416. [Google Scholar] [CrossRef]
- Varon, R.; Garcia, M.; Garcia-Canovas, F.; Tudela, J. Transient-phase kinetics of enzyme inactivation induced by suicide substrates: Enzymes involving two substrates. J. Mol. Catal. 1990, 59, 97–118. [Google Scholar] [CrossRef]
- Galvez, J.; Varon, R.I. Transient phase kinetics of enzyme reactions. J. Theor. Biol. 1981, 89, 1–17. [Google Scholar] [CrossRef]
- Feuers, R.J.; Pattillo, F.M.; Osborn, C.K.; Adams, K.L.; DeLuca, D.; Grady Smith, W. Application of an integrated rate equation to the inactivation of catalase. Free Radic. Biol. Med. 1993, 15, 223–226. [Google Scholar] [CrossRef]
- Moosavi Movahedi, A.A.; Nazari, K.; Ghadermarzi, M. Suicide inactivation of peroxidase by H2O2: Kinetic equations for peroxidatic oxidation reaction of guaiacol and determination of the kinetic parameters. Ital. J. Biochem. 1999, 48, 9–17. [Google Scholar]
- Nazari, K.; Mahmoudi, A.; Khosraneh, M.; Haghighian, Z.; Moosavi-Movahedi, A.A. Kinetic analysis for suicide-substrate inactivation of microperoxidase-11: A modified model for bisubstrate enzymes in the presence of reversible inhibitors. J. Mol. Catal. B Enzym. 2009, 56, 61–69. [Google Scholar] [CrossRef]
- Nicholls, P.; Fita, I.; Loewen, P.C. Enzymology and structure of catalases. In Advances in Inorganic Chemistry; Sykes, A.G., Mauk, G., Eds.; Academic Press: San Diego, CA, USA, 2001; Volume 51, pp. 51–106. ISBN 0-12-023651-6. [Google Scholar]
- Trawczyńska, I. New method of determining kinetic parameters for decomposition of hydrogen peroxide by catalase. Catalysts 2020, 10, 323. [Google Scholar] [CrossRef]
- Miłek, J. Estimation of the kinetic parameters for H2O2 enzymatic decomposition and for catalase deactivation. Braz. J. Chem. Eng. 2018, 35, 995–1004. [Google Scholar] [CrossRef] [Green Version]
- DeLuca, D.C.; Dennis, R.; Smith, W.G. Inactivation of an animal and a fungal catalase by hydrogen peroxide. Arch. Biochem. Biophys. 1995, 320, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Bendou, O.; Gutiérrez-Fernández, I.; Marcos-Barbero, E.L.; Bueno-Ramos, N.; González-Hernández, A.I.; Morcuende, R.; Arellano, J.B. Theoretical and experimental considerations for a rapid and high throughput measurement of catalase in vitro. Antioxidants 2022, 11, 21. [Google Scholar] [CrossRef] [PubMed]
- Wolfram Research, Inc. Mathematica, Version 12.2; Wolfram Research, Inc.: Champaign, IL, USA, 2020. [Google Scholar]
- Schnell, S.; Mendoza, C. Closed form solution for time-dependent enzyme kinetics. J. Theor. Biol. 1997, 187, 207–212. [Google Scholar] [CrossRef]
- Fridovich, I. Superoxide dismutase. In Encyclopedia of Biological Chemistry, 2nd ed.; Lennarz, W., Lane, M., Eds.; Academic Press: Amsterdam, The Netherlands, 2013; pp. 352–354. ISBN 9780123786319. [Google Scholar]
- Galvez, J.; Varon, R.; Garcia-Canovas, F.; Carmona, F.G., IV. Transient phase of the uni-bi mechanisms. J. Theor. Biol. 1982, 94, 413–420. [Google Scholar] [CrossRef]
- Maguire, R.J.; Hijazi, N.H.; Laidler, K.J. Transient-phase kinetics of α-chymotrypsin and other enzyme systems. Biochim. Biophys. Acta 1974, 341, 1–14. [Google Scholar] [CrossRef]
- Alberty, R.A. The relationship between Michaelis constants, maximum velocities and the equilibrium constant for an enzyme-catalyzed reaction. J. Am. Chem. Soc. 1953, 75, 1928–1932. [Google Scholar] [CrossRef]
