
Multiscale Modelling of β-Adrenergic Stimulation in Cardiac Electromechanical Function



Abstract
:1. Introduction
2. Mathematical Models of β-ARS

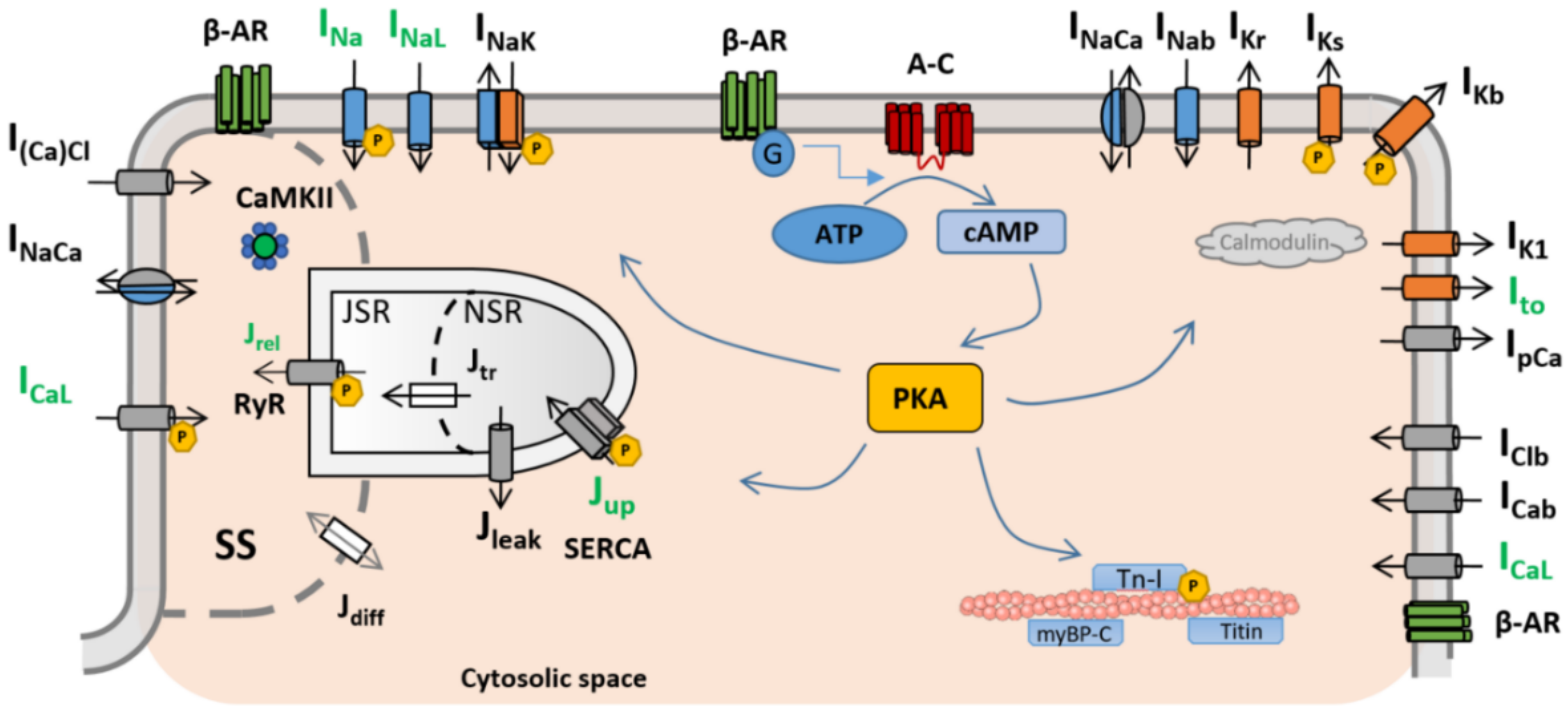{kind=link}
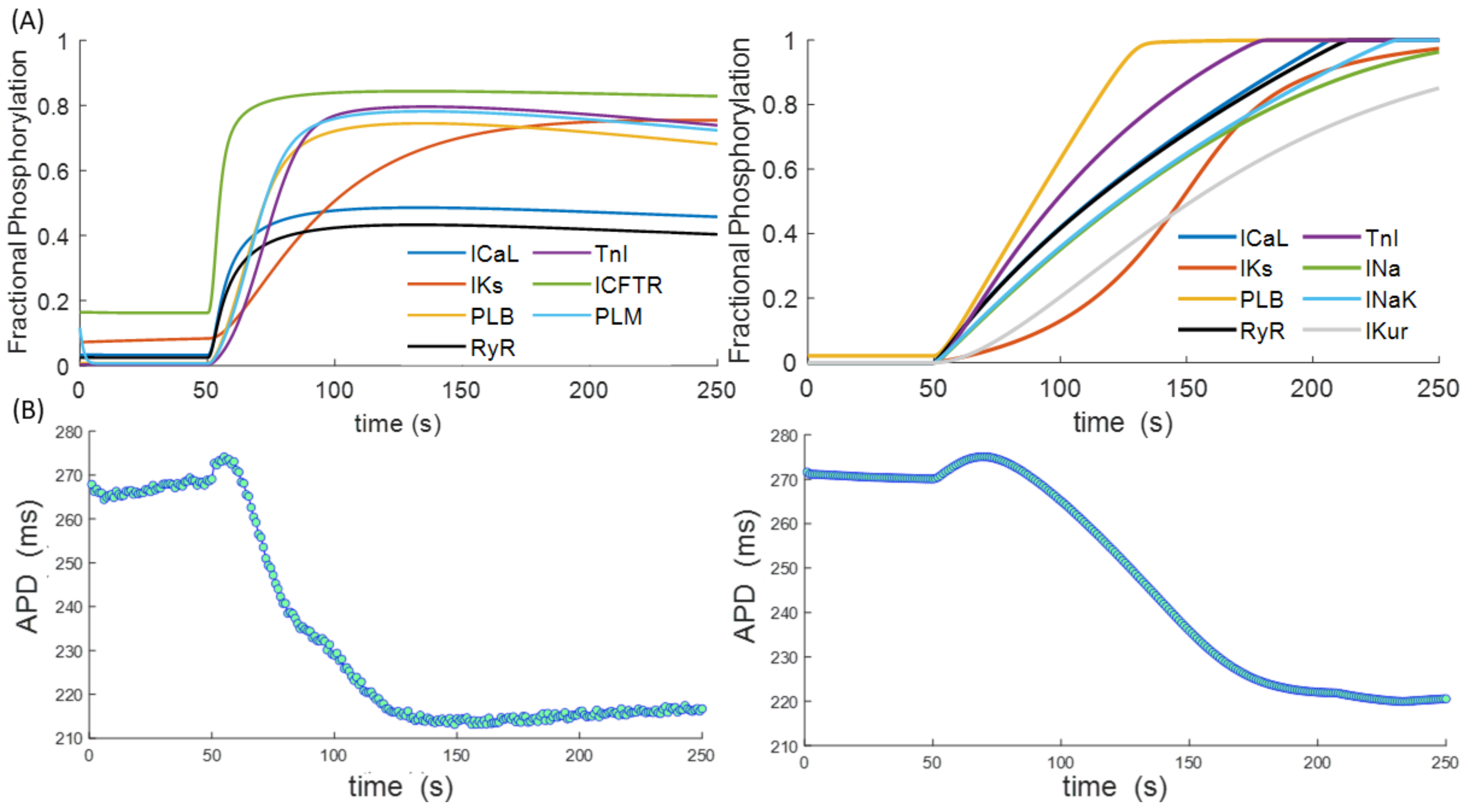{kind=link}
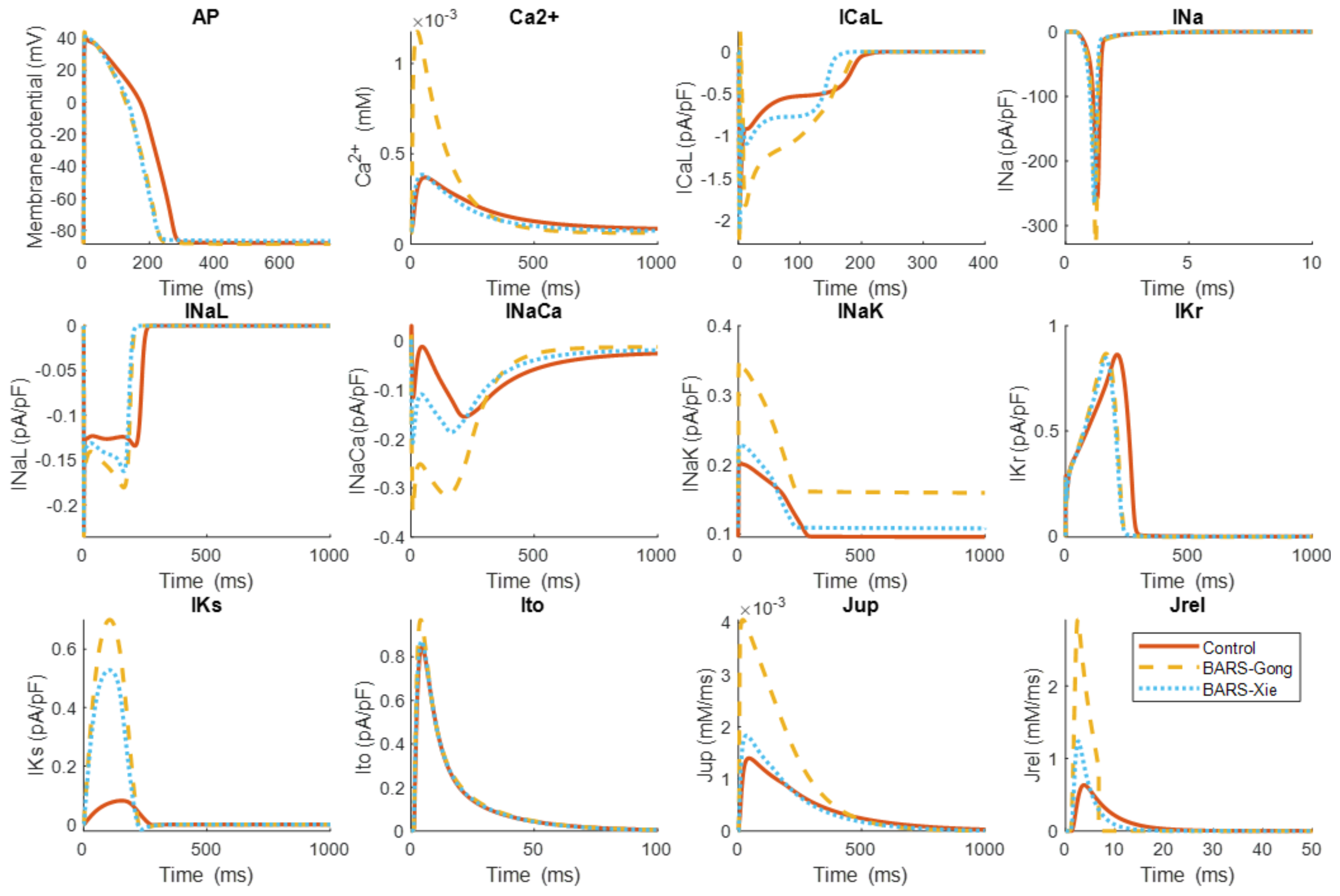{kind=link}
{kind=link}
| Model (Year) | Species | β-ARS Isoform | Signalling | Substrates | Main Model Advances |
|---|---|---|---|---|---|
| Zeng and Rudy [35] (1995) | Guinea Pig | Generic | None | ICaL; IK; PLB; INaK; INa | Simulation of the isoproterenol effect by increasing conductances and parameter shift |
| Saucerman et al. [36] (2003) | Rat | β1 | cAMP; PKA | ICaL; PLB | Dynamic target phosphorylation integrated with cell signalling |
| Greenstein et al. [37] (2004) | Dog | Generic | None | ICaL; PLB; IKr; IKs | Introduction of a binary population-based approach for target phosphorylation |
| Iancu et al. * [38] (2007) | Guinea Pig | β1 | cAMP | N/A | Cellular signalling compartmentation |
| Soltis & Saucerman [29] (2010) | Rabbit | β1 | cAMP; PKA | ICaL; IKs; PLB; RyR; TnI; ICFTR | Integration with dynamic CaMKII regulation |
| Hiejman et al. [39] (2011) | Dog | β1, β2 | cAMP; PKA | ICaL; IKs; IKur; PLB; INaK; INa; RyR; TnI | Two different β isoforms; population-based approach with four different populations |
| Bondarenko [40] (2014) | Mouse | β1 | cAMP; PKA | ICaL; INa; INaK; RyR; IKur; Ito; IK1; PLB; TnI | Compartmentalised mouse model with new L-type calcium channel subpopulations |
| Khalilimeybodi et al. * [41] (2018) | Mouse | β1, β2 | cAMP; PKA; GSK3β; ERK | N/A | Addition of new molecular signalling pathways (GSK3β and ERK) |
3. Differences in Formulation between Leading Mathematical Models of β-ARS
4. Multiscale Studies including β-ARS
4.1. Effects of β-ARS in Electromechanical Coupling
4.2. Long QT Syndrome
4.3. Heart Failure
4.4. Other Cardiac Pathologies
5. Discussion and Perspectives
5.1. Future Work in β-ARS Modelling
5.2. β-ARS and 3D Modelling
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lands, A.M.; Arnold, A.; McAuliff, J.P.; Luduena, F.P.; Brown, T.G. Differentiation of receptor systems activated by sympathomimetic amines. Nature 1967, 214, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Lands, A.M.; Luduena, F.P.; Buzzo, H.J. Differentiation of receptors responsive to isoproterenol. Life Sci. 1967, 6, 2241–2249. [Google Scholar] [CrossRef]
- Coppini, R.; Ferrantini, C.; Yao, L.; Fan, P.; Del Lungo, M.; Stillitano, F.; Sartiani, L.; Tosi, B.; Suffredini, S.; Tesi, C.; et al. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation 2013, 127, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.R.; Chen, L.; Rao, M.; Chen, K.; Song, J.P.; Hu, S.S. A modified method for isolation of human cardiomyocytes to model cardiac diseases. J. Transl. Med. 2018, 16, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandi, E.; Ripplinger, C.M. Antiarrhythmic mechanisms of beta blocker therapy. Pharmacol. Res. 2019, 146, 104274. [Google Scholar] [CrossRef] [PubMed]
- Pogwizd, S.M.; Schlotthauer, K.; Li, L.; Yuan, W.; Bers, D.M. Arrhythmogenesis and Contractile Dysfunction in Heart Failure. Circ. Res. 2001, 88, 1159–1167. [Google Scholar] [CrossRef] [Green Version]
- Desantiago, J.; Ai, X.; Islam, M.; Acuna, G.; Ziolo, M.T.; Bers, D.M.; Pogwizd, S.M. Arrhythmogenic effects of β2-adrenergic stimulation in the failing heart are attributable to enhanced sarcoplasmic reticulum Ca load. Circ. Res. 2008, 102, 1389–1397. [Google Scholar] [CrossRef] [Green Version]
- Myles, R.C.; Wang, L.; Kang, C.; Bers, D.M.; Ripplinger, C.M. Local β-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circ. Res. 2012, 110, 1454–1464. [Google Scholar] [CrossRef] [Green Version]
