
Microwave-Assisted Synthesis of Schiff Base Metal–Ligand Complexes with Copper and Nickel Centres for Electrochemical In Vitro Sensing of Nitric Oxide in an Aqueous Solution


Abstract
:1. Introduction
2. Materials and Methods
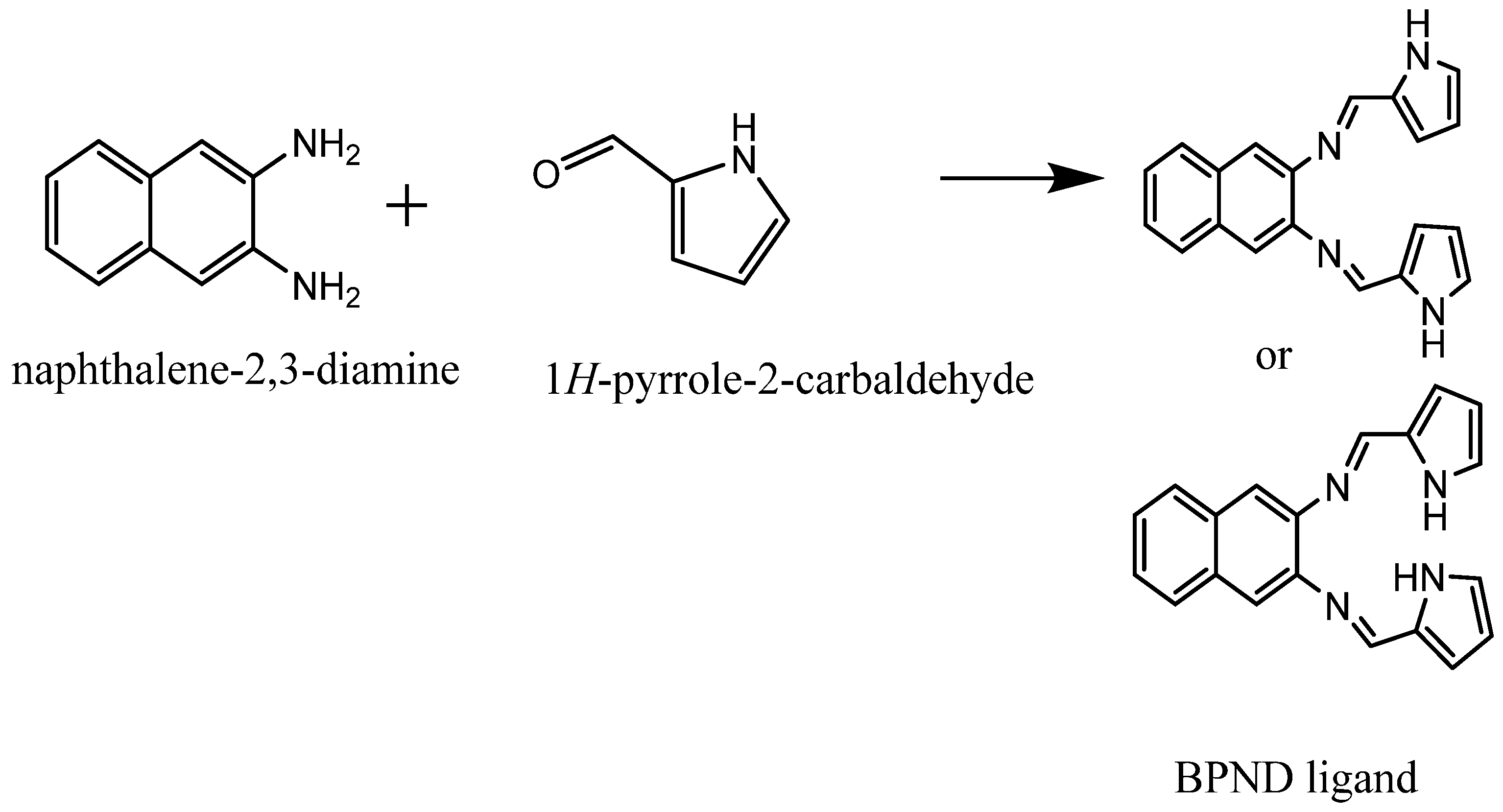
2.1. Synthesis of a Schiff Base Ligand (N,N′-bis(1H-pyrrole-2yl)methylene naphthalene-2,3-diimine)
2.2. Synthesis of Schiff Base Metal Complexes
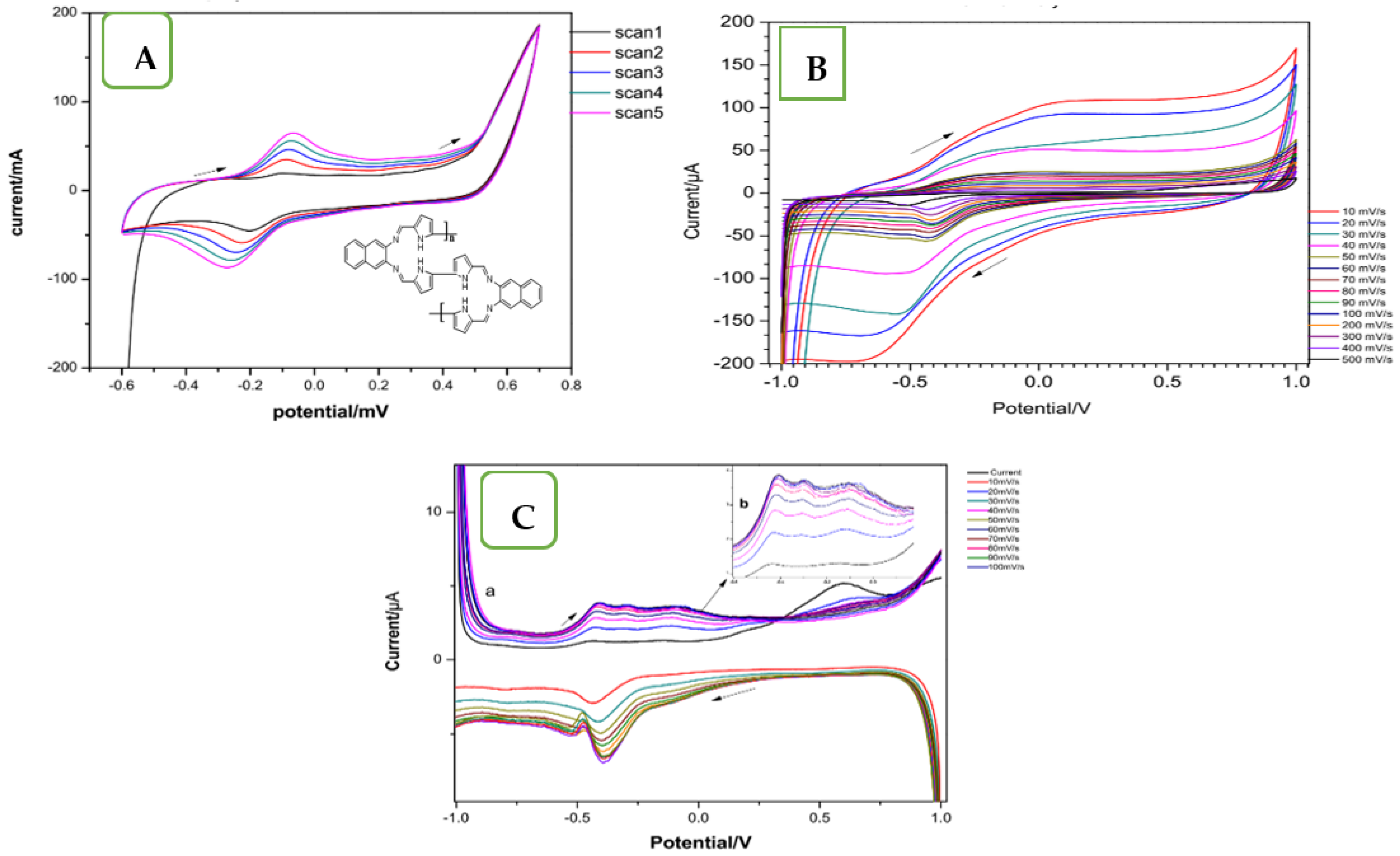
2.3. Electrochemical Synthesis of the BPND Ligand onto SPCE
2.4. Electrodeposition of the Metal Complexes onto the Screen-Printed Carbon Electrodes: SPCE/BPND-Ni/Cu
3. Results
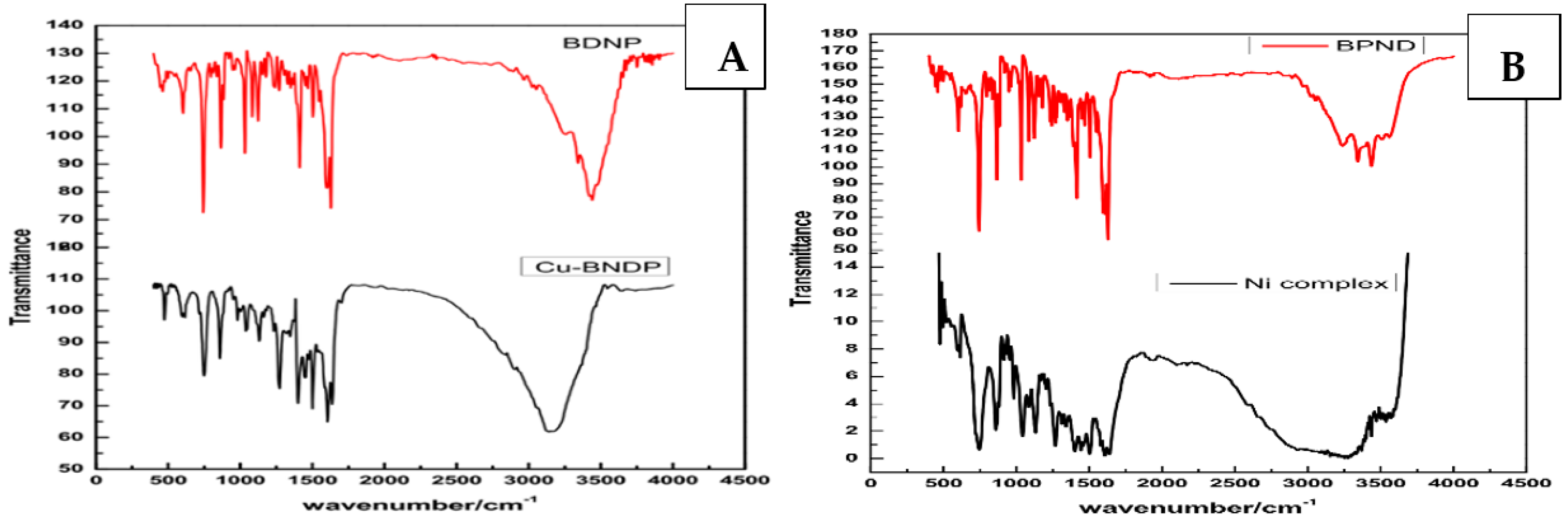
3.1. Structural Analysis of the M–Ligand Complexes
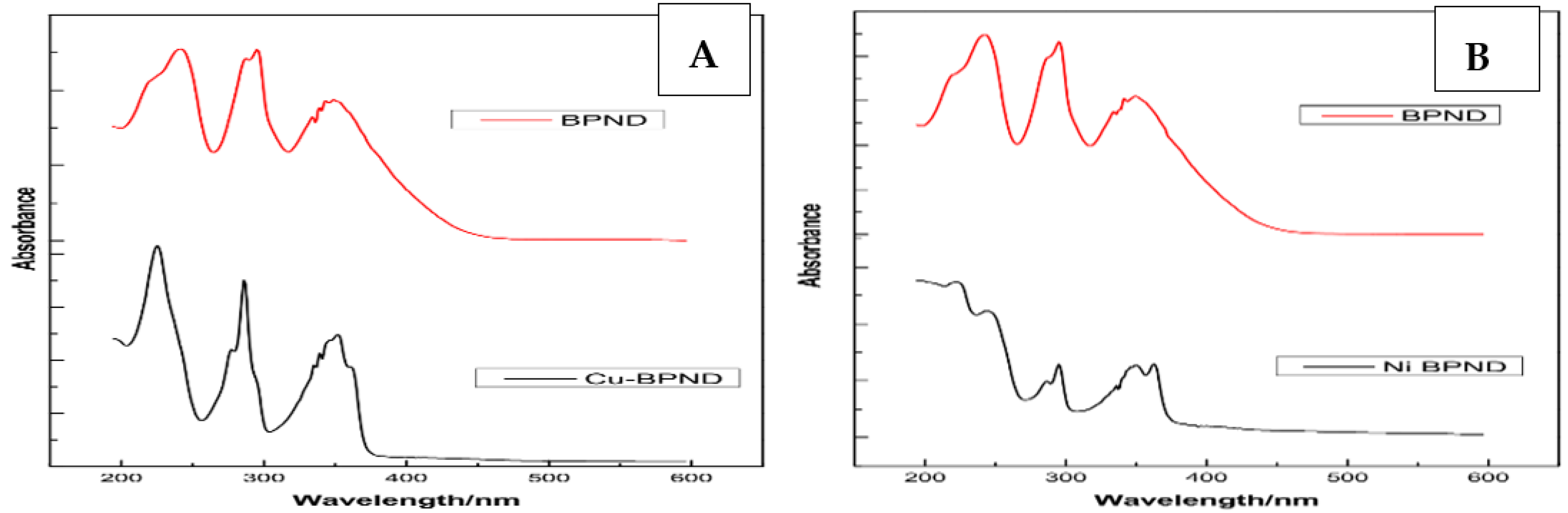
3.2. UV-Vis Spectral Studies
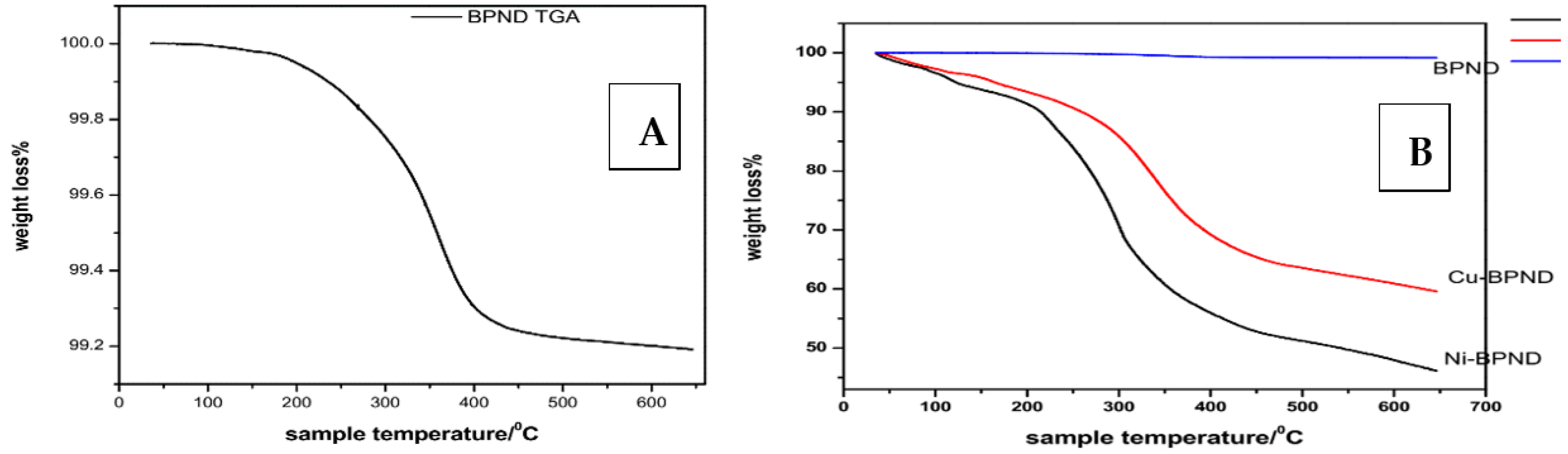
3.3. Thermogravimetric Analysis
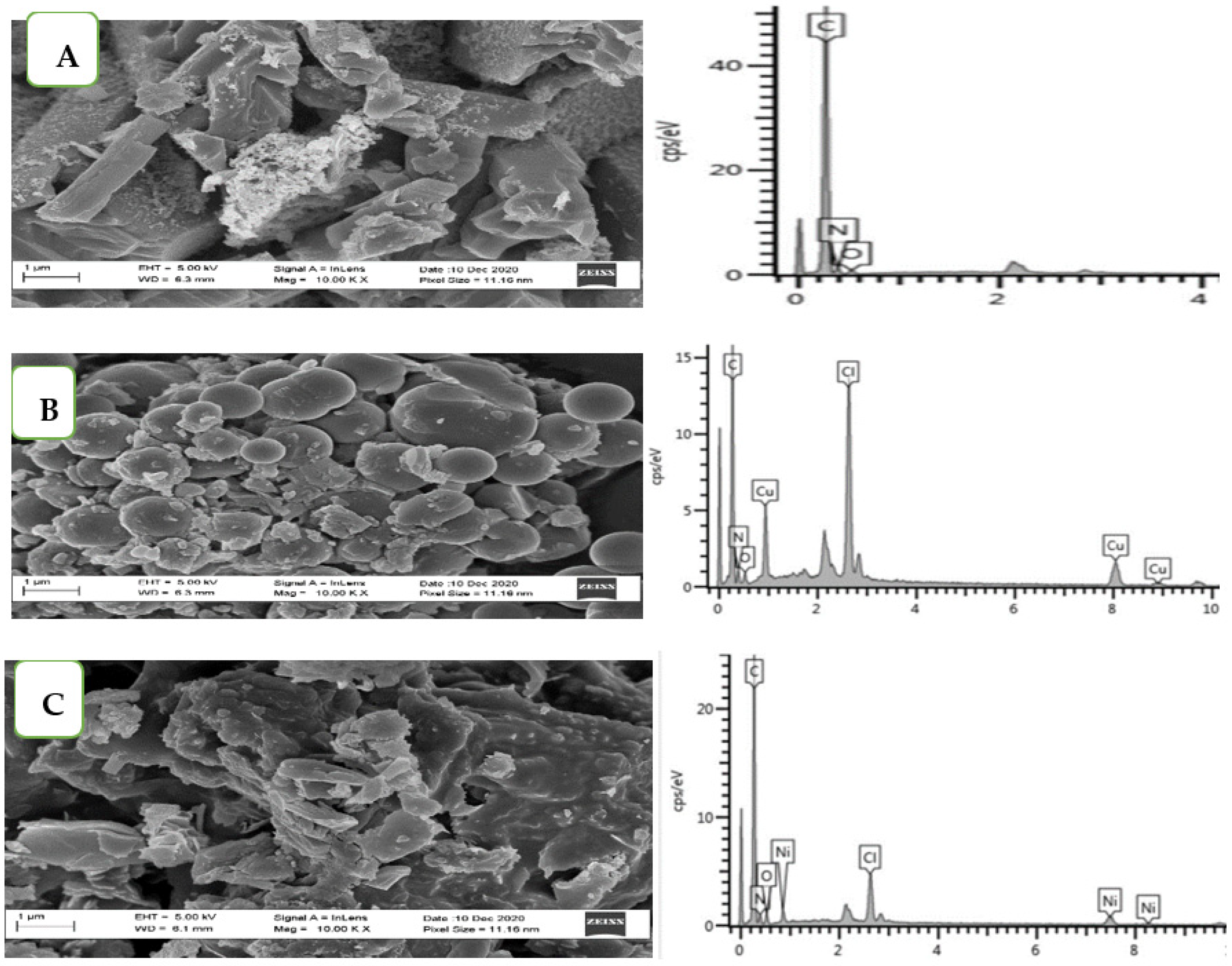
3.4. Morphology of the Ligand and the Metal Complexes
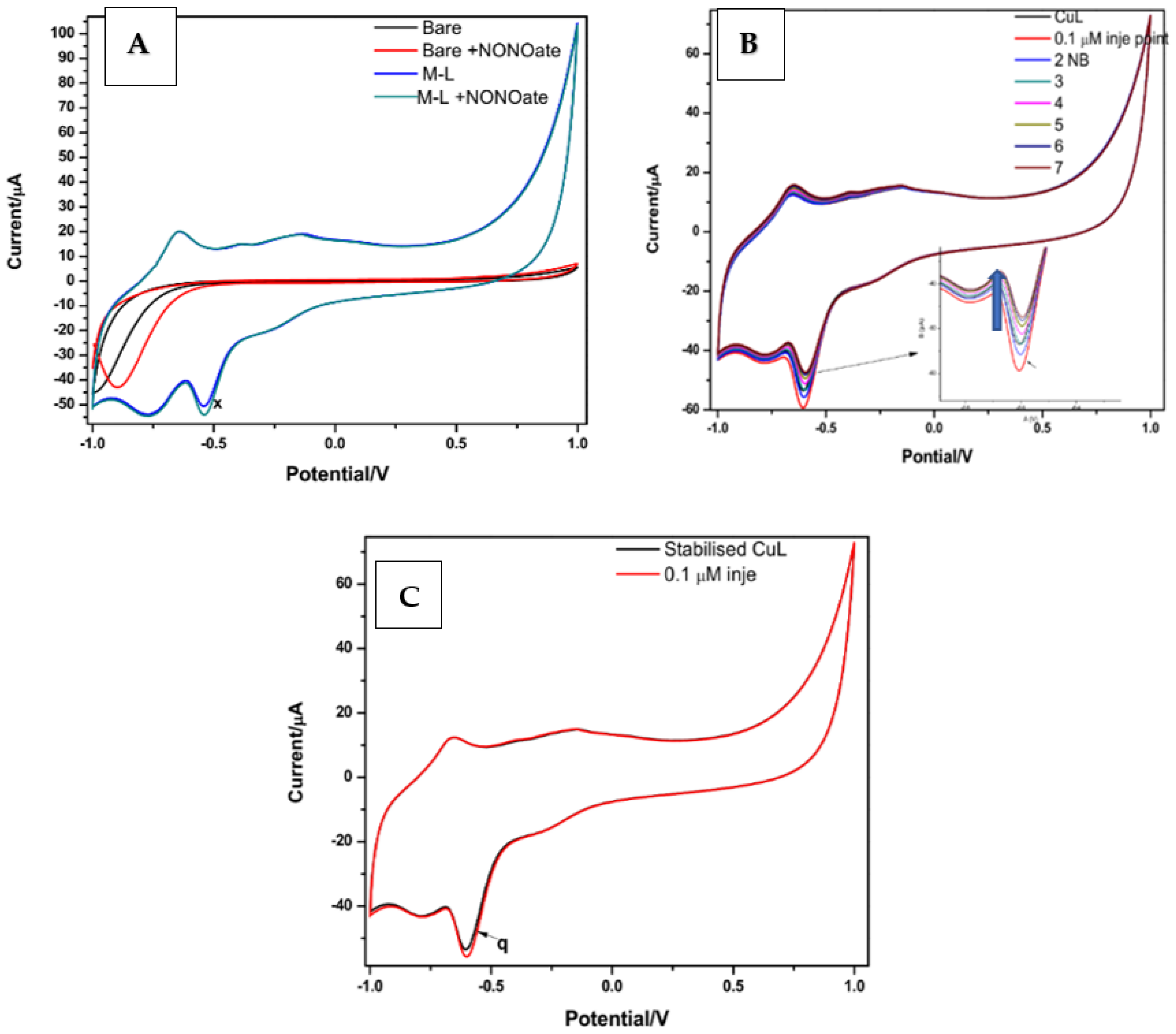
4. Electrochemical Evaluation of BPND and M-BPND Prepared as Thin Films onto SPCEs
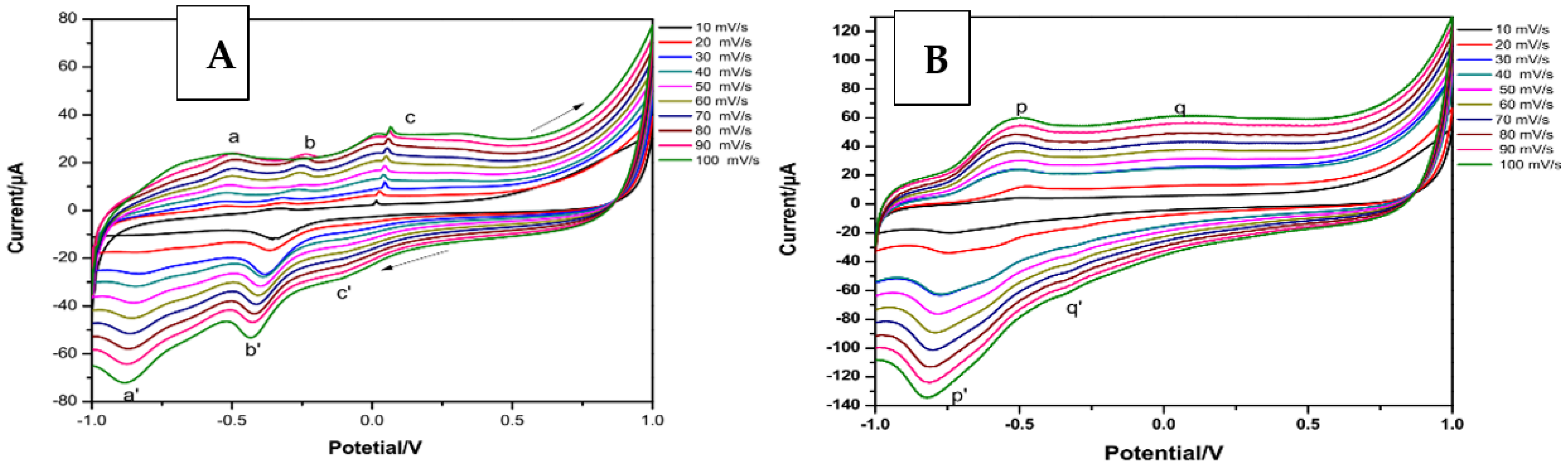
4.1. Morphology of Electrode Systems
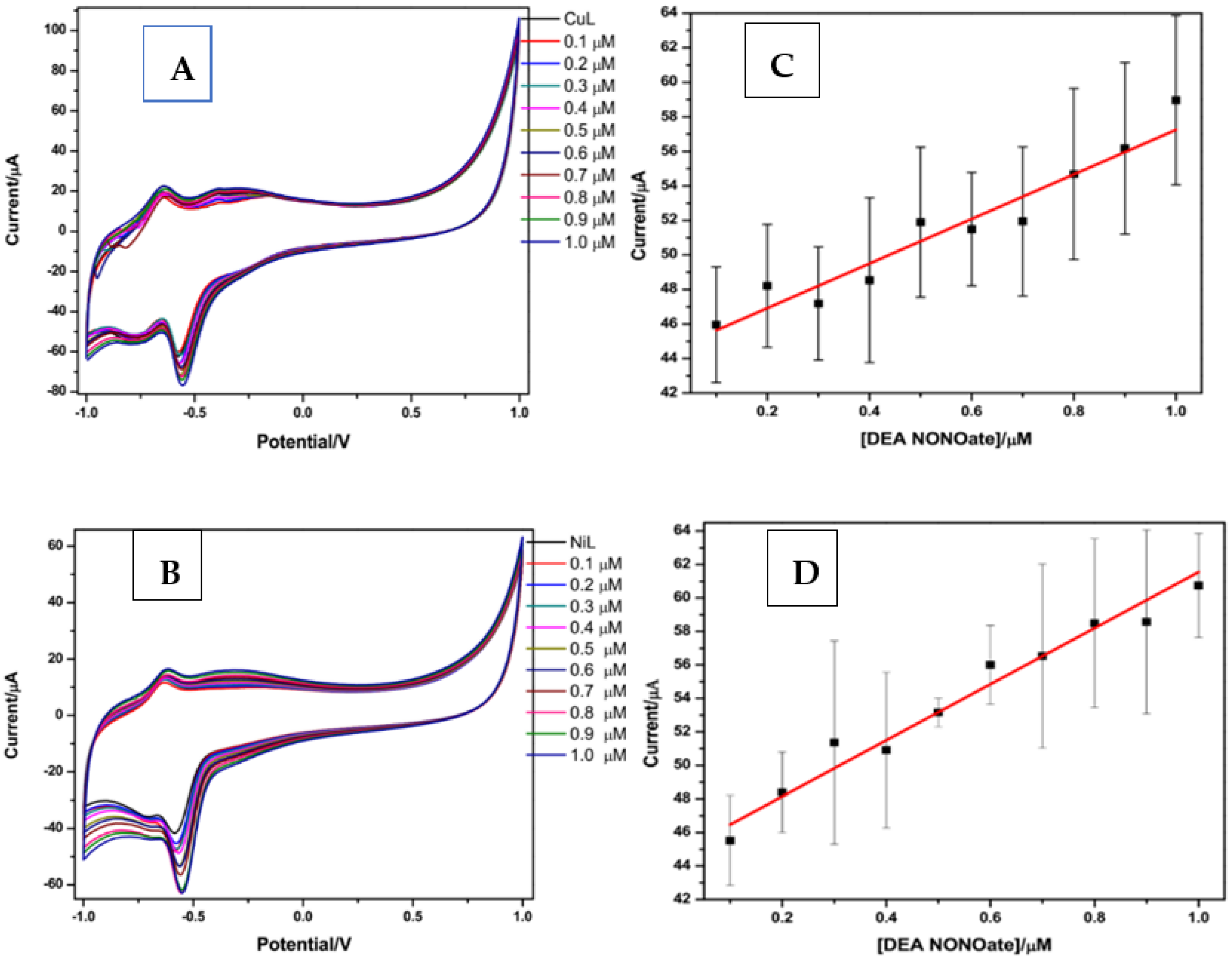
4.2. Analytical Response of Chemical Sensors towards NO Detection
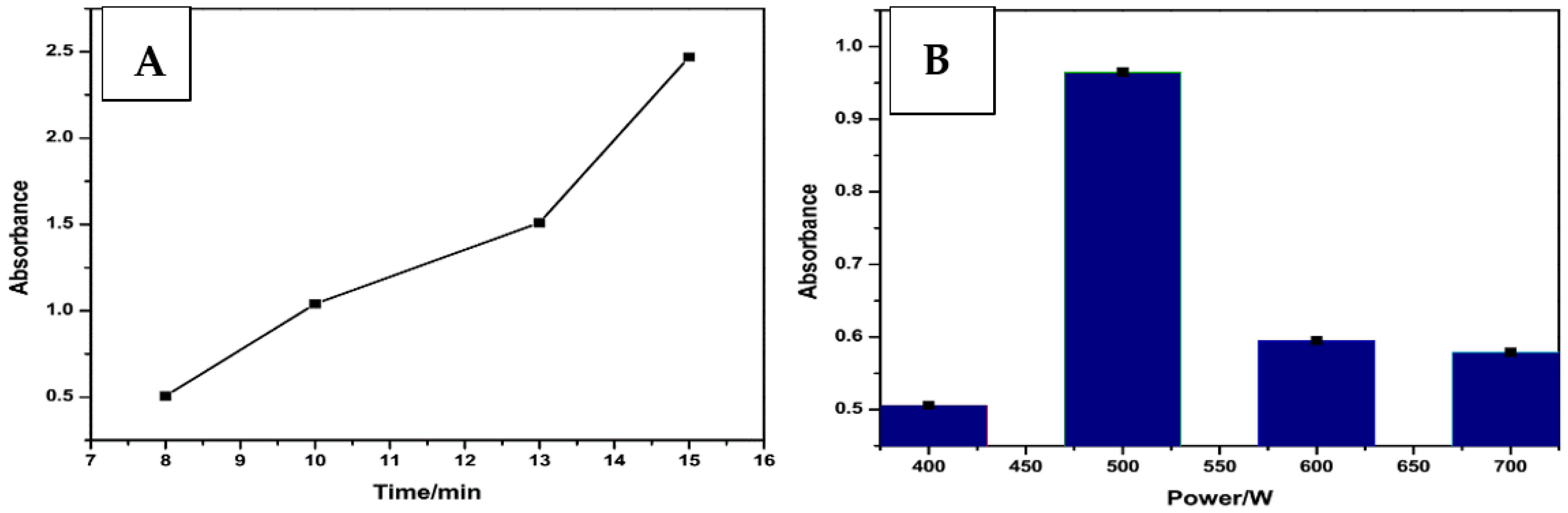
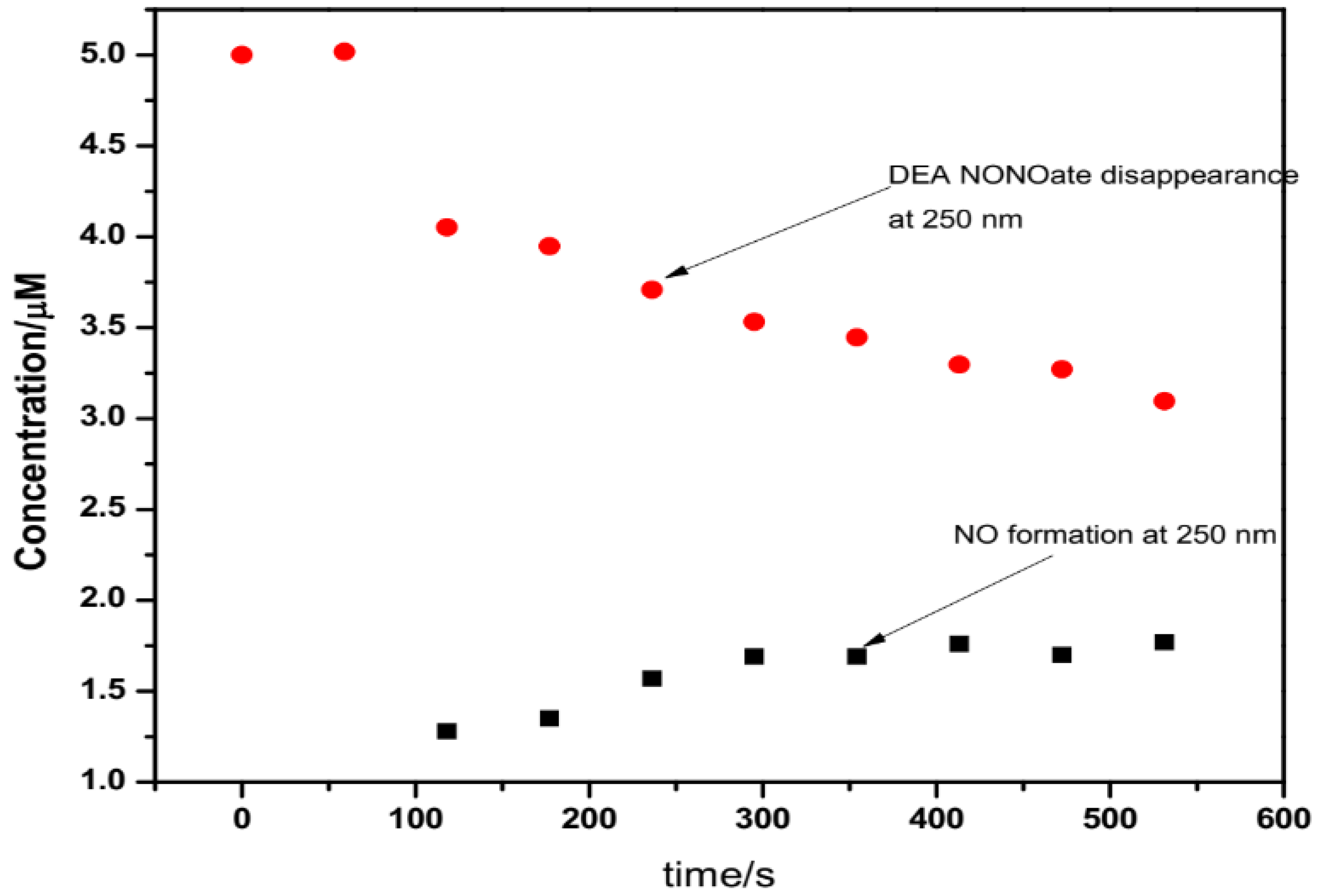
4.2.1. DEA NONOate Standard Solutions
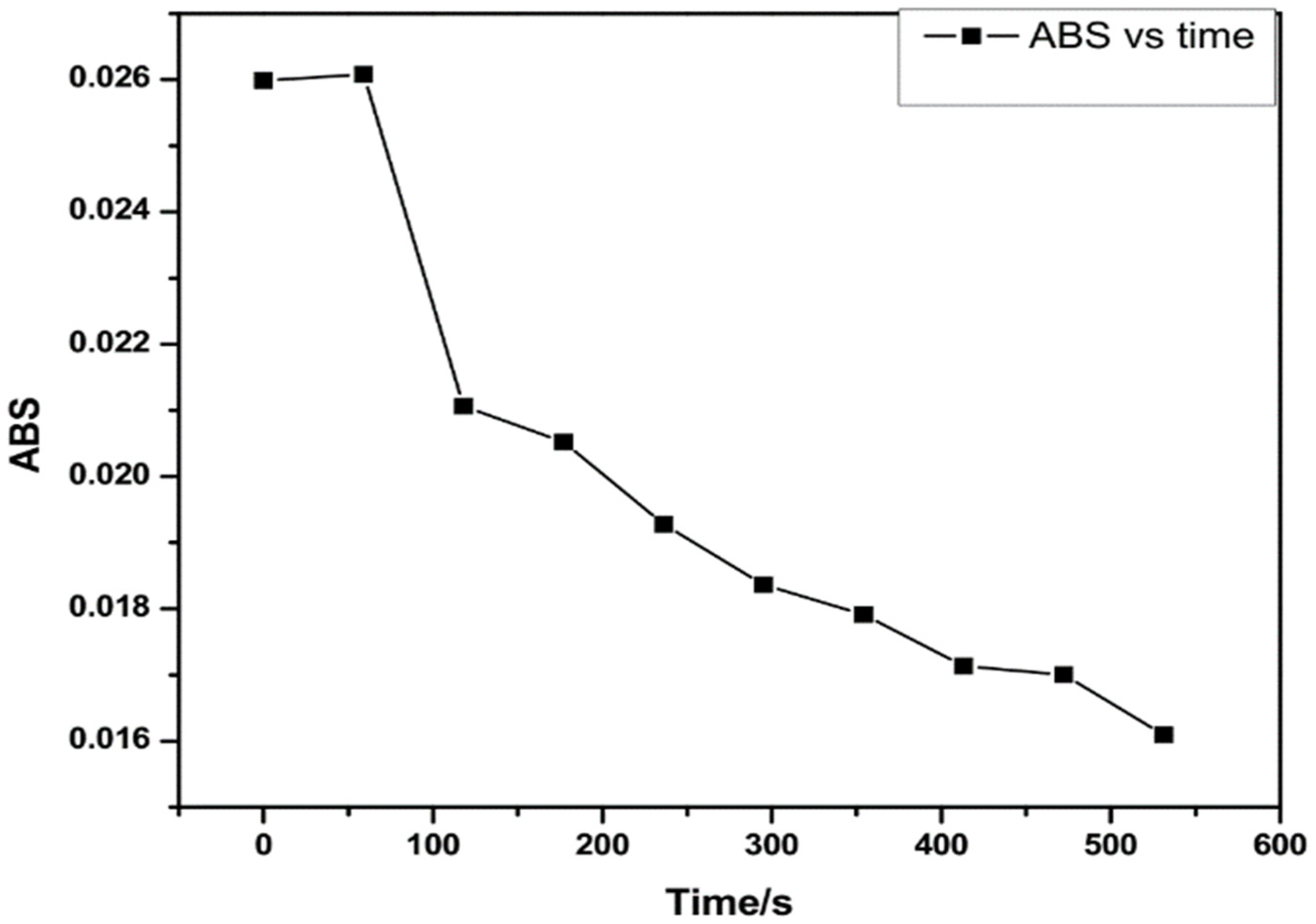
4.2.2. UV/Vis Analysis of DEA NONOate Dissociation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taha, Z.H. Nitric oxide measurements in biological samples. Talanta 2003, 61, 3–10. [Google Scholar] [CrossRef]
- Thatcher, G.R.J.; Nicolescu, A.C.; Bennett, B.M.; Toader, V. Nitrates and no release: Contemporary aspects in biological and medicinal chemistry. Free Radic. Biol. Med. 2004, 37, 1122–1143. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.; Stapleton, J.; Hofferber, E.; Haworth, A.; Kachman, S.; Iverson, N.M. Quantification of nitric oxide concentration using single-walled carbon nanotube sensors. Nanomaterials 2021, 11, 243. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Zhang, Y.; Bai, J.; Li, J.; Wang, J.; Li, L.; Zhou, T.; Chen, S.; Rahim, M.; Zhou, B. Efficient denitrification and removal of natural organic matter, emerging pollutants simultaneously for RO concentrate based on photoelectrocatalytic radical reaction. Sep. Purif. Technol. 2020, 234, 116032. [Google Scholar] [CrossRef]
- Chandran, B.; Janakiraman, K. New Disposable Nitric Oxide Sensor Fabrication Using GaN Nanowires. ACS Omega 2019, 4, 17171–17176. [Google Scholar] [CrossRef] [Green Version]
