

Persistent Homology Identifies Pathways Associated with Hepatocellular Carcinoma from Peripheral Blood Samples
 , , and
, , and
Abstract
:1. Introduction
2. Topological Data Analysis: Background
2.1. Simplicial Complex and Homology
2.2. Persistent Homology
3. Materials and Methods
| Algorithm 1: Persistent Homology Analysis of Gene Expression Data |
Input:
|
3.1. PBMC Gene Expression Data
3.2. KEGG Pathways
3.3. PH Implementation
3.4. Topological Descriptors
3.5. Differential Expression Analysis
3.6. Enrichment Analysis of Pathways
3.7. Significance of Topological Descriptors
4. Results
4.1. Genome-Wide Persistent Homology Analysis
4.2. Choice of Relevant Topological Descriptors
4.3. Classical Differential Gene Expression Analysis and Enrichment-Based Pathway Analysis
4.4. Persistent Homology Analysis for Pathway-Specific Gene Sets
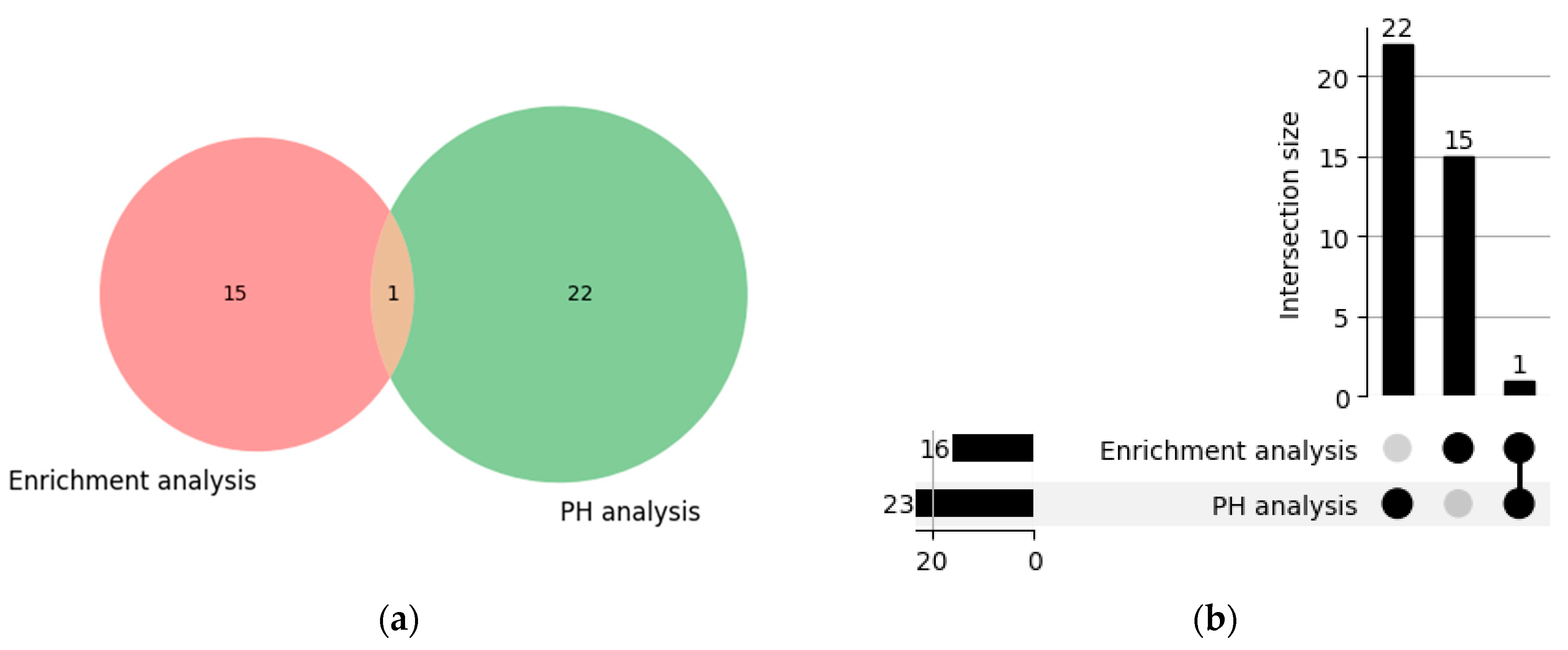
4.5. Comparison of Pathways Identified by the PH Method and the Enrichment Analysis
5. Discussion
6. Conclusions and Future Research
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FDR | False discovery rate |
| HCC | Hepatocellular carcinoma |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| PBMC | Peripheral blood mononuclear cell |
| PH | Persistent homology |
| RNA-seq | RNA sequencing |
| TDA | Topological data analysis |
| TPM | Transcripts per million |
| VR | Vietoris–Rips |
Appendix A. Simplicial Complex and Homology
- for any simplex , all its faces must be in ;
- if for any two simplices then either or is a common face of and .