- Ferrantini, C.; Pioner, J.M.; Mazzoni, L.; Gentile, F.; Tosi, B.; Rossi, A.; Belardinelli, L.; Tesi, C.; Palandri, C.; Matucci, R.; et al. Late sodium current inhibitors to treat exercise-induced obstruction in hypertrophic cardiomyopathy: An in vitro study in human myocardium. Br. J. Pharmacol. 2018, 175, 2635–2652. [Google Scholar] [CrossRef] [Green Version]
- Goldenberg, I.; Thottathil, P.; Lopes, C.M.; Moss, A.J.; McNitt, S.; Jin, O.U.; Robinson, J.L.; Zareba, W.; Ackerman, M.J.; Kaufman, E.S.; et al. Trigger-specific ion-channel mechanisms, risk factors, and response to therapy in type 1 long QT syndrome. Heart Rhythm 2012, 9, 49–56. [Google Scholar] [CrossRef]
- Uchi, J.; Rice, J.J.; Ruwald, M.H.; Parks, X.X.; Ronzier, E.; Moss, A.J.; Zareba, W.; Lopes, C.M. Impaired IKs channel activation by Ca2+-dependent PKC shows correlation with emotion/arousal-triggered events in LQT1. J. Mol. Cell. Cardiol. 2015, 79, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Danielsen, T.K.; Manotheepan, R.; Sadredini, M.; Leren, I.S.; Edwards, A.G.; Vincent, K.P.; Lehnart, S.E.; Sejersted, O.M.; Sjaastad, I.; Haugaa, K.H.; et al. Arrhythmia initiation in catecholaminergic polymorphic ventricular tachycardia type 1 depends on both heart rate and sympathetic stimulation. PLoS ONE 2018, 13, e0207100. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.; Ko, K.S.; Rhee, B.D.; Han, J.; Kim, N. Different effects of prolonged β-adrenergic stimulation on heart and cerebral artery. Integr. Med. Res. 2014, 3, 204–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelhardt, S.; Hein, L.; Wiesmann, F.; Lohse, M.J. Progressive hypertrophy and heart failure in β1-adrenergic receptor transgenic mice. Proc. Natl. Acad. Sci. USA 1999, 96, 7059–7064. [Google Scholar] [CrossRef] [Green Version]
- Ostrom, R.S.; Naugle, J.E.; Hase, M.; Gregorian, C.; Swaney, J.S.; Insel, P.A.; Brunton, L.L.; Meszaros, J.G. Angiotensin II enhances adenylyl cyclase signaling via Ca2+/calmodulin: Gq-Ga cross-talk regulates collagen production in cardiac fibroblasts. J. Biol. Chem. 2003, 278, 24461–24468. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.M.; Antoons, G. Arrhythmogenic Mechanisms in Heart Failure: Linking β-Adrenergic Stimulation, Stretch, and Calcium. Front. Physiol. 2018, 9, 1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beuckelmann, D.J.; Erdmann, E. Ca2+-currents and intracellular [Ca2+]i-transients in single ventricular myocytes isolated from terminally failing human myocardium. In Cellular and Molecular Alterations in the Failing Human Heart; Steinkopff: Heidelberg, Germany, 1992; pp. 235–243. [Google Scholar] [CrossRef]
- Harding, S.E.; Jones, S.M.; O’Gara, P.; Vescovo, G.; Poole-Wilson, P.A. Reduced β-agonist sensitivity in single atrial cells from failing human hearts. Am. J. Physiol. Heart Circ. Physiol. 1990, 259. [Google Scholar] [CrossRef]
- Leosco, D.; Rengo, G.; Iaccarino, G.; Filippelli, A.; Lymperopoulos, A.; Zincarelli, C.; Fortunato, F.; Golino, L.; Marchese, M.; Esposito, G.; et al. Exercise training and β-blocker treatment ameliorate age-dependent impairment of β-adrenergic receptor signaling and enhance cardiac responsiveness to adrenergic stimulation. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, 1596–1603. [Google Scholar] [CrossRef] [Green Version]
- Lucia, C.D.; Eguchi, A.; Koch, W.J. New Insights in Cardiac β-Adrenergic Signaling During Heart Failure and Aging. Front. Pharmacol. 2018, 9, 904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlik-Feldmann, R.; Kramer, H.H.; Wicht, H.; Feldmann, R.; Netz, H.; Reinhardt, D. Distribution of Myocardial β-Adrenoceptor Subtypes and Coupling to the Adenylate Cyclase in Children With Congenital Heart Disease and Implications for Treatment. J. Clin. Pharmacol. 1993, 33, 588–595. [Google Scholar] [CrossRef]