- Liao, W.; Ye, T.; Liu, H. Prognostic value of inducible nitric oxide synthase (iNOS) in human cancer: A systematic review and meta-analysis. BioMed Res. Int. 2019, 2019, 6304851. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Qi, H.; Ma, Y.; Deng, Y.; Liu, S.; Jie, Y.; Jing, J.; He, J.; Zhang, X.; Wheatley, L.; et al. A flexible and physically transient electrochemical sensor for real-time wireless nitric oxide monitoring. Nat. Commun. 2020, 11, 1. [Google Scholar] [CrossRef]
- Griveau, S.; Dumézy, C.; Goldner, P.; Bedioui, F. Electrochemical analysis of the kinetics of nitric oxide release from two diazeniumdiolates in buffered aqueous solutions. Electrochem. Commun. 2007, 9, 2551–2556. [Google Scholar] [CrossRef]
- Bedioui, F.; Griveau, S. Electrochemical Detection of Nitric Oxide: Assessement of Twenty Years of Strategies. Electroanalysis 2013, 25, 587–600. [Google Scholar] [CrossRef]
- Glynn, S.A. Emerging novel mechanisms of action for nitric oxide in cancer progression. Curr. Opin. Physiol. 2019, 9, 18–25. [Google Scholar] [CrossRef]
- Xu, W.; Liu, L.Z.; Loizidou, M.; Ahmed, M.; Charles, I.G. The role of nitric oxide in cancer. Cell Res. 2002, 12, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Choudhari, S.K.; Chaudhary, M.; Bagde, S.; Gadbail, A.R.; Joshi, V. Nitric oxide and cancer: A review. World J. Surg. Oncol. 2013, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.Y.; Liaw, W.F. Nitric oxide reduction forming hyponitrite triggered by metal-containing complexes. J. Chin. Chem. Soc. 2020, 67, 206–212. [Google Scholar] [CrossRef]
- Housein, Z.; Kareem, T.S.; Salihi, A. In vitro anticancer activity of hydrogen sulfide and nitric oxide alongside nickel nanoparticle and novel mutations in their genes in CRC patients. Sci. Rep. 2021, 11, 2536. [Google Scholar] [CrossRef]
- Ragsdale, S.W. Nickel-based enzyme systems. J. Biol. Chem. 2009, 284, 18571–18575. [Google Scholar] [CrossRef] [Green Version]
- Torres, J.; Svistunenko, D.; Karlsson, B.; Cooper, C.E.; Wilson, M.T. Fast reduction of a copper center in laccase by nitric oxide and formation of a peroxide intermediate. J. Am. Chem. Soc. 2002, 124, 963–967. [Google Scholar] [CrossRef]
- Brown, M.D.; Schoenfisch, M.H. Electrochemical Nitric Oxide Sensors: Principles of Design and Characterization. Chem. Rev. 2019, 119, 11551–11575. [Google Scholar] [CrossRef]
- Jeong, D.W.; Kim, K.H.; Kim, B.S.; Byun, Y.T. Characteristics of highly sensitive and selective nitric oxide gas sensors using defect-functionalized single-walled carbon nanotubes at room temperature. Appl. Surf. Sci. 2021, 550, 149250. [Google Scholar] [CrossRef]
- Luo, P.; Xie, M.; Luo, J.; Kan, H.; Wei, Q. Nitric oxide sensors using nanospiral ZnO thin film deposited by GLAD for application to exhaled human breath. RSC Adv. 2020, 10, 14877–14884. [Google Scholar] [CrossRef] [Green Version]
- Khandar, A.A.; Cardin, C.; Hosseini-Yazdi, S.A.; McGrady, J.; Abedi, M.; Zarei, S.A.; Gan, Y. Nickel(II) and copper(II) complexes of Schiff base ligands containing N4O2 and N4S2 donors with pyrrole terminal binding groups: Synthesis, characterization, X-ray structures, DFT and electrochemical studies. Inorg. Chim. Acta 2010, 363, 4080–4087. [Google Scholar] [CrossRef]
