Robust Linear Trend Test for Low-Coverage Next-Generation Sequence Data Controlling for Covariates


Abstract
:1. Introduction
2. Materials and Methods
2.1. Mixture Model Accounting for Covariates
2.2. Derivation of EM Algorithm under
2.3. Hypothesis Tests of Genetic Association Controlling for Covariates
3. Results
3.1. Simulation Study
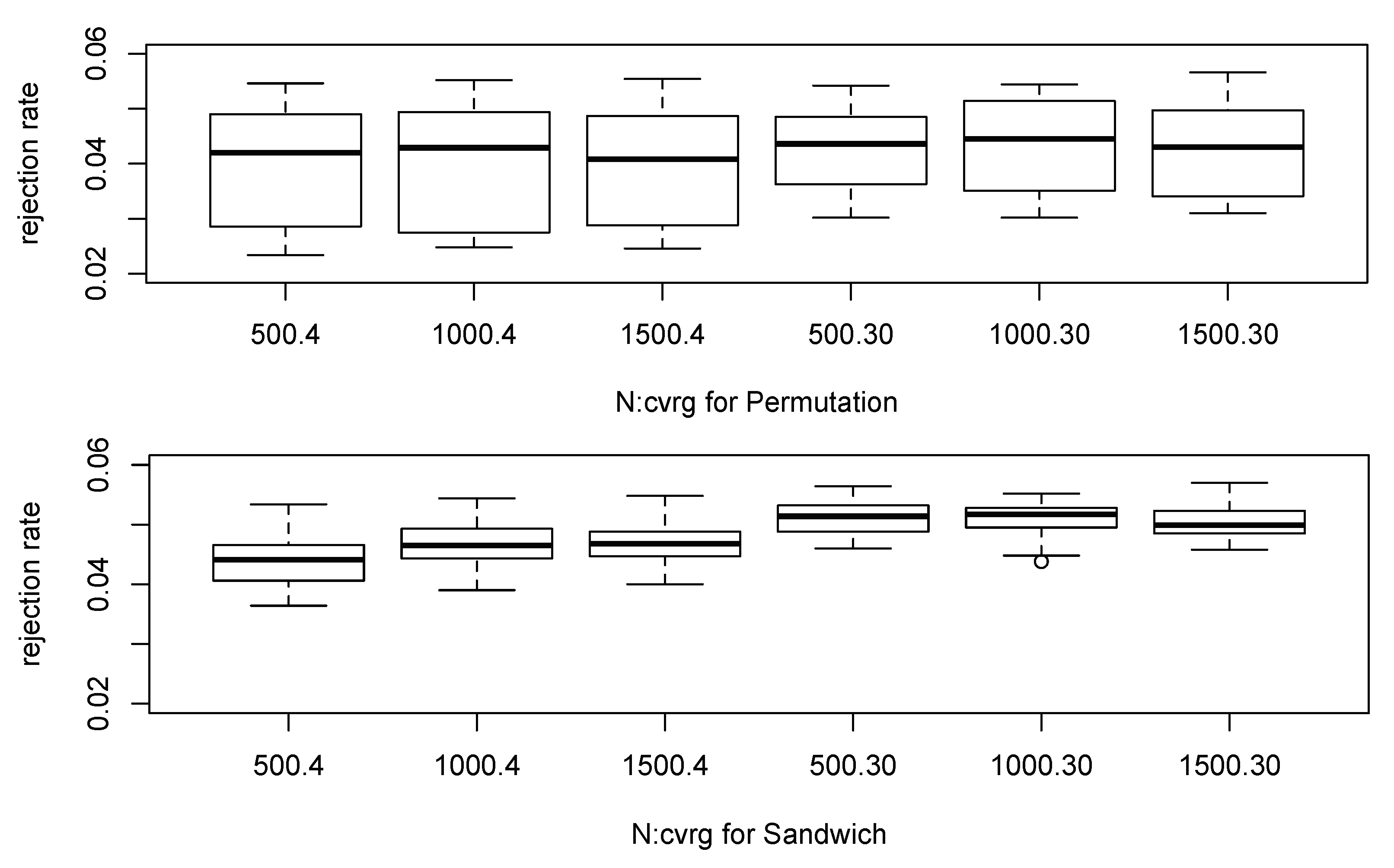
3.1.1. Simulation Study for Null Distribution
- (i)
- Prevalence (): 0.1, 0.3
- (ii)
- Coverage (v): 4, 30
- (iii)
- Minor allele frequency (q): 0.05, 0.3
- (iv)
- Total sample size (n): 500, 1000, 1500
- (v)
- Covariate (): single normal or normal mixture with mean given genotype
- (vi)
- Regression coefficient : 0, 1
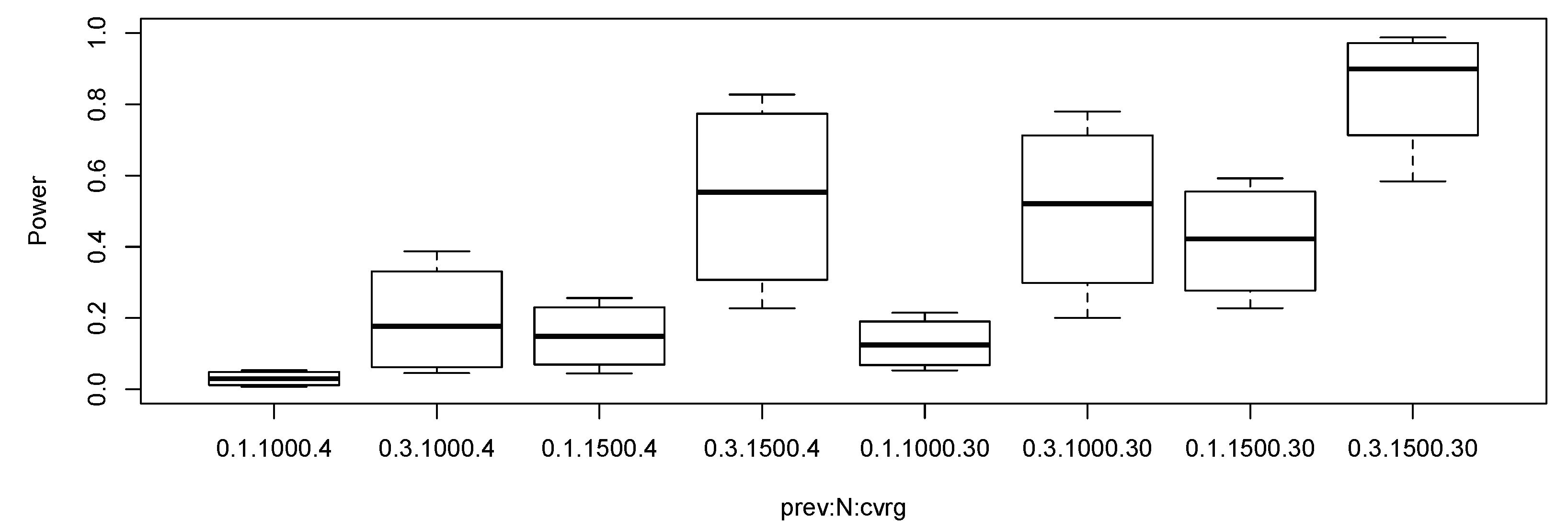
3.1.2. Simulation Study for Statistical Power
3.2. Real Data Analysis
4. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| EM | Expectation–Maximization |
| GWAS | Genome-wide association study |
| HWE | Hardy–Weinberg equilibrium |
| maf | Minor allele frequency |
| NGS | Next-generation sequence |
| SNP | Single nucleotide polymorphism |
| TDT | Transmission disequilibrium test |
References
- Wu, M.C.; Lee, S.; Cai, T.; Li, Y.; Boehnke, M.; Lin, X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 2011, 89, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Cirulli, E.T.; White, S.; Read, R.W.; Elhanan, G.; Metcalf, W.J.; Tanudjaja, F.; Fath, D.M.; Sandoval, E.; Isaksson, M.; Schlauch, K.A.; et al. Genome-wide rare variant analysis for thousands of phenotypes in over 70,000 exomes from two cohorts. Nat. Commun. 2020, 11, 542. [Google Scholar] [CrossRef]
- Lakiotaki, K.; Kanterakis, A.; Kartsaki, E.; Katsila, T.; Patrinos, G.P.; Potamias, G. Exploring public genomics data for population pharmacogenomics. PLoS ONE 2017, 12, e0182138. [Google Scholar] [CrossRef] [PubMed]
- Patrinos, G.P.; Giannopoulou, E.; Katsila, T.; Tsermpini, E.E.; Mitropoulou, C. Integrating next-generation sequencing in the clinical pharmacogenomics workflow. Front. Pharmacol. 2019, 10, 384. [Google Scholar]
- Celesti, F.; Celesti, A.; Wan, J.; Villari, M. Why Deep Learning Is Changing the Way to Approach NGS Data Processing: A Review. IEEE Rev. Biomed. Eng. 2018, 11, 68–76. [Google Scholar] [CrossRef]