- Lazzerini, P.E.; Boutjdir, M.; Capecchi, P.L. COVID-19, Arrhythmic Risk, and Inflammation. Circulation 2020, 142, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Sutanto, H.; Heijman, J. Beta-Adrenergic Receptor Stimulation Modulates the Cellular Proarrhythmic Effects of Chloroquine and Azithromycin. Front. Physiol. 2020, 11, 587709. [Google Scholar] [CrossRef]
- Corral-Acero, J.; Margara, F.; Marciniak, M.; Rodero, C.; Loncaric, F.; Feng, Y.; Gilbert, A.; Fernandes, J.F.; Bukhari, H.A.; Wajdan, A.; et al. The “Digital Twin” to enable the vision of precision cardiology. Eur. Heart J. 2020, 41, 4556–4564. [Google Scholar] [CrossRef]
- Woo, A.Y.H.; Xiao, R.P. β-Adrenergic receptor subtype signaling in heart: From bench to bedside. Acta Pharmacol. Sin. 2012, 33, 335–341. [Google Scholar] [CrossRef] [Green Version]
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volders, P.G.A.; Stengl, M.; Van Opstal, J.M.; Gerlach, U.; Spätjens, R.L.H.M.G.; Beekman, J.D.M.; Sipido, K.R.; Vos, M.A. Probing the contribution of IKs to canine ventricular repolarization: Key role for β-adrenergic receptor stimulation. Circulation 2003, 107, 2753–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, S.; Dun, W.; Boyden, P.A. Can PKA activators rescue Na+ channel function in epicardial border zone cells that survive in the infarcted canine heart? Cardiovasc. Res. 2004, 64, 260–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soltis, A.R.; Saucerman, J.J. Synergy between CaMKII substrates and β-adrenergic signaling in regulation of cardiac myocyte Ca2+ handling. Biophys. J. 2010, 99, 2038–2047. [Google Scholar] [CrossRef] [Green Version]
- Negroni, J.A.; Morotti, S.; Lascano, E.C.; Gomes, A.V.; Grandi, E.; Puglisi, J.L.; Bers, D.M. Β-Adrenergic Effects on Cardiac Myofilaments and Contraction in an Integrated Rabbit Ventricular Myocyte Model. J. Mol. Cell. Cardiol. 2015, 81, 162–175. [Google Scholar] [CrossRef] [Green Version]
- Despa, S.; Bossuyt, J.; Han, F.; Ginsburg, K.S.; Jia, L.G.; Kutchai, H.; Tucker, A.L.; Bers, D.M. Phospholemman-phosphorylation mediates the β-adrenergic effects on Na/K pump function in cardiac myocytes. Circ. Res. 2005, 97, 252–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginsburg, K.S.; Bers, D.M. Modulation of excitation-contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ current trigger. J. Physiol. 2004, 556, 463–480. [Google Scholar] [CrossRef]
- Stelzer, J.E.; Patel, J.R.; Walker, J.W.; Moss, R.L. Differential roles of cardiac myosin-binding protein C and cardiac troponin I in the myofibrillar force responses to protein kinase A phosphorylation. Circ. Res. 2007, 101, 503–511. [Google Scholar] [CrossRef] [Green Version]
- Krüger, M.; Linke, W.A. Protein kinase-A phosphorylates titin in human heart muscle and reduces myofibrillar passive tension. J. Muscle Res. Cell Motil. 2006, 27, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Rudy, Y. Early afterdepolarizations in cardiac myocytes: Mechanism and rate dependence. Biophys. J. 1995, 68, 949–964. [Google Scholar] [CrossRef] [Green Version]
- Saucerman, J.J.; Brunton, L.L.; Michailova, A.P.; McCulloch, A.D. Modeling β-Adrenergic Control of Cardiac Myocyte Contractility in Silico. J. Biol. Chem. 2003, 278, 47997–48003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenstein, J.L.; Tanskanen, A.J.; Winslow, R.L. Modeling the actions of β-adrenergic signaling on excitation- contraction coupling processes. Ann. N. Y. Acad. Sci. 2004, 1015, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iancu, R.V.; Jones, S.W.; Harvey, R.D. Compartmentation of cAMP signaling in cardiac myocytes: A computational study. Biophys. J. 2007, 92, 3317–3331. [Google Scholar] [CrossRef] [Green Version]