Appendix B. Kernel Density Estimator (KDE) Plots

References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Sun, J.; Guo, R.; Bi, X.; Wu, M.; Tang, Z.; Lau, W.Y.; Zheng, S.; Wang, X.; Yu, J.; Chen, X.; et al. Guidelines for Diagnosis and Treatment of Hepatocellular Carcinoma with Portal Vein Tumor Thrombus in China (2021 Edition). Liver Cancer 2022, 11, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Shahini, E.; Pasculli, G.; Solimando, A.G.; Tiribelli, C.; Cozzolongo, R.; Giannelli, G. Updating the Clinical Application of Blood Biomarkers and Their Algorithms in the Diagnosis and Surveillance of Hepatocellular Carcinoma: A Critical Review. Int. J. Mol. Sci. 2023, 24, 4286. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO International Programme on Chemical Safety Biomarkers and Risk Assessment: Concepts and Principles; World Health Organization: Geneva, Switzerland, 1993; Available online: http://www.inchem.org/documents/ehc/ehc/ehc155.htm (accessed on 4 January 2024).
- Lok, A.S.; Sterling, R.K.; Everhart, J.E.; Wright, E.C.; Hoefs, J.C.; Di Bisceglie, A.M.; Morgan, T.R.; Kim, H.; Lee, W.M.; Bonkovsky, H.L.; et al. Des-γ-Carboxy Prothrombin and α-Fetoprotein as Biomarkers for the Early Detection of Hepatocellular Carcinoma. Gastroenterology 2010, 138, 493–502. [Google Scholar] [CrossRef]
- Marrero, J.A.; Feng, Z.; Wang, Y.; Nguyen, M.H.; Befeler, A.S.; Roberts, L.R.; Reddy, K.R.; Harnois, D.; Llovet, J.M.; Normolle, D.; et al. α-Fetoprotein, Des-γ Carboxyprothrombin, and Lectin-Bound α-Fetoprotein in Early Hepatocellular Carcinoma. Gastroenterology 2009, 137, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Thomas London, W.; Petrick, J.L.; McGlynn, K.A. Liver Cancer. In Cancer Epidemiology and Prevention, 4th ed.; Thun, M.J., Linet, M.S., Cerhan, J.R., Haiman, C.A., Schittenfeld, D., Eds.; Oxford University Press: New York, NY, USA, 2017; pp. 635–660. ISBN 9780190238667. [Google Scholar]
- Chazal, F.; Michel, B. An Introduction to Topological Data Analysis: Fundamental and Practical Aspects for Data Scientists. Front. Artif. Intell. 2021, 4, 667963. [Google Scholar] [CrossRef]
- Carlsson, G. Topology and Data. Bull. Am. Math. Soc. 2009, 46, 255–308. [Google Scholar] [CrossRef]
- Edelsbrunner, H.; Harer, J. Computational Topology: An Introduction; American Mathematical Society (AMS): Providence, RI, USA, 2010. [Google Scholar] [CrossRef]
- Skaf, Y.; Laubenbacher, R. Topological Data Analysis in Biomedicine: A Review. J. Biomed. Inform. 2022, 130, 104082. [Google Scholar] [CrossRef]
- Conti, F.; Moroni, D.; Pascali, M.A. A Topological Machine Learning Pipeline for Classification. Mathematics 2022, 10, 3086. [Google Scholar] [CrossRef]
- Du, Y.; Zhang, M.; Stonis, G.; Juan, S. Topological Data Analysis on Magnetic Resonance Image Biomarkers. In Proceedings of the 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), San Diego, CA, USA, 18–21 November 2019; pp. 1185–1187. [Google Scholar]
- Nielson, J.L.; Cooper, S.R.; Yue, J.K.; Sorani, M.D.; Inoue, T.; Yuh, E.L.; Mukherjee, P.; Petrossian, T.C.; Paquette, J.; Lum, P.Y.; et al. Uncovering Precision Phenotype-Biomarker Associations in Traumatic Brain Injury Using Topological Data Analysis. PLoS ONE 2017, 12, e0169490. [Google Scholar] [CrossRef]
- Asaad, A.; Ali, D.; Majeed, T.; Rashid, R. Persistent Homology for Breast Tumor Classification Using Mammogram Scans. Mathematics 2022, 10, 4039. [Google Scholar] [CrossRef]