- Meghdadi, S.; Amirnasr, M.; Mereiter, K.; Molaee, H.; Amiri, A. Synthesis, structure and electrochemistry of Co(III) complexes with an unsymmetrical Schiff base ligand derived from 2-aminobenzylamine and pyrrole-2-carboxaldehyde. Polyhedron 2011, 30, 1651–1656. [Google Scholar] [CrossRef]
- Adsule, S.; Barve, V.; Chen, D.; Ahmed, F.; Dou, Q.P.; Padhye, S.; Sarkar, F.H. Novel Schiff base copper complexes of quinoline-2 carboxaldehyde as proteasome inhibitors in human prostate cancer cells. J. Med. Chem. 2006, 49, 7242–7246. [Google Scholar] [CrossRef]
- Kaczmarek, M.T.; Zabiszak, M.; Nowak, M.; Jastrzab, R. Lanthanides: Schiff base complexes, applications in cancer diagnosis, therapy, and antibacterial activity. Coord. Chem. Rev. 2018, 370, 42–54. [Google Scholar] [CrossRef]
- Khuhawar, M.Y.; Mughal, M.A.; Channar, A.H. Synthesis and characterization of some new Schiff base polymers. Eur. Polym. J. 2004, 40, 805–809. [Google Scholar] [CrossRef]
- Bilici, A.; Kaya, I.; Saçak, M. Oxidative polymerization of N202 type schiff base monomer and its metal complexes: Synthesis and thermal, optical and electrochemical properties. J. Inorg. Organomet. Polym. Mater. 2010, 20, 124–133. [Google Scholar] [CrossRef]
- Ayinde, W.B.; Gitari, W.M.; Samie, A. Optimization of microwave-assisted synthesis of silver nanoparticle by Citrus paradisi peel and its application against pathogenic water strain. Green Chem. Lett. Rev. 2019, 12, 225–234. [Google Scholar]
- Cantón-Díaz, A.M.; Muñoz-Flores, B.M.; Moggio, I.; Arias, E.; Turlakov, G.; Del Angel-Mosqueda, C.; Ramirez-Montes, P.I.; Jiménez-Péreza, V.M. Molecular structures, DFT studies and their photophysical properties in solution and solid state. Microwave-assisted multicomponent synthesis of organotin bearing Schiff bases. J. Mol. Struct. 2019, 1180, 642–650. [Google Scholar] [CrossRef]
- Chen, Y. Microwave-assisted synthesis of a novel steroid-derived Schiff base chemosensor for detection of Al3+ in aqueous media. J. Chem. Res. 2020, 44, 750–755. [Google Scholar] [CrossRef]
- More, M.S.; Joshi, P.G.; Mishra, Y.K.; Khanna, P.K. Metal complexes driven from Schiff bases and semicarbazones for biomedical and allied applications: A review. Mater. Today Chem. 2019, 14, 100195. [Google Scholar] [CrossRef]
- Fahim, A.M.; Shalaby, M.A.; Ibrahim, M.A. Microwave-assisted synthesis of novel 5-aminouracil-based compound with DFT calculations. J. Mol. Struct. 2019, 1194, 211–226. [Google Scholar] [CrossRef]
- Shaikh, M.; Wagare, D.; Farooqui, M.; Durrani, A. Microwave Assisted Synthesis of Novel Schiff Bases of Pyrazolyl Carbaldehyde and Triazole in PEG-400. Polycycl. Aromat. Compd. 2020, 40, 1315–1320. [Google Scholar] [CrossRef]