- Heijman, J.; Volders, P.G.A.; Westra, R.L.; Rudy, Y. Local control of β-adrenergic stimulation: Effects on ventricular myocyte electrophysiology and Ca2+-transient. J. Mol. Cell. Cardiol. 2011, 50, 863–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondarenko, V.E. A Compartmentalized Mathematical Model of the β1-Adrenergic Signaling System in Mouse Ventricular Myocytes. PLoS ONE 2014, 9, e89113. [Google Scholar] [CrossRef] [Green Version]
- Khalilimeybodi, A.; Daneshmehr, A.; Sharif-Kashani, B. Investigating β-adrenergic-induced cardiac hypertrophy through computational approach: Classical and non-classical pathways. J. Physiol. Sci. 2018, 68, 503–520. [Google Scholar] [CrossRef]
- Tomek, J.; Bueno-Orovio, A.; Passini, E.; Zhou, X.; Minchole, A.; Britton, O.; Bartolucci, C.; Severi, S.; Shrier, A.; Virag, L.; et al. Development, calibration, and validation of a novel human ventricular myocyte model in health, disease, and drug block. eLife 2019, 8, e48890. [Google Scholar] [CrossRef]
- Tanskanen, A.J.; Greenstein, J.L.; O’Rourke, B.; Winslow, R.L. The role of stochastic and modal gating of cardiac L-type Ca2+ channels on early after-depolarizations. Biophys. J. 2005, 88, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Saucerman, J.J.; McCulloch, A.D. Mechanistic systems models of cell signaling networks: A case study of myocyte adrenergic regulation. Prog. Biophys. Mol. Biol. 2004, 85, 261–278. [Google Scholar] [CrossRef]
- Zaccolo, M.; Pozzan, T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 2002, 295, 1711–1715. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, V.; Pak, E.; Robinson, R.B.; Steinberg, S.F. β2-Adrenergic receptor actions in neonatal and adult rat ventricular myocytes. Circ. Res. 1995, 76, 40–52. [Google Scholar] [CrossRef]
- Vittone, L.; Mundiña-Weilenmann, C.; Said, M.; Mattiazzi, A. Mechanisms involved in the acidosis enhancement of the isoproterenol-induced phosphorylation of phospholamban in the intact heart. J. Biol. Chem. 1998, 273, 9804–9811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, R.P.; Lakatta, E.G. β1-Adrenoceptor stimulation and β2-adrenoceptor stimulation differ in their effects on contraction, cytosolic Ca2+, and Ca2+ current in single rat ventricular cells. Circ. Res. 1993, 73, 286–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.H.; Saucerman, J.J. Phospholemman is a negative feed-forward regulator of Ca2+ in β-adrenergic signaling, accelerating β-adrenergic inotropy. J. Mol. Cell. Cardiol. 2012, 52, 1048–1055. [Google Scholar] [CrossRef] [Green Version]
- Meyer, E.E.; Clancy, C.E.; Lewis, T.J. Dynamics of adrenergic signaling in cardiac myocytes and implications for pharmacological treatment. J. Theor. Biol. 2021, 519, 110619. [Google Scholar] [CrossRef]
- Warrier, S.; Belevych, A.E.; Ruse, M.; Eckert, R.L.; Zaccolo, M.; Pozzan, T.; Harvey, R.D. β-adrenergic- and muscarinic receptor-induced changes in cAMP activity in adult cardiac myocytes detected with FRET-based biosensor. Am. J. Physiol. Cell Physiol. 2005, 289, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Hohl, C.M.; Li, Q. Compartmentation of cAMP in adult canine ventricular myocytes: Relation to single-cell free Ca2+ transients. Circ. Res. 1991, 69, 1369–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagykaldi, Z.; Kem, D.; Lazzara, R.; Szabo, B. Canine ventricular myocyte β2-adrenoceptors are not functionally coupled to L-type calcium current. J. Cardiovasc. Electrophysiol. 1999, 10, 1240–1251. [Google Scholar] [CrossRef]