- Malek, A.A.; Alias, M.A.; Razak, F.A.; Noorani, M.S.M.; Mahmud, R.; Zulkepli, N.F.S. Persistent Homology-Based Machine Learning Method for Filtering and Classifying Mammographic Microcalcification Images in Early Cancer Detection. Cancers 2023, 15, 2606. [Google Scholar] [CrossRef] [PubMed]
- Aslam, J.; Ardanza-Trevijano, S.; Xiong, J.; Arsuaga, J.; Sazdanovic, R. TAaCGH Suite for Detecting Cancer—Specific Copy Number Changes Using Topological Signatures. Entropy 2022, 24, 896. [Google Scholar] [CrossRef]
- Penrice-Randal, R.; Dong, X.; Shapanis, A.; Gardener, A.I.; Harding, N.; Legebeke, J.; Lord, J.; Vallejo Pulido, A.; Poole, S.; Brendish, N.J.; et al. Blood Gene Expression Predicts Intensive Care Unit Admission in Hospitalised Patients with COVID-19. Front. Immunol. 2022, 13, 988685. [Google Scholar] [CrossRef]
- Blair, P.W.; Brandsma, J.; Chenoweth, J.; Richard, S.A.; Epsi, N.J.; Mehta, R.; Striegel, D.; Clemens, E.G.; Alharthi, S.; Lindholm, D.A.; et al. Distinct Blood Inflammatory Biomarker Clusters Stratify Host Phenotypes during the Middle Phase of COVID-19. Sci. Rep. 2022, 12, 22471. [Google Scholar] [CrossRef] [PubMed]
- Shapanis, A.; Jones, M.G.; Schofield, J.; Skipp, P. Topological Data Analysis Identifies Molecular Phenotypes of Idiopathic Pulmonary Fibrosis. Thorax 2023, 78, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Feng, W.; Hu, R.; Ge, Q.; Ma, W.; Zhang, W.; Xu, S.; Zhan, B.; Zhang, L.; Sun, X.; et al. RNA-Seq Profiling Reveals PBMC RNA as a Potential Biomarker for Hepatocellular Carcinoma. Sci. Rep. 2021, 11, 17797. [Google Scholar] [CrossRef]
- Fell, D.A. Increasing the Flux in Metabolic Pathways: A Metabolic Control Analysis Perspective. Biotechnol. Bioeng. 1998, 58, 121–124. [Google Scholar] [CrossRef]
- Ryu, H.; Chung, M.; Dobrzyński, M.; Fey, D.; Blum, Y.; Lee, S.S.; Peter, M.; Kholodenko, B.N.; Jeon, N.L.; Pertz, O. Frequency Modulation of ERK Activation Dynamics Rewires Cell Fate. Mol. Syst. Biol. 2015, 11, 838. [Google Scholar] [CrossRef]
- Blüthgen, N. Signaling Output: It’s All about Timing and Feedbacks. Mol. Syst. Biol. 2015, 11, 843. [Google Scholar] [CrossRef]
- Hatcher, A. Algebraic Topology; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Ghrist, R. Barcodes: The Persistent Topology of Data. Bull. Am. Math. Soc. 2008, 45, 61–75. [Google Scholar] [CrossRef]
- Edelsbrunner, H.; Letscher, D.; Zomorodian, A. Topological Persistence and Simplification. Discret. Comput. Geom. 2002, 28, 511–533. [Google Scholar] [CrossRef]
- Mileyko, Y.; Mukherjee, S.; Harer, J. Probability Measures on the Space of Persistence Diagrams. Inverse Probl. 2011, 27, 124007. [Google Scholar] [CrossRef]
- Bubenik, P. The Persistence Landscape and Some of Its Properties. In Topological Data Analysis: The Abel Symposium 2018; Springer International Publishing: Berlin/Heidelberg, Germany, 2020; pp. 97–117. [Google Scholar]
- Bubenik, P. Statistical Topological Data Analysis Using Persistence Landscapes. J. Mach. Learn. Res. 2015, 16, 77–102. [Google Scholar]
- Adams, H.; Emerson, T.; Kirby, M.; Neville, R.; Peterson, C.; Shipman, P.; Chepushtanova, S.; Hanson, E.; Motta, F.; Ziegelmeier, L. Persistence Images: A Stable Vector Representation of Persistent Homology. J. Mach. Learn. Res. 2017, 18, 1–35. [Google Scholar]
- Chung, Y.-M.; Lawson, A. Persistence Curves: A Canonical Framework for Summarizing Persistence Diagrams. Adv. Comput. Math. 2022, 48, 6. [Google Scholar] [CrossRef]