- Shi, Y.; Jiang, K.; Zhen, R.; Fu, J.; Yan, L.; Gu, Q.; Zhang, Y.; Ling, F. Design, Microwave-Assisted Synthesis and in Vitro Antibacterial and Antifungal Activity of 2,5-Disubstituted Benzimidazole. Chem. Biodivers. 2019, 16, 3. [Google Scholar] [CrossRef]
- Kang, K.; Nanda, S.; Sun, G.; Qiu, L.; Gu, Y.; Zhang, T.; Zhu, M.; Sun, R. Microwave-assisted hydrothermal carbonization of corn stalk for solid biofuel production: Optimization of process parameters and characterization of hydrochar. Energy 2019, 186, 115795. [Google Scholar] [CrossRef]
- Kumar, A.; Kuang, Y.; Liang, Z.; Sun, X. Microwave chemistry, recent advancements, and eco-friendly microwave-assisted synthesis of nanoarchitectures and their applications: A review. Mater. Today Nano 2020, 11, 100076. [Google Scholar] [CrossRef]
- Sharma, A.; Kodgire, P.; Kachhwaha, S.S. Biodiesel production from waste cotton-seed cooking oil using microwave-assisted transesterification: Optimization and kinetic modeling. Renew. Sustain. Energy Rev. 2019, 116, 109394. [Google Scholar] [CrossRef]
- Bordei (Telehoiu), A.T.; Nuță, D.C.; Muşat, G.C.; Missir, A.V.; Căproiu, M.T.; Dumitraşcu, F.; Zarafu, I.; Ioniță, P.; Bădiceanu, C.L.; Limban, C.; et al. Microwave assisted synthesis and spectroscopic characterization of some novel schiff bases of carprofen hydrazide. Farmacia 2019, 67, 955–962. [Google Scholar] [CrossRef]
- Pagadala, R.; Ali, P.; Meshram, J.S. Microwave assisted synthesis and characterization of N,N′-bis(salicylaldehydo)ethylenediimine complexes of Mn(II), Co(II), Ni(II), and Zn(II). J. Coord. Chem. 2009, 62, 4009–4017. [Google Scholar] [CrossRef]
- Ward, M.; Botha, S.; Iwuoha, E.; Baker, P. Actuation Behaviour of a Derivatized Pyrrole Accordion Type Polymer. Int. J. Electrochem. Sci. 2014, 9, 4776–4792. [Google Scholar]
- Monzani, E.; Anthony, G.J.; Koolhaas, A.; Spandre, A.; Leggieri, E.; Casella, L.; Gullotti, M.; Nardin, G.; Randaccio, L.; Fontani, M.; et al. Binding of nitrite and its reductive activation to nitric oxide at biomimetic copper centers. J. Biol. Inorg. Chem. 2000, 5, 251–261. [Google Scholar] [CrossRef]
- Ermler, U.; Grabarse, W.; Shima, S.; Goubeaud, M.; Thauer, R.K. Active sites of transition-metal enzymes with a focus on nickel Abbreviations ACS acetyI-CoA synthase CoA coenzyme A CoB coenzyrne B CODH carbon monoxide dehydrogenase CoM coenzyme M EPR spectroscopy electron paramagnetic resonance spectroscopy MCR methyI-CoM reductase SOD superoxide dismutase. Curr. Opin. Struct. Biol. 1998, 8, 749–758. [Google Scholar]
- BSingh, K.; Prakash, A.; Rajour, H.K.; Bhojak, N.; Adhikari, D. Spectroscopic characterization and biological activity of Zn(II), Cd(II), Sn(II) and Pb(II) complexes with Schiff base derived from pyrrole-2-carboxaldehyde and 2-amino phenol. Spectrochim. Acta–Part A Mol. Biomol. Spectrosc. 2010, 76, 376–383. [Google Scholar] [CrossRef]