- Johnson, D.M.; Heijman, J.; Pollard, C.E.; Valentin, J.P.; Crijns, H.J.G.M.; Abi-Gerges, N.; Volders, P.G.A. IKs restricts excessive beat-to-beat variability of repolarization during beta-adrenergic receptor stimulation. J. Mol. Cell. Cardiol. 2010, 48, 122–130. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, T.; Rudy, Y. Arrhythmia formation in subclinical (“silent”) long QT syndrome requires multiple insults: Quantitative mechanistic study using the KCNQ1 mutation Q357R as example. Heart Rhythm 2012, 9, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.Q.X.; Susilo, M.E.; Sher, A.; Musante, C.J.; Sobie, E.A. Quantitative analysis of variability in an integrated model of human ventricular electrophysiology and β-adrenergic signaling. J. Mol. Cell. Cardiol. 2020, 143, 96–106. [Google Scholar] [CrossRef]
- Robertson, S.P.; Johnson, J.D.; Holroyde, M.J.; Kraniass, E.G.; Potter, J.D.; Solaro, R.J. The Effect of Troponin I Phosphorylation on the Ca2+-binding of Bovine Cardiac Troponin * Properties of the Ca2+-regulatory Site. J. Biol. Chem. 1982, 257, 260–263. [Google Scholar] [CrossRef]
- Xie, Y.; Grandi, E.; Puglisi, J.L.; Sato, D.; Bers, D.M. β-adrenergic stimulation activates early afterdepolarizations transiently via kinetic mismatch of PKA targets. J. Mol. Cell. Cardiol. 2013, 58, 153–161. [Google Scholar] [CrossRef] [Green Version]
- O’Hara, T.; Virág, L.; Varró, A.; Rudy, Y. Simulation of the Undiseased Human Cardiac Ventricular Action Potential: Model Formulation and Experimental Validation. PLoS Comput. Biol. 2011, 7, e1002061. [Google Scholar] [CrossRef] [Green Version]
- Pueyo, E.; Orini, M.; Rodríguez, J.F.; Taggart, P. Interactive effect of beta-adrenergic stimulation and mechanical stretch on low-frequency oscillations of ventricular action potential duration in humans. J. Mol. Cell. Cardiol. 2016, 97, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Dai, L.; Zang, Y.; Zheng, D.; Luo, R.; Xia, L. Modelling the integrated regulation role of β-adrenergic signaling and CaMKII in human myocyte electrophysiological properties. Comput. Cardiol. 2013, 40, 389–392. [Google Scholar]
- Kuzumoto, M.; Takeuchi, A.; Nakai, H.; Oka, C.; Noma, A.; Matsuoka, S. Simulation analysis of intracellular Na+ and Cl− homeostasis during β1-adrenergic stimulation of cardiac myocyte. Prog. Biophys. Mol. Biol. 2008, 96, 171–186. [Google Scholar] [CrossRef]
- Surdo, N.C.; Berrera, M.; Koschinski, A.; Brescia, M.; Machado, M.R.; Carr, C.; Wright, P.; Gorelik, J.; Morotti, S.; Grandi, E.; et al. FRET biosensor uncovers cAMP nano-domains at β-adrenergic targets that dictate precise tuning of cardiac contractility. Nat. Commun. 2017, 8, 15031. [Google Scholar] [CrossRef]
- Lyon, A.; Dupuis, L.J.; Arts, T.; Crijns, H.J.G.M.; Prinzen, F.W.; Delhaas, T.; Heijman, J.; Lumens, J. Differentiating the effects of β-adrenergic stimulation and stretch on calcium and force dynamics using a novel electromechanical cardiomyocyte model. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H519–H530. [Google Scholar] [CrossRef]
- Dupuis, L.J.; Lumens, J.; Arts, T.; Delhaas, T. High tension in sarcomeres hinders myocardial relaxation: A computational study. PLoS ONE 2018, 13, e0204642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, A.J.; Zareba, W. Long QT syndrome: Therapeutic considerations. In Cardiac Electrophysiology: From Cell to Bedside; Elsevier Inc.: Amsterdam, The Netherlands, 2004; pp. 660–667. ISBN 9780721603230. [Google Scholar]
- Terrenoire, C.; Clancy, C.E.; Cormier, J.W.; Sampson, K.J.; Kass, R.S. Autonomic control of cardiac action potentials: Role of potassium channel kinetics in response to sympathetic stimulation. Circ. Res. 2005, 96, e25–e34. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.; Badiceanu, A.; Brennan, J.A.; Gloschat, C.; Qiao, Y.; Trayanova, N.A.; Efimov, I.R. β-adrenergic stimulation augments transmural dispersion of repolarization via modulation of delayed rectifier currents IKs and IKr in the human ventricle. Sci. Rep. 2017, 7, 15922. [Google Scholar] [CrossRef]