- Abrams, Z.B.; Johnson, T.S.; Huang, K.; Payne, P.R.O.; Coombes, K. A Protocol to Evaluate RNA Sequencing Normalization Methods. BMC Bioinform. 2019, 20, 679. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of MRNA Abundance Using RNA-Seq Data: RPKM Measure Is Inconsistent among Samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Maria, C.; Boissonnat, J.-D.; Glisse, M.; Yvinec, M. The Gudhi Library: Simplicial Complexes and Persistent Homology. In Mathematical Software–ICMS 2014: 4th International Congress, Seoul, Republic of Korea, 5–9 August 2014. Proceedings 4; Springer: Berlin/Heidelberg, Germany, 2014; pp. 167–174. [Google Scholar]
- Muzellec, B.; Teleńczuk, M.; Cabeli, V.; Andreux, M. PyDESeq2: A Python Package for Bulk RNA-Seq Differential Expression Analysis. Bioinformatics 2023, 39, btad547. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Seabold, S.; Perktold, J. Statsmodels: Econometric and Statistical Modeling with Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; pp. 92–96. [Google Scholar]
- Shnier, D.; Voineagu, M.A.; Voineagu, I. Persistent Homology Analysis of Brain Transcriptome Data in Autism. J. R. Soc. Interface 2019, 16, 20190531. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Cao, Y.; Qi, G.; Huang, J.; Liu, Z.-P. Discovering Pathway Biomarkers of Hepatocellular Carcinoma Occurrence and Development by Dynamic Network Entropy Analysis. Gene 2023, 873, 147467. [Google Scholar] [CrossRef]
- Liu, Y.; Deng, M.; Wang, Y.; Wang, H.; Li, C.; Wu, H. Identification of Differentially Expressed Genes and Biological Pathways in Para-Carcinoma Tissues of HCC with Different Metastatic Potentials. Oncol. Lett. 2020, 19, 3799–3814. [Google Scholar] [CrossRef] [PubMed]
- Ghaderi-Zefrehi, H.; Rezaei, M.; Sadeghi, F.; Heiat, M. Genetic Polymorphisms in DNA Repair Genes and Hepatocellular Carcinoma Risk. DNA Repair. 2021, 107, 103196. [Google Scholar] [CrossRef] [PubMed]
- Mohan, V.; Madhusudan, S. DNA Base Excision Repair: Evolving Biomarkers for Personalized Therapies in Cancer. In New Research Directions in DNA Repair; Chen, C., Ed.; IntechOpen: Rijeka, Croatia, 2013. [Google Scholar]
- Ceballos, M.P.; Rigalli, J.P.; Ceré, L.I.; Semeniuk, M.; Catania, V.A.; Ruiz, M.L. ABC Transporters: Regulation and Association with Multidrug Resistance in Hepatocellular Carcinoma and Colorectal Carcinoma. Curr. Med. Chem. 2019, 26, 1224–1250. [Google Scholar] [CrossRef]
- Fan, L.; Zhang, Y.; Zhou, Y.; Wang, Z.; Zhang, Y.; Chen, H. Clinical Significance of ABC Transporter Expression in Patients with Hepatocellular Carcinoma. J. Hard Tissue Biol. 2016, 25, 81–88. [Google Scholar] [CrossRef]
- Nwosu, Z.C.; Megger, D.A.; Hammad, S.; Sitek, B.; Roessler, S.; Ebert, M.P.; Meyer, C.; Dooley, S. Identification of the Consistently Altered Metabolic Targets in Human Hepatocellular Carcinoma. Cell Mol. Gastroenterol. Hepatol. 2017, 4, 303–323. [Google Scholar] [CrossRef]
- Farid, R.M.; Abu-Zeid, R.M.; El-Tawil, A. Emerging Role of Adipokine Apelin in Hepatic Remodelling and Initiation of Carcinogensis in Chronic Hepatitis C Patients. Int. J. Clin. Exp. Pathol. 2014, 7, 2707. [Google Scholar]
- Zhang, X.; Ma, L.; Zhai, L.; Chen, D.; Li, Y.; Shang, Z.; Zhang, Z.; Gao, Y.; Yang, W.; Li, Y.; et al. Construction and Validation of a Three-MicroRNA Signature as Prognostic Biomarker in Patients with Hepatocellular Carcinoma. Int. J. Med. Sci. 2021, 18, 984–999. [Google Scholar] [CrossRef]
- Kunst, C.; Haderer, M.; Heckel, S.; Schlosser, S.; Müller, M. The P53 Family in Hepatocellular Carcinoma. Transl. Cancer Res. 2016, 5, 632–638. [Google Scholar] [CrossRef]