- Rana, S.; Mittal, S.K.; Singh, N.; Singh, J.; Banks, C.E. Schiff base modified screen printed electrode for selective determination of aluminium(III) at trace level. Sens. Actuators B Chem. 2017, 239, 17–27. [Google Scholar] [CrossRef]
- Vogt, A.; Prasad, L.; Gupta, A. Synthesis and characterization of nickel (ll), copper (II), manganese (III) and iron (III) complexes with new chiral salen-type ligand. Science 1998, 17, 8. [Google Scholar]
- Percy, G.C.; Thornton, D.A. Infrared spectra of N-aryl salicylaldimine complexes substituted in both aryl rings. J. Inorg. Nucl. Chem. 1973, 35, 2319–2327. [Google Scholar] [CrossRef]
- Kumar, S.P.; Suresh, R.; Giribabu, K.; Manigandan, R.; Munusamy, S.; Muthamizh, S.; Narayanan, V. Synthesis and characterization of chromium(III) Schiff base complexes: Antimicrobial activity and its electrocatalytic sensing ability of catechol. Spectrochim. Acta–Part A Mol. Biomol. Spectrosc. 2015, 139, 431–441. [Google Scholar] [CrossRef]
- Çelik, C.; Tümer, M.; Serin, S. Complexes of tetradentate Schiff base ligands with divalent transition metals. Synth. React. Inorg. Met. Chem. 2002, 32, 1839–1854. [Google Scholar] [CrossRef]
- Mohanan, K.; Kumari, B.S.; Rijulal, G. Microwave assisted synthesis, spectroscopic, thermal, and antifungal studies of some lanthanide(III) complexes with a heterocyclic bishydrazone. J. Rare Earths 2008, 26, 16–21. [Google Scholar] [CrossRef]
- Kaushik, A.; Kumar, R.; Arya, S.K.; Nair, M.; Malhotra, B.D.; Bhansali, S. Organic-Inorganic Hybrid Nanocomposite-Based Gas Sensors for Environmental Monitoring. Chem. Rev. 2015, 115, 4571–4606. [Google Scholar] [CrossRef]
- Ourari, A.; Aggoun, D.; Ouahab, L. A novel copper(II)-Schiff base complex containing pyrrole ring: Synthesis, characterization and its modified electrodes applied in oxidation of aliphatic alcohols. Inorg. Chem. Commun. 2013, 33, 118–124. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, K.; Drew, M.G.B.; Chattopadhyay, S. Synthesis and structure of a cobalt(III) complex containing pendant Schiff base ligand: Exploration of its catechol oxidase and phenoxazinone synthase like activity. Inorg. Chim. Acta 2018, 482, 23–33. [Google Scholar] [CrossRef]
- Mehta, P.K.; Oh, E.T.; Park, H.J.; Lee, K.H. Ratiometric detection of Cu+ in aqueous buffered solutions and in live cells using fluorescent peptidyl probe to mimic the binding site of the metalloprotein for Cu+. Sens. Actuators B Chem. 2018, 256, 393–401. [Google Scholar] [CrossRef]
- Fekri, R.; Shaabani, B. Synthesis and Characterization of Ni (II) Complex with Tridentate Ligand of Schiff ’ s Bases of Salicylaldehyde with Thiocyanate Ligand. Appl. Environ. Biol. Sci. 2013, 3, 75–80. [Google Scholar]