- Le, N.Q.K.; Yapp, E.K.Y.; Nagasundaram, N.; Chua, M.C.H.; Yeh, H.Y. Computational identification of vesicular transport proteins from sequences using deep gated recurrent units architecture. Comput. Struct. Biotechnol. J. 2019, 17, 1245–1254. [Google Scholar] [CrossRef]
- Tripathi, R.; Sharma, P.; Chakraborty, P.; Varadwaj, P.K. Next-generation sequencing revolution through big data analytics. Front. Life Sci. 2016, 9, 119–149. [Google Scholar] [CrossRef]
- Cirillo, D.; Valencia, A. Big data analytics for personalized medicine. Curr. Opin. Biotechnol. 2019, 58, 161–167. [Google Scholar] [CrossRef]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef]
- Song, K.; Li, L.; Zhang, G. Coverage recommendation for genotyping analysis of highly heterologous species using next-generation sequencing technology. Sci. Rep. 2016, 6, 35736. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.; Finch, S.J.; Nothnagel, M.; Ott, J. Power and sample size calculations for case-control genetic association tests when errors are present: Application to single nucleotide polymorphisms. Hum. Hered. 2002, 54, 22–33. [Google Scholar] [CrossRef]
- Ahn, K.; Haynes, C.; Kim, W.; Fleur, R.S.; Gordon, D.; Finch, S.J. The effects of SNP genotyping errors on the power of the Cochran-Armitage linear trend test for case/control association studies. Ann. Hum. Genet. 2007, 71, 249–261. [Google Scholar] [CrossRef]
- Kim, W.; Londono, D.; Zhou, L.; Xing, J.; Nato, A.Q.; Musolf, A.; Matise, T.C.; Finch, S.J.; Gordon, D. Single-variant and multi-variant trend tests for genetic association with next-generation sequencing that are robust to sequencing error. Hum. Hered. 2012, 74, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Sun, N.; Mane, S.; Sayward, F.; Rajeevan, N.; Cheung, K.; Cho, K.; Pyarajan, S.; Aslan, M.; Miller, P. Impact of genotyping errors on statistical power of association tests in genomic analyses: A case study. Genet. Epidemiol. 2017, 41, 152–162. [Google Scholar] [CrossRef] [Green Version]
- Consortium, G.P. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56. [Google Scholar]
- Le, S.Q.; Durbin, R. SNP detection and genotyping from low-coverage sequencing data on multiple diploid samples. Genome Res. 2011, 21, 952–960. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Sidore, C.; Kang, H.M.; Boehnke, M.; Abecasis, G.R. Low-coverage sequencing: Implications for design of complex trait association studies. Genome Res. 2011, 21, 940–951. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Gordon, D.; Sebat, J.; Kenny, Q.Y.; Finch, S.J. Computing power and sample size for case-control association studies with copy number polymorphism: Application of mixture-based likelihood ratio test. PLoS ONE 2008, 3, e3475. [Google Scholar] [CrossRef]