- Yu, M.; Xu, W.; Jie, Y.; Pang, J.; Huang, S.; Cao, J.; Gong, J.; Li, X.; Chong, Y. Identification and Validation of Three Core Genes in P53 Signaling Pathway in Hepatitis B Virus-Related Hepatocellular Carcinoma. World J. Surg. Oncol. 2021, 19, 66. [Google Scholar] [CrossRef]
- Zhen, L.I.; Jiangkai, L.I.U. The Mechanism of P53 Signaling Pathway Regulating Ferroptosis in Hepatocellular Carcinoma. J. Clin. Hepatol. 2023, 39, 956–960. [Google Scholar]
- Wu, M.; Liu, Z.; Zhang, A.; Li, N. Identification of Key Genes and Pathways in Hepatocellular Carcinoma: A Preliminary Bioinformatics Analysis. Medicine 2019, 98, e14287. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zeng, M.; Yan, K.; Yang, Y.; Li, H.; Xu, X. IL-17 Promotes Hepatocellular Carcinoma through Inhibiting Apoptosis Induced by IFN-γ. Biochem. Biophys. Res. Commun. 2020, 522, 525–531. [Google Scholar] [CrossRef]
- Liao, R.; Sun, J.; Wu, H.; Yi, Y.; Wang, J.-X.; He, H.-W.; Cai, X.-Y.; Zhou, J.; Cheng, Y.-F.; Fan, J.; et al. High Expression of IL-17 and IL-17RE Associate with Poor Prognosis of Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. 2013, 32, 3. [Google Scholar] [CrossRef]
- Zhang, Y.; Qiu, Z.; Wei, L.; Tang, R.; Lian, B.; Zhao, Y.; He, X.; Xie, L. Integrated Analysis of Mutation Data from Various Sources Identifies Key Genes and Signaling Pathways in Hepatocellular Carcinoma. PLoS ONE 2014, 9, e100854. [Google Scholar] [CrossRef]
- Agren, R.; Mardinoglu, A.; Asplund, A.; Kampf, C.; Uhlen, M.; Nielsen, J. Identification of Anticancer Drugs for Hepatocellular Carcinoma through Personalized Genome-Scale Metabolic Modeling. Mol. Syst. Biol. 2014, 10, 721. [Google Scholar] [CrossRef] [PubMed]
- Tenen, D.G.; Chai, L.; Tan, J.L. Metabolic Alterations and Vulnerabilities in Hepatocellular Carcinoma. Gastroenterol. Rep. 2021, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Miao, P.; Sheng, S.; Sun, X.; Liu, J.; Huang, G. Lactate Dehydrogenase a in Cancer: A Promising Target for Diagnosis and Therapy. IUBMB Life 2013, 65, 904–910. [Google Scholar] [CrossRef]
- Sheng, S.L.; Liu, J.J.; Dai, Y.H.; Sun, X.G.; Xiong, X.P.; Huang, G. Knockdown of Lactate Dehydrogenase A Suppresses Tumor Growth and Metastasis of Human Hepatocellular Carcinoma. FEBS J. 2012, 279, 3898–3910. [Google Scholar] [CrossRef]
- Zheng, R.; Weng, S.; Xu, J.; Li, Z.; Wang, Y.; Aizimuaji, Z.; Ma, S.; Zheng, L.; Li, H.; Ying, W.; et al. Autophagy and Biotransformation Affect Sorafenib Resistance in Hepatocellular Carcinoma. Comput. Struct. Biotechnol. J. 2023, 21, 3564–3574. [Google Scholar] [CrossRef]
- Shen, M.; Xu, M.; Zhong, F.; Crist, M.C.; Prior, A.B.; Yang, K.; Allaire, D.M.; Choueiry, F.; Zhu, J.; Shi, H. A Multi-Omics Study Revealing the Metabolic Effects of Estrogen in Liver Cancer Cells HepG2. Cells 2021, 10, 455. [Google Scholar] [CrossRef]
- Tian, Y.; Lu, J.; Qiao, Y. A Metabolism-Associated Gene Signature for Prognosis Prediction of Hepatocellular Carcinoma. Front. Mol. Biosci. 2022, 9, 988323. [Google Scholar] [CrossRef]
- Eun, H.S.; Cho, S.Y.; Lee, B.S.; Seong, I.-O.; Kim, K.-H. Profiling Cytochrome P450 Family 4 Gene Expression in Human Hepatocellular Carcinoma. Mol. Med. Rep. 2018, 18, 4865–4876. [Google Scholar] [CrossRef]