- Elango, G.; Arumugam, A.; Guhanathan, S. Co (II), Ni (II) and Cu (II) Complexes with Schiff Base Ligand: Syntheses, Characterization, Antimicrobial Studies and Molecular Docking Studies. SOJ Mater. Sci. Eng. 2017, 5, 1–12. [Google Scholar] [CrossRef]
- Mangamamba, T.; Ganorkar, M.C.; Swarnabala, G. Characterization of Complexes Synthesized Using Schiff Base Ligands and Their Screening for Toxicity Two Fungal and One Bacterial Species on Rice Pathogens. Int. J. Inorg. Chem. 2014, 2014, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Júnior, G.C.; Silva, A.P.S.; Guinesi, L.S. Synthesis, characterization and electropolymerization of a new polypyrrole iron(II) Schiff-base complex. Polyhedron 2004, 23, 1953–1960. [Google Scholar] [CrossRef]
- Tümer, M. Polydentate Schiff-base ligands and their Cd(II) and Cu(II) metal complexes: Synthesis, characterization, biological activity and electrochemical properties. J. Coord. Chem. 2007, 60, 2051–2065. [Google Scholar] [CrossRef]
- Hassaan, A.M.A.; Khalifa, M.A. Metal chelates of some transition and non-transition metal ions with Schiff base derived from isatin with o-phenylenediamine. Monatshefte für Chem. Chem. Mon. 1993, 124, 803–808. [Google Scholar] [CrossRef]
- Sumrra, S.H.; Ibrahim, M.; Ambreen, S.; Imran, M.; Danish, M.; Rehmani, F.S. Synthesis, Spectral Characterization, and Biological Evaluation of Transition Metal Complexes of Bidentate N, O Donor Schiff Bases. Bioinorg. Chem. Appl. 2014, 2014, 812924. [Google Scholar] [CrossRef]
- Yousif, E.; Majeed, A.; Al-Sammarrae, K.; Salih, N.; Salimon, J.; Abdullah, B. Metal complexes of Schiff base: Preparation, characterization and antibacterial activity. Arab. J. Chem. 2017, 10, S1639–S1644. [Google Scholar] [CrossRef] [Green Version]
- Kavitha, N.; Lakshmi, P.V.A. Synthesis, characterization and thermogravimetric analysis of Co(II), Ni(II), Cu(II) and Zn(II) complexes supported by ONNO tetradentate Schiff base ligand derived from hydrazino benzoxazine. J. Saudi Chem. Soc. 2017, 21, S457–S466. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, F.; Yilmaz, V.T.; Topcu, S.; Menek, N. Syntheses, spectral, thermal and electrochemical studies of 3-carboxylacetonehydroxamic acid and its iron(II), cobalt(II), nickel(II), copper(II) and zinc(II) complexes. J. Coord. Chem. 2003, 56, 903–911. [Google Scholar] [CrossRef]
- Nejo, A.A.; Kolawole, G.A.; Nejo, A.O. Synthesis, characterization, antibacterial, and thermal studies of unsymmetrical Schiff-base complexes of cobalt(II). J. Coord. Chem. 2010, 63, 4398–4410. [Google Scholar] [CrossRef]
- Amer, A.A.; Ilikti, H.; Beyens, C.; Lyskawa, J.; Maschke, U. Elaboration of new modified electrodes (MEs) by electropolymerization of Cu(II)-Schiff base complexes bearing pyrrole moieties: Application in electroreduction of acetophenone and carbon dioxide. Eur. Polym. J. 2019, 112, 569–580. [Google Scholar] [CrossRef]
