
Voltammetric Determination of Active Pharmaceutical Ingredients Using Screen-Printed Electrodes





Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Apparatus
2.3. Differential Pulse Voltammetric Measurements
3. Results and Discussion
3.1. Optimization
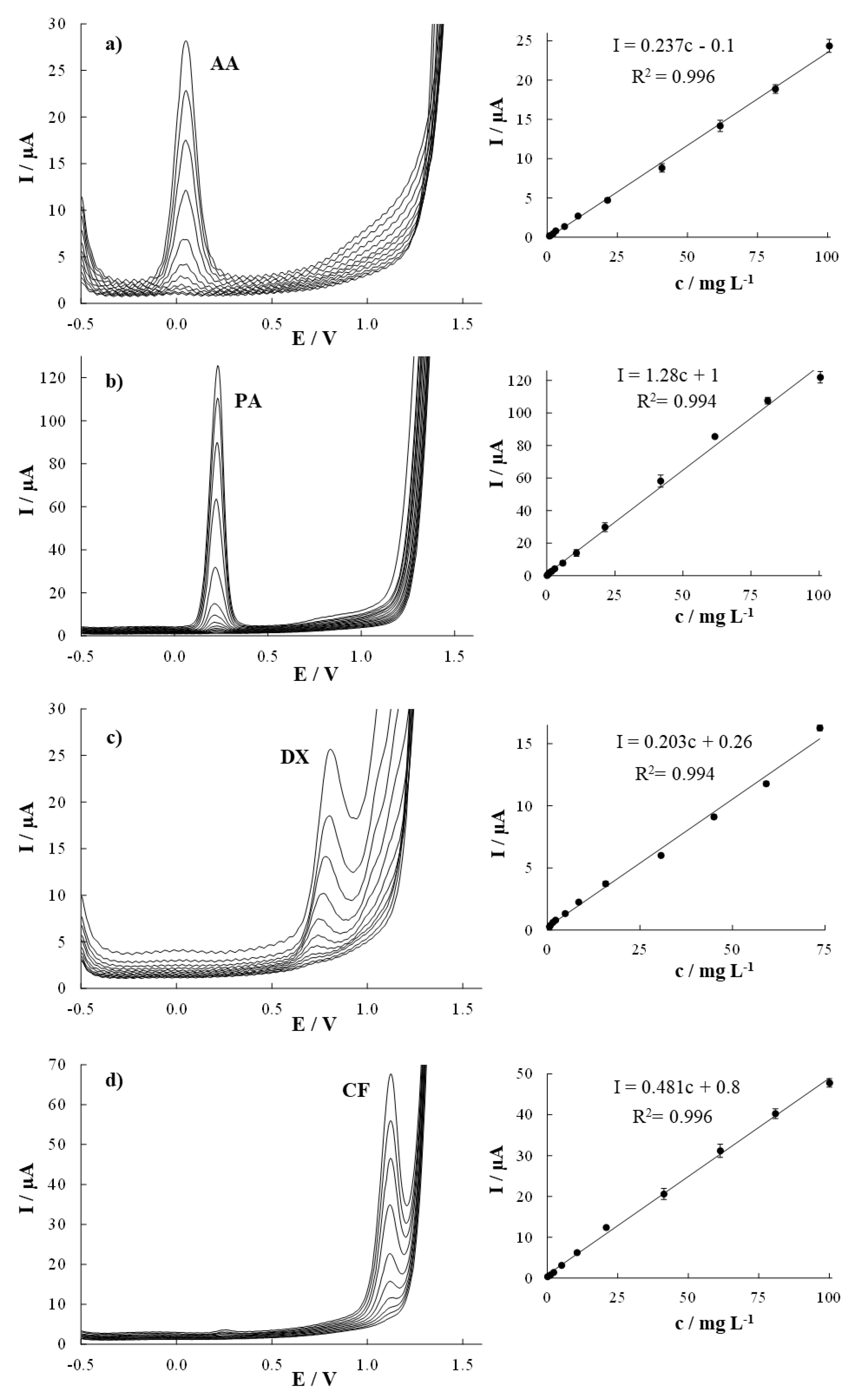
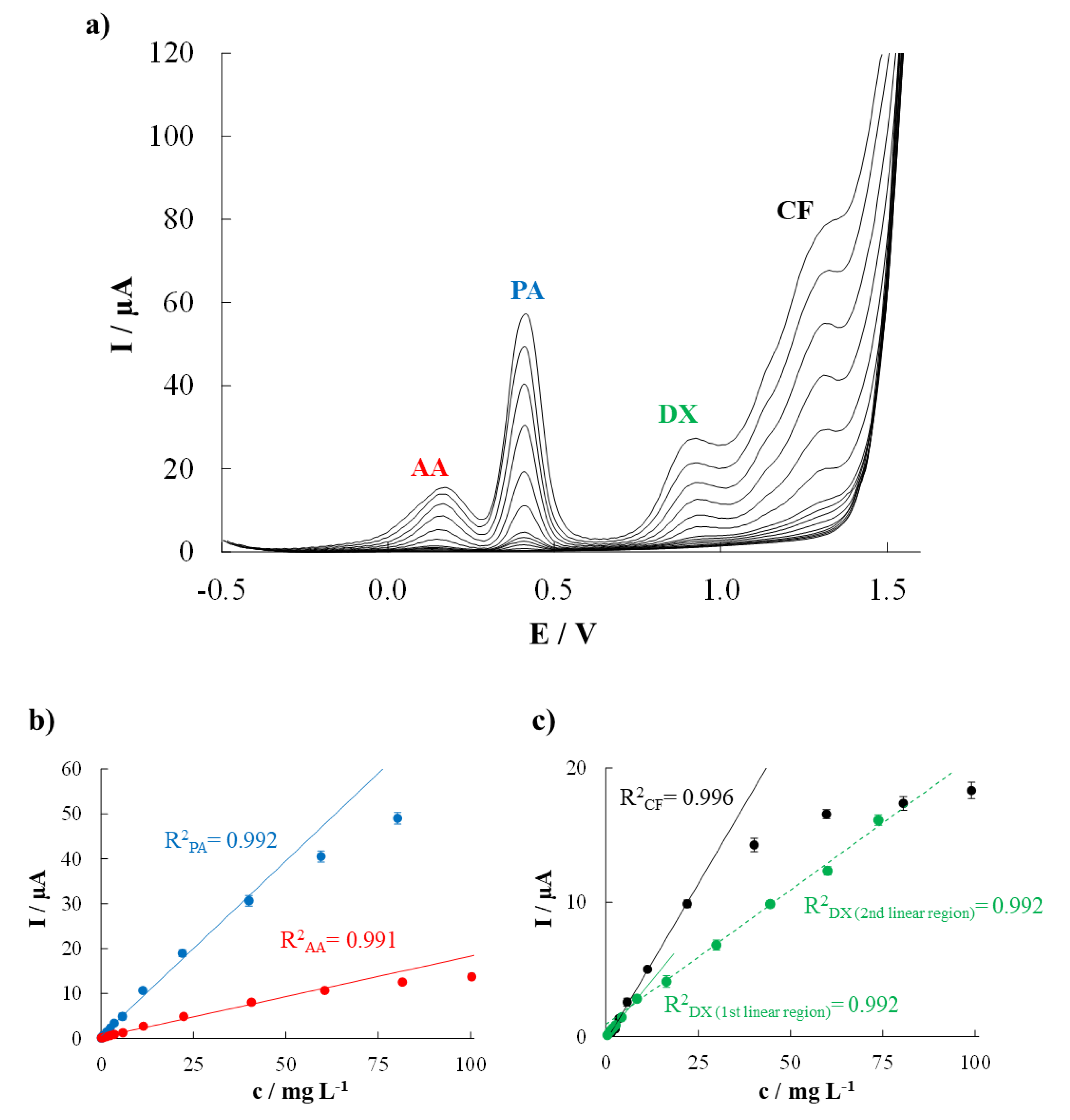
3.2. Analytical Performance Evaluation
3.3. Application to Real Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- González Peña, O.I.; López Zavala, M.Á.; Cabral Ruelas, H. Pharmaceuticals Market, Consumption Trends and Disease Incidence Are Not Driving the Pharmaceutical Research on Water and Wastewater. Int. J. Environ. Res. Public Health 2021, 18, 2532. [Google Scholar] [CrossRef] [PubMed]
- Global Pharmaceutical Industry. Statista. Available online: https://www.statista.com/topics/1764/global-pharmaceutical-industry/ (accessed on 25 October 2021).