- King, E.L.; Altman, C. A schematic method of deriving the rate laws for enzyme-catalyzed reactions. J. Phys. Chem. 1956, 60, 1375–1378. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nomenclature | Definition |
|---|---|
| Compounds | |
| A | Reaction (suicide) substrate |
| E | Active enzyme |
| EA | Intermediate substrate–enzyme complex |
| EX | Second intermediate substrate–enzyme complex in the Waley model |
| F | Intermediate active enzyme of a ping-pong reaction |
| FA | Intermediate substrate–enzyme complex of a ping-pong reaction |
| Inactive enzyme of the Waley model | |
| Inactive enzyme of a one-step suicide substrate inactivation reaction | |
| P | (First) reaction product (of a ping-pong reaction) |
| Q | Second reaction product of a ping-pong reaction |
| Constants | |
| , mol·m−3 | |
| , mol·m−3 | |
| , mol·m−3 | |
| Second-order forward rate constant, m3·(mol·s)−1 | |
| Second-order rate constant for H2O2 decomposition, m3·(mol·s)−1 | |
| First-order backward rate constant, s−1 | |
| First-order forward rate constant, s−1 | |
| Second-order forward rate constant, m3·(mol·s)−1 | |
| First-order backward rate constant, s−1 | |
| First-order forward rate constant, s−1 | |
| , s−1 | |
| Second-order forward rate constant for the formation of I, m3·(mol·s)−1 | |
| Second-order rate constant for catalase inactivation, m3·(mol·s)−1 | |
| First-order forward rate constant for the decomposition of EX, s−1 | |
| First-order forward rate constant for the formation of EX, s−1 | |
| , m3·(mol·s)−1 | |
| , m3·(mol·s)−1 | |
| , m3·(mol·s)−1 | |
| Subscripts | |
| E | Enzyme state inactivated by suicide substrate |
| F | Intermediate enzyme state inactivated by suicide substrate |
| max | Maximum |
| (n)ss | (Non)quasi-steady state |
| n | Grade of the polynomial of a power expansion series |
| ssi | Suicide substrate inactivation |
| 0 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutiérrez-Fernández, I.; Bendou, O.; Bueno-Ramos, N.; Marcos-Barbero, E.L.; Morcuende, R.; Arellano, J.B. One-Step Suicide Substrate Inactivation Kinetics of a Ping-Pong Reaction with One Substrate Undergoing Disproportionation: A Theoretical Approach with Approximate Solutions. Mathematics 2022, 10, 4240. https://doi.org/10.3390/math10224240
Gutiérrez-Fernández I, Bendou O, Bueno-Ramos N, Marcos-Barbero EL, Morcuende R, Arellano JB. One-Step Suicide Substrate Inactivation Kinetics of a Ping-Pong Reaction with One Substrate Undergoing Disproportionation: A Theoretical Approach with Approximate Solutions. Mathematics. 2022; 10(22):4240. https://doi.org/10.3390/math10224240
Chicago/Turabian StyleGutiérrez-Fernández, Ismael, Ouardia Bendou, Nara Bueno-Ramos, Emilio L. Marcos-Barbero, Rosa Morcuende, and Juan B. Arellano. 2022. "One-Step Suicide Substrate Inactivation Kinetics of a Ping-Pong Reaction with One Substrate Undergoing Disproportionation: A Theoretical Approach with Approximate Solutions" Mathematics 10, no. 22: 4240. https://doi.org/10.3390/math10224240
APA StyleGutiérrez-Fernández, I., Bendou, O., Bueno-Ramos, N., Marcos-Barbero, E. L., Morcuende, R., & Arellano, J. B. (2022). One-Step Suicide Substrate Inactivation Kinetics of a Ping-Pong Reaction with One Substrate Undergoing Disproportionation: A Theoretical Approach with Approximate Solutions. Mathematics, 10(22), 4240. https://doi.org/10.3390/math10224240