- Kilfoil, P.J.; Lotteau, S.; Zhang, R.; Yue, X.; Aynaszyan, S.; Solymani, R.E.; Cingolani, E.; Marbán, E.; Goldhaber, J.I. Distinct features of calcium handling and β-adrenergic sensitivity in heart failure with preserved versus reduced ejection fraction. J. Physiol. 2020, 598, 5091–5108. [Google Scholar] [CrossRef]
- Morotti, S.; Edwards, A.G.; Mcculloch, A.D.; Bers, D.M.; Grandi, E. A novel computational model of mouse myocyte electrophysiology to assess the synergy between Na+ loading and CaMKII. J. Physiol. 2014, 592, 1181–1197. [Google Scholar] [CrossRef] [PubMed]
- Passini, E.; Mincholé, A.; Coppini, R.; Cerbai, E.; Rodriguez, B.; Severi, S.; Bueno-Orovio, A. Mechanisms of pro-arrhythmic abnormalities in ventricular repolarisation and anti-arrhythmic therapies in human hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2016, 96, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Bueno-Orovio, A.; Orini, M.; Hanson, B.; Hayward, M.; Taggart, P.; Lambiase, P.D.; Burrage, K.; Rodriguez, B. In Vivo and in Silico Investigation into Mechanisms of Frequency Dependence of Repolarization Alternans in Human Ventricular Cardiomyocytes. Circ. Res. 2016, 118, 266–278. [Google Scholar] [CrossRef]
- Mora, M.T.; Gong, J.Q.X.; Sobie, E.A.; Trenor, B. The role of β-adrenergic system remodeling in human heart failure: A mechanistic investigation. J. Mol. Cell. Cardiol. 2021, 153, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Loucks, A.D.; O’Hara, T.; Trayanova, N.A. Degradation of T-Tubular Microdomains and Altered cAMP Compartmentation Lead to Emergence of Arrhythmogenic Triggers in Heart Failure Myocytes: An in silico Study. Front. Physiol. 2018, 9, 1737. [Google Scholar] [CrossRef]
- Sanchez-Alonso, J.L.; Loucks, A.; Schobesberger, S.; van Cromvoirt, A.M.; Poulet, C.; Chowdhury, R.A.; Trayanova, N.; Gorelik, J. Nanoscale regulation of L-type calcium channels differentiates between ischemic and dilated cardiomyopathies. EBioMedicine 2020, 57, 102845. [Google Scholar] [CrossRef]
- Iyer, V.; Hajjar, R.J.; Armoundas, A.A. Mechanisms of abnormal calcium homeostasis in mutations responsible for catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2007, 100, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Tomek, J.; Rodriguez, B.; Bub, G.; Heijman, J. β-Adrenergic receptor stimulation inhibits proarrhythmic alternans in postinfarction border zone cardiomyocytes: A computational analysis. Am. J. Physiol. Heart. Circ. Physiol. 2017, 313, H338–H353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomek, J.; Hao, G.; Tomková, M.; Lewis, A.; Carr, C.; Paterson, D.J.; Rodriguez, B.; Bub, G.; Herring, N. β-Adrenergic Receptor Stimulation and Alternans in the Border Zone of a Healed Infarct: An ex vivo Study and Computational Investigation of Arrhythmogenesis. Front. Physiol. 2019, 10, 350. [Google Scholar] [CrossRef] [Green Version]
- Tsumoto, K.; Ashihara, T.; Naito, N.; Shimamoto, T.; Amano, A.; Kurata, Y.; Kurachi, Y. Specific decreasing of Na+ channel expression on the lateral membrane of cardiomyocytes causes fatal arrhythmias in Brugada syndrome. Sci. Rep. 2020, 10, 19964. [Google Scholar] [CrossRef]
- Sampedro-Puente, D.A.; Fernandez-Bes, J.; Szentandrássy, N.; Nánási, P.; Taggart, P.; Pueyo, E. Time Course of Low-Frequency Oscillatory Behavior in Human Ventricular Repolarization Following Enhanced Sympathetic Activity and Relation to Arrhythmogenesis. Front. Physiol. 2020, 10, 1547. [Google Scholar] [CrossRef] [Green Version]
- Brodde, O.E.; Bruck, H.; Leineweber, K.; Seyfarth, T. Presence, distribution and physiological function of adrenergic and muscarinic receptor subtypes in the human heart. Basic Res. Cardiol. 2001, 96, 528–538. [Google Scholar] [CrossRef] [PubMed]