- Scott, T.M.; Phillips, P.J.; Kolpin, D.W.; Colella, K.M.; Furlong, E.T.; Foreman, W.T.; Gray, J.L. Pharmaceutical Manufacturing Facility Discharges Can Substantially Increase the Pharmaceutical Load to U.S. Wastewaters. Sci. Total Environ. 2018, 636, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Li, W.C. Occurrence, Sources, and Fate of Pharmaceuticals in Aquatic Environment and Soil. Environ. Pollut. 2014, 187, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.M.P.T.; Silva, L.J.G.; Laranjeiro, C.S.M.; Meisel, L.M.; Lino, C.M.; Pena, A. Human Pharmaceuticals in Portuguese Rivers: The Impact of Water Scarcity in the Environmental Risk. Sci. Total Environ. 2017, 609, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Lutz, A.; Carroll, R.; Keteles, K.; Dahlin, K.; Murphy, M.; Nguyen, D. Occurrence, Distribution, and Seasonality of Emerging Contaminants in Urban Watersheds. Chemosphere 2018, 200, 133–142. [Google Scholar] [CrossRef]
- Sharma, B.M.; Bečanová, J.; Scheringer, M.; Sharma, A.; Bharat, G.K.; Whitehead, P.G.; Klánová, J.; Nizzetto, L. Health and Ecological Risk Assessment of Emerging Contaminants (Pharmaceuticals, Personal Care Products, and Artificial Sweeteners) in Surface and Groundwater (Drinking Water) in the Ganges River Basin, India. Sci. Total Environ. 2019, 646, 1459–1467. [Google Scholar] [CrossRef]
- Rivera-Utrilla, J.; Sánchez-Polo, M.; Ferro-García, M.Á.; Prados-Joya, G.; Ocampo-Pérez, R. Pharmaceuticals as Emerging Contaminants and Their Removal from Water. A Review. Chemosphere 2013, 93, 1268–1287. [Google Scholar] [CrossRef]
- European Union Strategic Approach to Pharmaceuticals in the Environment; European Commission: Brussels, Belgium, 2019.
- Arrigoni, O.; de Tullio, M.C. Ascorbic Acid: Much More than Just an Antioxidant. Biochim. Biophys. Acta 2002, 1569, 1–9. [Google Scholar] [CrossRef]
- Devaki, S.J.; Raveendran, R.L. Vitamin C: Sources, Functions, Sensing and Analysis. In Vitamin C; Hamza, A.H., Ed.; InTech Open: London, UK, 2017. [Google Scholar]
- Brunton, L.L.; Hilal-Dandan, R.; Knollmann, B.C. Goodman & Gilman’s: The Pharmacological Basis of Therapeutics, 13th ed.; McGraw-Hill Education: New York, NY, USA, 2006. [Google Scholar]
- Fekadu, S.; Alemayehu, E.; Dewil, R.; van der Bruggen, B. Pharmaceuticals in Freshwater Aquatic Environments: A Comparison of the African and European Challenge. Sci. Total Environ. 2019, 654, 324–337. [Google Scholar] [CrossRef]
- Thurman, E.M.; Ferrer, I. Liquid Chromatography/Quadrupole-Time-of-Flight Mass Spectrometry with Metabolic Profiling of Human Urine as a Tool for Environmental Analysis of Dextromethorphan. J. Chromatogr. A 2012, 1259, 158–166. [Google Scholar] [CrossRef]
- Ternes, T.A. Analytical Methods for the Determination of Pharmaceuticals in Aqueous Environmental Samples. Trends Anal. Chem. 2001, 20, 419–434. [Google Scholar] [CrossRef]
- Petrović, M.; Hernando, M.D.; Díaz-Cruz, M.S.; Barceló, D. Liquid Chromatography-Tandem Mass Spectrometry for the Analysis of Pharmaceutical Residues in Environmental Samples: A Review. J. Chromatogr. A 2005, 1067, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ryu, H.D.; Chung, E.G.; Kim, Y.; Lee, J.K. A Review of Analytical Procedures for the Simultaneous Determination of Medically Important Veterinary Antibiotics in Environmental Water: Sample Preparation, Liquid Chromatography, and Mass Spectrometry. J. Environ. Manag. 2018, 217, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.K.; Jain, R.; Radhapyari, K.; Jadon, N.; Agarwal, S. Voltammetric Techniques for the Assay of Pharmaceuticals—A Review. Anal. Biochem. 2011, 408, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Smart, A.; Crew, A.; Pemberton, R.; Hughes, G.; Doran, O.; Hart, J.P. Screen-Printed Carbon Based Biosensors and Their Applications in Agri-Food Safety. TrAC-Trends Anal. Chem. 2020, 127, 115898. [Google Scholar] [CrossRef]