- Banasz, R.; Wałęsa-Chorab, M. Polymeric complexes of transition metal ions as electrochromic materials: Synthesis and properties. Coord. Chem. Rev. 2019, 389, 1–18. [Google Scholar] [CrossRef]
- Fuku, X.; Iftikar, F.; Hess, E.; Iwuoha, E.; Baker, P. Cytochrome c biosensor for determination of trace levels of cyanide and arsenic compounds. Anal. Chim. Acta 2012, 730, 49–59. [Google Scholar] [CrossRef]
- Tabares, J.S.F.; Blas, M.L.; Sereno, L.E.; Silber, J.J.; Correa, N.M.; Molina, P.G. Electrochemistry in large unilamellar vesicles. the distribution of 1-naphthol studied by square wave voltammetry. Electrochim. Acta 2011, 56, 10231–10237. [Google Scholar] [CrossRef]
- Khandar, A.A.; Hosseini-Yazdi, S.A.; Zarei, S.A. Synthesis, characterization and X-ray crystal structures of copper(II) and nickel(II) complexes with potentially hexadentate Schiff base ligands. Inorg. Chim. Acta 2005, 358, 3211–3217. [Google Scholar] [CrossRef]
- Zolezzi, S.; Spodine, E.; Decinti, A. Electrochemical studies of copper(II) complexes with Schiff-base ligands. Polyhedron 2002, 21, 55–59. [Google Scholar] [CrossRef]
- Bulut, I. Study of binary complexes of nickel(II), copper(II), and vanadium(V) with acetazolamide in aqueous medium by voltammetry. J. Chem. 2009, 33, 507–520. [Google Scholar]
- Bott, R.C.; Bowmaker, G.A.; Davis, C.A.; Hope, G.A.; Jones, B.E. Crystal Structure of [ Cu 4 (tu) 7 ](SO 4) 2 ] ‚ H 2 O and Vibrational Spectroscopic Studies of Some Copper (I) Thiourea Complexes. Inorg. Chem. 1998, 4, 651–657. [Google Scholar] [CrossRef]
- Rizvi, M.A. Complexation Modulated Redox Behavior of Transition Metal Systems (Review). J. Gen. Chem. 2015, 85, 959–973. [Google Scholar] [CrossRef]
- Ourari, A.; Ouennoughi, Y.; Aggoun, D.; Mubarak, M.S.; Pasciak, E.M.; Peters, D.G. Synthesis, characterization, and electrochemical study of a new tetradentate nickel(II)-Schiff base complex derived from ethylenediamine and 5′-(N-methyl-N-phenylaminomethyl)-2′-hydroxyacetophenone. Polyhedron 2014, 67, 59–64. [Google Scholar] [CrossRef]
- Losada, J.; del Peso, I.; Beyer, L. Electrochemical and spectroelectrochemical properties of copper(II) Schiff-base complexes. Inorg. Chim. Acta 2001, 321, 107–115. [Google Scholar]
- Lovecchio, F.V.; Gore, E.S.; Busch, D.H. The Oxidation and Reduction Behavior of Macrocyclic Complexes of Nickel. Electrochemical and Electron Spin Resonance Studies. J. Am. Chem. Soc. 1974, 96, 3109–3118. [Google Scholar] [CrossRef]
- Kadish, K.M.; Sazou, D.; Maiya, G.B.; Han, B.C.; Liu, Y.M.; Saoiabi, A.; Ferhat, M.; Guilard, R. Electrochemistry of Nickel ‘Picket Fence’ Porphyrin: Electrogeneration and Spectral Characterization of Nickel Complexes In Unusual Oxidation States. Inorg. Chem. 1989, 28, 2542–2547. [Google Scholar] [CrossRef]