- McCabe, K.J.; Rangamani, P. Computational modeling approaches to cAMP/PKA signaling in cardiomyocytes. J. Mol. Cell. Cardiol. 2021, 154, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Judina, A.; Gorelik, J.; Wright, P.T. Studying signal compartmentation in adult cardiomyocytes. Biochem. Soc. Trans. 2020, 48, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Schobesberger, S.; Wright, P.; Tokar, S.; Bhargava, A.; Mansfield, C.; Glukhov, A.V.; Poulet, C.; Buzuk, A.; Monszpart, A.; Sikkel, M.; et al. T-tubule remodelling disturbs localized β2-adrenergic signalling in rat ventricular myocytes during the progression of heart failure. Cardiovasc. Res. 2017, 113, 770–782. [Google Scholar] [CrossRef]
- Ruzsnavszky, F.; Hegyi, B.; Kistamás, K.; Váczi, K.; Horváth, B.; Szentandrássy, N.; Bányász, T.; Nánási, P.P.; Magyar, J. Asynchronous activation of calcium and potassium currents by isoproterenol in canine ventricular myocytes. Naunyn. Schmiedebergs. Arch. Pharmacol. 2014, 387, 457–467. [Google Scholar] [CrossRef]
- Liu, G.X.; Choi, B.R.; Ziv, O.; Li, W.; de Lange, E.; Qu, Z.; Koren, G. Differential conditions for early after-depolarizations and triggered activity in cardiomyocytes derived from transgenic LQT1 and LQT2 rabbits. J. Physiol. 2012, 590, 1171–1180. [Google Scholar] [CrossRef]
- Bers, D.M.; Zaccolo, M. Whole-Cell cAMP and PKA Activity are Epiphenomena, Nanodomain Signaling Matters. Physiology 2019, 34, 240–249. [Google Scholar] [CrossRef]
- Shugg, T.; Hudmon, A.; Overholser, B.R. Neurohormonal Regulation of IKs in Heart Failure: Implications for Ventricular Arrhythmogenesis and Sudden Cardiac Death. J. Am. Heart Assoc. 2020, 9, e016900. [Google Scholar] [CrossRef] [PubMed]
- Morales, D.; Hermosilla, T.; Varela, D. Calcium-dependent inactivation controls cardiac L-type Ca2+ currents under β-adrenergic stimulation. J. Gen. Physiol. 2018, 151, 786–797. [Google Scholar] [CrossRef] [Green Version]
- Treinys, R.; Kanaporis, G.; Fischmeister, R.; Jurevičius, J. Metabolic inhibition induces transient increase of L-type Ca2+ current in human and rat cardiac myocytes. Int. J. Mol. Sci. 2019, 20, 1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, A.G.; Louch, W.E. Species-Dependent Mechanisms of Cardiac Arrhythmia: A Cellular Focus. Clin. Med. Insights Cardiol. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Varshneya, M.; Devenyi, R.A.; Sobie, E.A. Slow Delayed Rectifier Current Protects Ventricular Myocytes From Arrhythmic Dynamics Across Multiple Species. Circ. Arrhythm. Electrophysiol. 2018, 11, e006558. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Morotti, S.; Tapa, S.; Stuart, S.D.F.; Jiang, Y.; Bers, D.M.; Wang, Z.; Myles, R.C.; Brack, K.E.; Andr, G.; et al. Different paths, same destination: Divergent action potential responses produce conserved cardiac fight-or-flight response in mouse and rabbit hearts. J. Physiol. 2019, 15, 3867–3883. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Shen, Y.T.; Kalthof, B.; Iwase, M.; Sato, N.; Uechi, M.; Vatner, S.F.; Vatner, D.E. Identification and functional role of β-adrenergic receptor subtypes in primate and rodent: In vivo versus isolated myocytes. J. Mol. Cell. Cardiol. 1996, 28, 1307–1317. [Google Scholar] [CrossRef]
- Chen, P.S.; Chen, L.S.; Fishbein, M.C.; Lin, S.F.; Nattel, S. Role of the autonomic nervous system in atrial fibrillation: Pathophysiology and therapy. Circ. Res. 2014, 114, 1500–1515. [Google Scholar] [CrossRef] [Green Version]
- Hindricks, G.; Potpara, T.; Dagres, N.; Bax, J.J.; Boriani, G.; Dan, G.A.; Fauchier, L.; Kalman, J.M.; Lane, D.A.; Lettino, M.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2021, 42, 373–498. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, S.; Wang, W.; Wang, K.; Shen, W. A Mathematical Model of the Mouse Atrial Myocyte With Inter-Atrial Electrophysiological Heterogeneity. Front. Physiol. 2020, 11, 972. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, W.; Wang, K.; Shen, W.; Zhang, H. Alternans in Mouse Atrial Cardiomyocytes: A Computational Study on the Influence of Cell-Cell Coupling and β-Adrenergic Stimulation. IEEE Access 2020, 8, 84806–84820. [Google Scholar] [CrossRef]