- Barton, J.; García, M.B.G.; Santos, D.H.; Fanjul-Bolado, P.; Ribotti, A.; McCaul, M.; Diamond, D.; Magni, P. Screen-Printed Electrodes for Environmental Monitoring of Heavy Metal Ions: A Review. Microchim. Acta 2016, 183, 503–517. [Google Scholar] [CrossRef]
- Abo El-Maali, N. Voltammetric Analysis of Drugs. Bioelectrochemistry 2004, 64, 99–107. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Upadhyay, S.S.; Rawool, C.R.; Punde, N.S.; Rajpurohit, A.S. Voltammetric Techniques for the Analysis of Drugs Using Nanomaterials Based Chemically Modified Electrodes. Curr. Anal. Chem. 2019, 15, 249–276. [Google Scholar] [CrossRef]
- Mohamed, H.M. Screen-Printed Disposable Electrodes: Pharmaceutical Applications and Recent Developments. TrAC-Trends Anal. Chem. 2016, 82, 1–11. [Google Scholar] [CrossRef]
- Khorshed, A.A.; Khairy, M.; Banks, C.E. Electrochemical Determination of Antihypertensive Drugs by Employing Costless and Portable Unmodified Screen-Printed Electrodes. Talanta 2019, 198, 447–456. [Google Scholar] [CrossRef]
- Serrano, N.; Castilla, Ò.; Ariño, C.; Diaz-Cruz, M.S.; Díaz-Cruz, J.M. Commercial Screen-Printed Electrodes Based on Carbon Nanomaterials for a Fast and Cost-Effective Voltammetric Determination of Paracetamol, Ibuprofen and Caffeine in Water Samples. Sensors 2019, 19, 4039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, D.C. Quantitative Chemical Analysis, 8th ed.; Clancy Marshal: Houndmills, UK, 2010; ISBN 978-1-4292-1815-3. [Google Scholar]
- Osial, M.; Warczak, M.; Kulesza, P.J.; Krysiński, P.; Gniadek, M. Hybrid Polyindole-gold Nanobrush for Electrochemical Oxidation of Ascorbic Acid. J. Electroanal. Chem. 2020, 877, 114664. [Google Scholar] [CrossRef]
- Amiri, M.; Rezapour, F.; Bezaatpour, A. Hydrophilic Carbon Nanoparticulates at the Surface of Carbon Paste Electrode Improve Determination of Paracetamol, Phenylephrine and Dextromethorphan. J. Electroanal. Chem. 2014, 735, 10–18. [Google Scholar] [CrossRef]
- Petrucci, R.; Zollo, G.; Curulli, A.; Marrosu, G. A New Insight into the Oxidative Mechanism of Caffeine and Related Methylxanthines in Aprotic Medium: May Caffeine Be Really Considered as an Antioxidant? Biochim. Biophys. Acta-Gen. Subj. 2018, 1862, 1781–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marín, J.; Serrano, N.; Ariño, C.; Díaz-Cruz, J.M. A Chemometric Survey about the Ability of Voltammetry to Discriminate Pharmaceutical Products from the Evolution of Signals as a Function of pH. Chemosensors 2020, 8, 46. [Google Scholar] [CrossRef]
- Heli, H.; Majdi, S.; Jabbari, A.; Sattarahmady, N.; Moosavi-Movahedi, A.A. Electrooxidation of Dextromethorphan on a Carbon Nanotube-Carbon Microparticle-Ionic Liquid Composite: Applied to Determination in Pharmaceutical Forms. J. Solid State Electrochem. 2010, 14, 1515–1523. [Google Scholar] [CrossRef]
- Moura, T.; Gaudy, D.; Jacob, M.; Cassanas, G. PH Influence on the Stability of Ascorbic Acid Spray-Drying Solutions. Pharm. Acta Helv. 1994, 69, 77–80. [Google Scholar] [CrossRef]
- Miller, J.; Miller, J. Calibration Methods in Instrumental Analysis: Regression and Correlation. In Statistics and Chemometrics for Analytical Chemistry; Weston, P., Ed.; Pearson Education Limited: Harlow, UK, 2010; pp. 110–153. ISBN 978-0-273-73042-2. [Google Scholar]
- Saciloto, T.R.; Cervini, P.; Cavalheiro, É.T.G. Simultaneous Voltammetric Determination of Acetaminophen and Caffeine at a Graphite and Polyurethane Screen-Printed Composite Electrode. J. Braz. Chem. Soc. 2013, 24, 1461–1468. [Google Scholar] [CrossRef]