- Nekvindova, J.; Mrkvicova, A.; Zubanova, V.; Vaculova, A.H.; Anzenbacher, P.; Soucek, P.; Radova, L.; Slaby, O.; Kiss, I.; Vondracek, J.; et al. Hepatocellular Carcinoma: Gene Expression Profiling and Regulation of Xenobiotic-Metabolizing Cytochromes P450. Biochem. Pharmacol. 2020, 177, 113912. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wen, Q.; Li, S.-F.; Zhang, Y.-F.; Gao, N.; Tian, X.; Fang, Y.; Gao, J.; Cui, M.-Z.; He, X.-P.; et al. Significant Change of Cytochrome P450s Activities in Patients with Hepatocellular Carcinoma. Oncotarget 2016, 7, 50612–50623. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Cho, E.J.; Yu, K.-S.; Jang, I.-J.; Yoon, J.-H.; Park, T.; Cho, J.-Y. Comprehensive Metabolomic Search for Biomarkers to Differentiate Early Stage Hepatocellular Carcinoma from Cirrhosis. Cancers 2019, 11, 1497. [Google Scholar] [CrossRef] [PubMed]
- Ai, Y.; Wang, B.; Xiao, S.; Luo, S.; Wang, Y. Tryptophan Side-Chain Oxidase Enzyme Suppresses Hepatocellular Carcinoma Growth through Degradation of Tryptophan. Int. J. Mol. Sci. 2021, 22, 12428. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Gu, X.; Zhao, Y.; Jia, J.; Zheng, Q.; Su, Y.; Bao, Z.; Lu, J.; Li, L. Prediction of Hepatocellular Carcinoma Prognosis and Immunotherapeutic Effects Based on Tryptophan Metabolism-Related Genes. Cancer Cell Int. 2022, 22, 308. [Google Scholar] [CrossRef]
- Shen, H.; Wu, H.; Sun, F.; Qi, J.; Zhu, Q. A Novel Four-Gene of Iron Metabolism-Related and Methylated for Prognosis Prediction of Hepatocellular Carcinoma. Bioengineered 2021, 12, 240–251. [Google Scholar] [CrossRef]
- Bin Goh, W.W.; Lee, Y.H.; Zubaidah, R.M.; Jin, J.; Dong, D.; Lin, Q.; Chung, M.C.M.; Wong, L. Network-Based Pipeline for Analyzing MS Data: An Application toward Liver Cancer. J. Proteome Res. 2011, 10, 2261–2272. [Google Scholar]
- Zi, Y.; Gao, J.; Wang, C.; Guan, Y.; Li, L.; Ren, X.; Zhu, L.; Mu, Y.; Chen, S.; Zeng, Z.; et al. Pantothenate Kinase 1 Inhibits the Progression of Hepatocellular Carcinoma by Negatively Regulating Wnt/β-Catenin Signaling. Int. J. Biol. Sci. 2022, 18, 1539–1554. [Google Scholar] [CrossRef]
- Nakano, T.; Moriya, K.; Koike, K.; Horie, T. Hepatitis C Virus Core Protein Triggers Abnormal Porphyrin Metabolism in Human Hepatocellular Carcinoma Cells. PLoS ONE 2018, 13, e0198345. [Google Scholar] [CrossRef]
- Udagawa, M.; Horie, Y.; Hirayama, C. Aberrant Porphyrin Metabolism in Hepatocellular Carcinoma. Biochem. Med. 1984, 31, 131–139. [Google Scholar] [CrossRef]
- Dey, T.K.; Mandal, S.; Mukherjee, S. Gene Expression Data Classification Using Topology and Machine Learning Models. BMC Bioinform. 2021, 22, 365. [Google Scholar] [CrossRef] [PubMed]
- Seemann, L.; Shulman, J.; Gunaratne, G.H. A Robust Topology-Based Algorithm for Gene Expression Profiling. Int. Sch. Res. Not. 2012, 2012, 381023. [Google Scholar] [CrossRef] [PubMed]
- Duman, A.N.; Pirim, H. Gene Coexpression Network Comparison via Persistent Homology. Int. J. Genom. 2018, 2018, 7329576. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Descriptor (Short Form) | Formula |
|---|---|
| Classical Euler characteristic (Classical EC) | |
| Persistence-wise Euler characteristic (Persistence EC) | |
| Sum of persistence of -dimensional features (Sum P-) | |
| Average persistence of -dimensional features (Average P-) | |
| Maximum persistence of -dimensional features (Max P-) | |
| Range of persistence of -dimensional features (Range P-) | |
| Sum of birth times of -dimensional features (Sum BT-) | |
| Average birth times of -dimensional features (Average BT-) | |
| Sum of death times of -dimensional features (Sum DT-) | |
| Average death times of -dimensional features (Average DT-) |
| Descriptors | Dataset | Difference (Disease—Control) | -Value | |
|---|---|---|---|---|
| Disease Class | Control Class | |||