- Kneller, J.; Zou, R.; Vigmond, E.J.; Wang, Z.; Leon, L.J.; Nattel, S. Cholinergic Atrial Fibrillation in a Computer Model of a Two-Dimensional Sheet of Canine Atrial Cells With Realistic Ionic Properties. Circ. Res. 2002, 90, e73–e87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matene, E.; Vinet, A.; Jacquemet, V. Dynamics of atrial arrhythmias modulated by time-dependent acetylcholine concentration: A simulation study. Europace 2014, 16, iv11–iv20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayer, J.D.; Boukens, B.J.; Krul, S.P.J.; Roney, C.H.; Driessen, A.H.G.; Berger, W.R.; van den Berg, N.W.E.; Verkerk, A.O.; Vigmond, E.J.; Coronel, R.; et al. Acetylcholine Delays Atrial Activation to Facilitate Atrial Fibrillation. Front. Physiol. 2019, 10, 1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawano, H.; Okada, R.; Yano, K. Histological study on the distribution of autonomic nerves in the human heart. Heart Vessels 2003, 18, 32–39. [Google Scholar] [CrossRef]
- Di Bona, A.; Vita, V.; Costantini, I.; Zaglia, T. Towards a clearer view of sympathetic innervation of cardiac and skeletal muscles. Prog. Biophys. Mol. Biol. 2020, 154, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Ajijola, O.A.; Lux, R.L.; Khahera, A.; Kwon, O.; Aliotta, E.; Ennis, D.B.; Fishbein, M.C.; Ardell, J.L.; Shivkumar, K. Sympathetic modulation of electrical activation in normal and infracted myocardium: Implications for arrhythmogenesis. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H608–H621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osanlouy, M.; Bandrowski, A.; de Bono, B.; Brooks, D.; Cassarà, A.M.; Christie, R.; Ebrahimi, N.; Gillespie, T.; Grethe, J.S.; Guercio, L.A.; et al. The SPARC DRC: Building a Resource for the Autonomic Nervous System Community. Front. Physiol. 2021, 12, 693735. [Google Scholar] [CrossRef] [PubMed]
- Niederer, S.A.; Lumens, J.; Trayanova, N.A. Computational models in cardiology. Nat. Rev. Cardiol. 2019, 16, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.J.; Santiago, A.; Zhou, X.; Wang, L.; Margara, F.; Levrero-Florencio, F.; Das, A.; Kelly, C.; Dallarmellina, E.; Vazquez, M.; et al. Human biventricular electromechanical simulations on the progression of electrocardiographic and mechanical abnormalities in post-myocardial infarction. Europace 2021, 23, I143–I152. [Google Scholar] [CrossRef] [PubMed]
- Levrero-Florencio, F.; Margara, F.; Zacur, E.; Bueno-Orovio, A.; Wang, Z.J.; Santiago, A.; Aguado-Sierra, J.; Houzeaux, G.; Grau, V.; Kay, D.; et al. Sensitivity analysis of a strongly-coupled human-based electromechanical cardiac model: Effect of mechanical parameters on physiologically relevant biomarkers. Comput. Methods Appl. Mech. Eng. 2020, 361, 112762. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doste, R.; Bueno-Orovio, A. Multiscale Modelling of β-Adrenergic Stimulation in Cardiac Electromechanical Function. Mathematics 2021, 9, 1785. https://doi.org/10.3390/math9151785
Doste R, Bueno-Orovio A. Multiscale Modelling of β-Adrenergic Stimulation in Cardiac Electromechanical Function. Mathematics. 2021; 9(15):1785. https://doi.org/10.3390/math9151785
Chicago/Turabian StyleDoste, Ruben, and Alfonso Bueno-Orovio. 2021. "Multiscale Modelling of β-Adrenergic Stimulation in Cardiac Electromechanical Function" Mathematics 9, no. 15: 1785. https://doi.org/10.3390/math9151785
APA StyleDoste, R., & Bueno-Orovio, A. (2021). Multiscale Modelling of β-Adrenergic Stimulation in Cardiac Electromechanical Function. Mathematics, 9(15), 1785. https://doi.org/10.3390/math9151785