- Barnes, C.; Plagnol, V.; Fitzgerald, T.; Redon, R.; Marchini, J.; Clayton, D.; Hurles, M.E. A robust statistical method for case-control association testing with copy number variation. Nat. Genet. 2008, 40, 1245. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Li, Y.; Guo, Y.; Li, R.; Holmkvist, J.; Hansen, T.; Pedersen, O.; Wang, J.; Nielsen, R. Design of association studies with pooled or un-pooled next-generation sequencing data. Genet. Epidemiol. 2010, 34, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.; Finch, S.J.; De La Vega, F. A new expectation-maximization statistical test for case-control association studies considering rare variants obtained by high-throughput sequencing. Hum. Hered. 2011, 71, 113–125. [Google Scholar] [CrossRef]
- Kim, W.; Kim, Y.H. Genetic association tests when a nuisance parameter is not identifiable under no association. Commun. Stat. Appl. Methods 2017, 24, 663–671. [Google Scholar] [CrossRef]
- Kim, W. Transmission Disequilibrium Tests Based on Read Counts for Low-Coverage Next,-Generation Sequence Data. Hum. Hered. 2015, 80, 36–49. [Google Scholar] [CrossRef]
- Chen, H.; Chen, J.; Kalbfleisch, J.D. A modified likelihood ratio test for homogeneity in finite mixture models. J. R. Stat. Soc. Ser. B 2001, 63, 19–29. [Google Scholar] [CrossRef]
- Zhou, H.; Pan, W. Binomial mixture model-based association tests under genetic heterogeneity. Ann. Hum. Genet. 2009, 73, 614–630. [Google Scholar] [CrossRef] [Green Version]
- Dempster, A.P.; Laird, N.M.; Rubin, D.B. Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. Ser. B 1977, 39, 1–22. [Google Scholar]
- White, H. Maximum Likelihood Estimation of Misspecified Models. Econometrica 1982, 50, 1–25. [Google Scholar] [CrossRef]
- Sidak, Z.; Sen, P.K.; Hajek, J. Theory of Rank Tests; Academic Press: San Diego, CA, USA, 1999. [Google Scholar]
- Anderson, T.W. An Introduction to Multivariate Statistical Analysis; Wiley: New York, NY, USA, 1962. [Google Scholar]
- Kang, K.W.; Kim, W.; Cho, Y.W.; Lee, S.K.; Jung, K.Y.; Shin, W.; Kim, D.W.; Kim, W.J.; Lee, H.W.; Kim, W. Genetic characteristics of non-familial epilepsy. PeerJ 2019, 7, e8278. [Google Scholar] [CrossRef]