| Persistence EC | 0.6252 | 0.3509 | 0.2743 | <0.00050 |
| Sum P-0 | 0.6305 | 0.3573 | 0.2732 | <0.00050 |
| Average P-0 | 0.0371 | 0.0210 | 0.0161 | <0.00050 |
| Range P-2 | 0.0000 | 0.0003 | −0.0003 | 0.03667 |
| Sum BT-2 | 0.0000 | 0.0569 | −0.0569 | 0.03667 |
| Sum DT-2 | 0.0000 | 0.0577 | −0.0577 | 0.03667 |
| Pathway (KEGG ID) | Descriptor | Difference (Disease—Control) | -Value |
|---|---|---|---|
| ABC transporters (02010) | Persistence EC | 1.0659 | <0.0005 |
| Sum P-0 | 1.0650 | <0.0005 | |
| Average P-0 | 0.0626 | <0.0005 | |
| Range P-2 | 0.0000 | <0.0005 | |
| Sum BT-2 | 0.0000 | 0.0240 | |
| Sum DT-2 | 0.0000 | 0.0240 | |
| Apelin signaling pathway (04371) | Persistence EC | 0.3427 | 0.0220 |
| Sum P-0 | 0.3429 | 0.0220 | |
| Average P-0 | 0.0202 | 0.0220 | |
| Range P-2 | 0.0000 | 0.0220 | |
| Sum BT-2 | 0.0383 | 0.0318 | |
| Sum DT-2 | 0.0385 | 0.0318 | |
| Ascorbate and aldarate metabolism (00053) | Persistence EC | 0.4311 | 0.0330 |
| Sum P-0 | 0.4293 | 0.0330 | |
| Average P-0 | 0.0253 | 0.0330 | |
| Range P-2 | 0.0000 | <0.0005 | |
| Sum BT-2 | 0.0000 | 0.0420 | |
| Sum DT-2 | 0.0000 | 0.0420 | |
| Base excision repair (03410) | Persistence EC | 0.6788 | <0.0005 |
| Sum P-0 | 0.6727 | <0.0005 | |
| Average P-0 | 0.0396 | <0.0005 | |
| Range P-2 | −0.0007 | 0.0220 | |
| Sum BT-2 | −0.0957 | 0.0283 | |
| Sum DT-2 | −0.0972 | 0.0283 | |
| beta-Alanine metabolism (00410) | Persistence EC | 0.3086 | 0.0440 |
| Sum P-0 | 0.3157 | 0.0440 | |
| Average P-0 | 0.0186 | 0.0440 | |
| Range P-2 | 0.0000 | <0.0005 | |
| Sum BT-2 | −0.0357 | 0.0440 | |
| Sum DT-2 | −0.0366 | 0.0440 | |
| Citrate cycle (TCA cycle) (00020) | Persistence EC | 1.1207 | 0.0110 |
| Sum P-0 | 1.1070 | 0.0110 | |
| Average P-0 | 0.0652 | 0.0110 | |
| Range P-2 | −0.0020 | 0.0110 | |
| Sum BT-2 | −0.1709 | 0.0477 | |
| Sum DT-2 | −0.1759 | 0.0477 | |
| Collecting duct acid secretion (04966) | Persistence EC | 1.1848 | <0.0005 |
| Sum P-0 | 1.1795 | <0.0005 | |
| Average P-0 | 0.0693 | <0.0005 | |
| Range P-2 | 0.0000 | <0.0005 | |
| Sum BT-2 | 0.0000 | 0.0040 | |
| Sum DT-2 | 0.0000 | 0.0040 | |
| Drug metabolism—cytochrome P450 (00982) | Persistence EC | 0.4315 | 0.0320 |
| Sum P-0 | 0.4568 | 0.0320 | |
| Average P-0 | 0.0268 | 0.0320 | |
| Range P-2 | 0.0000 | <0.0005 | |
| Sum BT-2 | 0.0000 | 0.0320 | |
| Sum DT-2 | 0.0000 | 0.0320 | |
| Glycine, serine, and threonine metabolism (00260) | Persistence EC | 1.6059 | <0.0005 |
| Sum P-0 | 1.5992 | <0.0005 | |
| Average P-0 | 0.0940 | <0.0005 | |
| Range P-2 | 0.0000 | <0.0005 | |
| Sum BT-2 | 0.0000 | 0.0260 | |
| Sum DT-2 | 0.0000 | 0.0260 | |
| Histidine metabolism (00340) | Persistence EC | 0.8008 | 0.0140 |
| Sum P-0 | 0.8045 | 0.0140 | |
| Average P-0 | 0.0473 | 0.0140 | |
| Range P-2 | 0.0000 | <0.0005 | |
| Sum BT-2 | 0.0000 | 0.0140 | |
| Sum DT-2 | 0.0000 | 0.0140 | |
| IL-17 signaling pathway (04657) | Persistence EC | 0.3977 | 0.0360 |
| Sum P-0 | 0.3962 | 0.0360 | |
| Average P-0 | 0.0233 | 0.0360 | |
| Range P-2 | 0.0000 | <0.0005 | |
| Sum BT-2 | 0.0000 | 0.0360 | |
| Sum DT-2 | 0.0000 | 0.0360 | |
| p53 signaling pathway (04115) | Persistence EC | 0.4171 | 0.0165 |
| Sum P-0 | 0.4117 | 0.0165 | |
| Average P-0 | 0.0242 | 0.0165 | |
| Range P-2 | 0.0000 | <0.0005 | |
| Sum BT-2 | 0.0000 | 0.0385 | |
| Sum DT-2 | 0.0000 | 0.0385 | |
| Pantothenate and CoA biosynthesis (00770) | Persistence EC | 0.3973 | 0.0400 |
| Sum P-0 | 0.3995 | 0.0400 | |
| Average P-0 | 0.0235 | 0.0400 | |
| Range P-2 | 0.0000 | <0.0005 | |
| Sum BT-2 | 0.0000 | 0.0400 | |