- Ping, J.; Wu, J.; Wang, Y.; Ying, Y. Simultaneous Determination of Ascorbic Acid, Dopamine and Uric Acid Using High-Performance Screen-Printed Graphene Electrode. Biosens. Bioelectron. 2012, 34, 70–76. [Google Scholar] [CrossRef]
- Khaled, E.; Hassan, H.N.A.; Mohamed, G.G.; Seleim, A.E.A. Towards Disposable Sensors for Drug Quality Control: Dextromethorphan Screen- Printed Electrodes. Drug Test. Anal. 2010, 2, 424–429. [Google Scholar] [CrossRef]
- Fanjul-Bolado, P.; Lamas-Ardisana, P.J.; Hernández-Santos, D.; Costa-García, A. Electrochemical Study and Flow Injection Analysis of Paracetamol in Pharmaceutical Formulations Based on Screen-Printed Electrodes and Carbon Nanotubes. Anal. Chim. Acta 2009, 638, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Serrano, N.; Cetó, X.; Núñez, O.; Aragó, M.; Gámez, A.; Ariño, C.; Díaz-Cruz, J.M. Characterization and Classification of Spanish Paprika (Capsicum annuum L.) by Liquid Chromatography Coupled to Electrochemical Detection with Screen-Printed Carbon-Based Nanomaterials Electrodes. Talanta 2018, 189, 296–301. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ascorbic Acid | Paracetamol | Dextromethorphan | Caffeine | |||||
|---|---|---|---|---|---|---|---|---|
| Individual Determination | Simultaneous Determination | Individual Determination | Simultaneous Determination | Individual Determination | Simultaneous Determination | Individual Determination | Simultaneous Determination | |
| Sensitivity (μA mg−1 L) | 0.237 (0.003) | 0.180 (0.003) | 1.28 (0.02) | 0.78 (0.02) | 0.203 (0.003) | 1st linear part 0.335 (0.007) | 0.481 (0.006) | 0.466 (0.008) |
| 2nd linear part 0.198 (0.005) | ||||||||
| Linear range a (mg L−1) | 1.3–100.6 | 1.7–60.5 | 0.5–100.5 | 0.6–40.0 | 1.1–73.6 | 1st linear part 0.9–8.4 | 1.2–100.0 | 1.8–22.0 |
| 2nd linear part 8.4–73.8 | ||||||||
| R2 | 0.996 | 0.991 | 0.994 | 0.992 | 0.994 | 1st linear part 0.992 | 0.996 | 0.996 |
| 2nd linear part 0.992 | ||||||||
| LOD (mg L−1) | 0.4 | 0.5 | 0.1 | 0.2 | 0.3 | 0.3 | 0.4 | 0.5 |
| Ascorbic Acid | Paracetamol | Dextromethorphan | Caffeine | |
|---|---|---|---|---|
| Repeatability (RSD, %) | 8.1 | 1.8 | 4.7 | 3.2 |
| Reproducibility (RSD, %) | 0.7 | 3.2 | 5.5 | 3.1 |
| Sample | Analyte | Cdetermined | RSD (%) | Relative Error (%) | Recovery (%) |
|---|---|---|---|---|---|
| Frenadol® Complex | AA | 260 (10) mg per sachet | 4.2 | 2.9 | N/A |
| PA | 644 (3) mg per sachet | 0.5 | 1.0 | N/A | |
| Spiked well water | AA | 51 (1) mg L−1 | 2.1 | N/A | 99.5 |
| PA | 19.0 (0.4) mg L−1 | 2.2 | N/A | 100.8 | |
| DX | 47.1 (0.5) mg L−1 | 1.0 | N/A | 100.1 | |
| CF | 14.1 (0.3) mg L−1 | 2.0 | N/A | 100.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clares, P.; Pérez-Ràfols, C.; Serrano, N.; Díaz-Cruz, J.M. Voltammetric Determination of Active Pharmaceutical Ingredients Using Screen-Printed Electrodes. Chemosensors 2022, 10, 95. https://doi.org/10.3390/chemosensors10030095
Clares P, Pérez-Ràfols C, Serrano N, Díaz-Cruz JM. Voltammetric Determination of Active Pharmaceutical Ingredients Using Screen-Printed Electrodes. Chemosensors. 2022; 10(3):95. https://doi.org/10.3390/chemosensors10030095
Chicago/Turabian StyleClares, Paula, Clara Pérez-Ràfols, Núria Serrano, and José Manuel Díaz-Cruz. 2022. "Voltammetric Determination of Active Pharmaceutical Ingredients Using Screen-Printed Electrodes" Chemosensors 10, no. 3: 95. https://doi.org/10.3390/chemosensors10030095
APA StyleClares, P., Pérez-Ràfols, C., Serrano, N., & Díaz-Cruz, J. M. (2022). Voltammetric Determination of Active Pharmaceutical Ingredients Using Screen-Printed Electrodes. Chemosensors, 10(3), 95. https://doi.org/10.3390/chemosensors10030095