- Kim, M.-K.K.; Moore, J.H.; Kim, J.K.; Cho, K.H.; Cho, Y.W.; Kim, Y.S.; Lee, M.C.; Kim, Y.O.; Shin, M.H. Evidence for epistatic interactions in antiepileptic drug resistance. J. Hum. Genet. 2011, 56, 71–76. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Method (cvrg) | ||||
|---|---|---|---|---|
| Permutation (4×) | 0 | |||
| Permutation (30×) | ||||
| Sandwich (4×) | ||||
| Sandwich (30×) |
| Coverage | Total Sample Size | Covariate | Naive | Proposed | |
|---|---|---|---|---|---|
| 4 | 1000 | Normal mixture | 0 | 0.102 | 0.113 |
| 4 | 1000 | Normal mixture | 1 | 0.233 | 0.261 |
| 4 | 1000 | Single normal | 0 | 0.190 | 0.277 |
| 4 | 1000 | Single normal | 1 | 0.269 | 0.374 |
| 4 | 1500 | Normal mixture | 0 | 0.398 | 0.429 |
| 4 | 1500 | Normal mixture | 1 | 0.657 | 0.701 |
| 4 | 1500 | Single normal | 0 | 0.626 | 0.741 |
| 4 | 1500 | Single normal | 1 | 0.736 | 0.840 |
| 30 | 1000 | Normal mixture | 0 | 0.384 | 0.355 |
| 30 | 1000 | Normal mixture | 1 | 0.617 | 0.603 |
| 30 | 1000 | Single normal | 0 | 0.622 | 0.637 |
| 30 | 1000 | Single normal | 1 | 0.734 | 0.760 |
| 30 | 1500 | Normal mixture | 0 | 0.792 | 0.761 |
| 30 | 1500 | Normal mixture | 1 | 0.959 | 0.954 |
| 30 | 1500 | Single normal | 0 | 0.933 | 0.939 |
| 30 | 1500 | Single normal | 1 | 0.978 | 0.978 |
| Read Depth v | Total | ||||
|---|---|---|---|---|---|
| Frequency | 43 | 86 | 95 | 176 | 400 |
| Proportion | 0.1075 | 0.215 | 0.2375 | 0.44 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.Y.; Kim, M.-K.; Kim, W. Robust Linear Trend Test for Low-Coverage Next-Generation Sequence Data Controlling for Covariates. Mathematics 2020, 8, 217. https://doi.org/10.3390/math8020217
Lee JY, Kim M-K, Kim W. Robust Linear Trend Test for Low-Coverage Next-Generation Sequence Data Controlling for Covariates. Mathematics. 2020; 8(2):217. https://doi.org/10.3390/math8020217
Chicago/Turabian StyleLee, Jung Yeon, Myeong-Kyu Kim, and Wonkuk Kim. 2020. "Robust Linear Trend Test for Low-Coverage Next-Generation Sequence Data Controlling for Covariates" Mathematics 8, no. 2: 217. https://doi.org/10.3390/math8020217
APA StyleLee, J. Y., Kim, M. -K., & Kim, W. (2020). Robust Linear Trend Test for Low-Coverage Next-Generation Sequence Data Controlling for Covariates. Mathematics, 8(2), 217. https://doi.org/10.3390/math8020217