| Sum DT-2 | 0.0000 | 0.0400 | |
| Phosphonate and phosphinate metabolism (00440) | Persistence EC | 3.2065 | 0.0200 |
| Sum P-0 | 3.2170 | 0.0154 | |
| Average P-0 | 0.1892 | 0.0154 | |
| Range P-2 | 0.0000 | <0.0005 | |
| Sum BT-2 | 0.0000 | 0.0055 | |
| Sum DT-2 | 0.0000 | 0.0055 | |
| Porphyrin metabolism (00860) | Persistence EC | 2.3922 | <0.0005 |
| Sum P-0 | 2.4252 | <0.0005 | |
| Average P-0 | 0.1426 | <0.0005 | |
| Range P-2 | 0.0000 | <0.0005 | |
| Sum BT-2 | 0.0000 | 0.0100 | |
| Sum DT-2 | 0.0000 | 0.0100 | |
| Primary bile acid biosynthesis (00120) | Persistence EC | 0.3730 | 0.0200 |
| Sum P-0 | 0.3737 | 0.0200 | |
| Average P-0 | 0.0220 | 0.0200 | |
| Range P-2 | 0.0000 | <0.0005 | |
| Sum BT-2 | 0.0000 | 0.0200 | |
| Sum DT-2 | 0.0000 | 0.0200 | |
| Protein processing in endoplasmic reticulum (04141) | Persistence EC | 0.5312 | <0.0005 |
| Sum P-0 | 0.5237 | <0.0005 | |
| Average P-0 | 0.0308 | <0.0005 | |
| Range P-2 | −0.0003 | 0.0176 | |
| Sum BT-2 | −0.0638 | 0.0220 | |
| Sum DT-2 | −0.0643 | 0.0220 | |
| Riboflavin metabolism (00740) | Persistence EC | 1.5118 | <0.0005 |
| Sum P-0 | 1.5413 | <0.0005 | |
| Average P-0 | 0.0906 | <0.0005 | |
| Range P-2 | 0.0000 | <0.0005 | |
| Sum BT-2 | 0.0000 | 0.0495 | |
| Sum DT-2 | 0.0000 | 0.0495 | |
| RNA polymerase (03020) | Persistence EC | 0.3368 | 0.0377 |
| Sum P-0 | 0.3691 | 0.0330 | |
| Average P-0 | 0.0218 | 0.0330 | |
| Range P-2 | 0.0000 | 0.0330 | |
| Sum BT-2 | 0.1141 | 0.0460 | |
| Sum DT-2 | 0.1171 | 0.0460 | |
| Sulfur metabolism (00920) | Persistence EC | 6.0523 | <0.0005 |
| Sum P-0 | 6.0452 | <0.0005 | |
| Average P-0 | 0.3556 | <0.0005 | |
| Range P-2 | 0.0000 | <0.0005 | |
| Sum BT-2 | 0.0000 | 0.0220 | |
| Sum DT-2 | 0.0000 | 0.0220 | |
| Synaptic vesicle cycle (04721) | Persistence EC | 0.3297 | <0.0005 |
| Sum P-0 | 0.3305 | <0.0005 | |
| Average P-0 | 0.0195 | <0.0005 | |
| Range P-2 | −0.0006 | 0.0385 | |
| Sum BT-2 | −0.0523 | 0.0403 | |
| Sum DT-2 | −0.0534 | 0.0403 | |
| Tryptophan metabolism (00380) | Persistence EC | 0.2584 | 0.0424 |
| Sum P-0 | 0.2530 | 0.0424 | |
| Average P-0 | 0.0148 | 0.0424 | |
| Range P-2 | 0.0000 | <0.0005 | |
| Sum BT-2 | 0.0000 | 0.0424 | |
| Sum DT-2 | 0.0000 | 0.0424 | |
| Virion—Herpesvirus (03266) | Persistence EC | 1.1037 | 0.0220 |
| Sum P-0 | 1.1240 | 0.0220 | |
| Average P-0 | 0.0661 | 0.0220 | |
| Range P-2 | 0.0000 | <0.0005 | |
| Sum BT-2 | 0.0000 | 0.0220 | |
| Sum DT-2 | 0.0000 | 0.0220 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdullahi, M.S.; Suratanee, A.; Piro, R.M.; Plaimas, K. Persistent Homology Identifies Pathways Associated with Hepatocellular Carcinoma from Peripheral Blood Samples. Mathematics 2024, 12, 725. https://doi.org/10.3390/math12050725
Abdullahi MS, Suratanee A, Piro RM, Plaimas K. Persistent Homology Identifies Pathways Associated with Hepatocellular Carcinoma from Peripheral Blood Samples. Mathematics. 2024; 12(5):725. https://doi.org/10.3390/math12050725
Chicago/Turabian StyleAbdullahi, Muhammad Sirajo, Apichat Suratanee, Rosario Michael Piro, and Kitiporn Plaimas. 2024. "Persistent Homology Identifies Pathways Associated with Hepatocellular Carcinoma from Peripheral Blood Samples" Mathematics 12, no. 5: 725. https://doi.org/10.3390/math12050725
APA StyleAbdullahi, M. S., Suratanee, A., Piro, R. M., & Plaimas, K. (2024). Persistent Homology Identifies Pathways Associated with Hepatocellular Carcinoma from Peripheral Blood Samples. Mathematics, 12(5), 725. https://doi.org/10.3390/math12050725