- Shi, W.; Li, J.; Wu, J.; Wei, Q.; Chen, C.; Bao, N.; Yu, C.; Gu, H. An electrochemical biosensor based on multi-wall carbon nanotube–modified screen-printed electrode immobilized by uricase for the detection of salivary uric acid. Anal. Bioanal. Chem. 2020, 412, 7275–7283. [Google Scholar] [CrossRef] [PubMed]
- da Cruz, A.G.B.; Wardell, J.L.; Simão, R.A.; Rocco, A.M. Preparation, structure and electrochemistry of a polypyrrole hybrid film with [Pd(dmit)2]2-, bis(1,3-dithiole-2-thione-4,5-dithiolate)palladate(II). Electrochim. Acta 2007, 52, 1899–1909. [Google Scholar] [CrossRef]
- Naghavi, N.; de Mel, A.; Alavijeh, O.S.; Cousins, B.G.; Seifalian, A.M. Nitric oxide donors for cardiovascular implant applications. Small 2013, 9, 22–35. [Google Scholar] [CrossRef]
- Willmot, M.; Gray, L.; Gibson, C.; Murphy, S.; Bath, P.M.W. A systematic review of nitric oxide donors and L-arginine in experimental stroke; effects on infarct size and cerebral blood flow. Nitric Oxide Biol. Chem. 2005, 12, 141–149. [Google Scholar] [CrossRef]
- Griveau, S.; Seguin, J.; Scherman, D.; Chabot, G.G.; Bedioui, F. In vivo electrochemical detection of nitroglycerin-derived nitric oxide in tumor-bearing mice. Electroanalysis 2009, 21, 631–634. [Google Scholar] [CrossRef]
- Brown, M.D.; Schoenfisch, M.H. Catalytic selectivity of metallophthalocyanines for electrochemical nitric oxide sensing. Electrochim. Acta 2018, 273, 98–104. [Google Scholar] [CrossRef]
- Mao, L.; Yamamoto, K.; Zhou, W.; Jin, L. Electrochemical nitric oxide sensors based on electropolymerized film of M(salen) with central ions of Fe, Co, Cu, and Mn. Electroanalysis 2000, 12, 72–77. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Temp Range (°C) | Sample Tmax (°C) | Mass Loss (%) | Total Mass Loss (%) |
|---|---|---|---|---|
| BPND ligand | 35.8–200.2 | 0.06 | ||
| 224.5–396.1 | 308.7 | 0.765 | 0.835 | |
| Cu-BPND | 35.8–155.1 | 11.11 | ||
| 230.3–35.2 | 288.2 | 32.66 | 43.8 | |
| Ni-BPND | 35.8–212.3 | 10.20 | ||
| 222.4–409.2 | 257.8 | 43.30 | 53.5 |
| Time (s) | DEA NONOate Concentration/µM | Absorbance | NO Concentration/µM |
|---|---|---|---|
| 0 | 5.0 | 0.0260 | 0.00 |
| 59.025 | 5.017 | 0.0261 | 0.00 |
| 118.050 | 4.053 | 0.0211 | 1.28 × 10−6 |
| 177.075 | 3.948 | 0.02052 | 1.35 × 10−6 |
| 236.100 | 3.709 | 0.0193 | 1.57 × 10−6 |
| 295.125 | 3.533 | 0.0184 | 1.69 × 10−6 |
| 354.150 | 3.446 | 0.0179 | 1.69 × 10−6 |
| 413.175 | 3.297 | 0.0171 | 1.76 × 10−6 |
| 472.200 | 3.271 | 0.0170 | 1.70 × 10−6 |
| 531.225 | 3.096 | 0.0161 | 1.77 × 10−6 |
| Material | R2 | Slope (A/M) | LOD (µM) | LOQ (µM) | Linear Range/µM | STD (n = 10) |
|---|---|---|---|---|---|---|
| SPCE/Cu-BPND | 0.924 | 13.1 | 0.53 | 1.61 | 0.1–1 | 1.25 |
| SPCE/Ni-BPND | 0.964 | 16.3 | 0.21 | 0.64 | 0.1–1 | 1.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moeketse, T.N.; Baker, P.G.; Farao, A.C.; Iwuoha, E.I. Microwave-Assisted Synthesis of Schiff Base Metal–Ligand Complexes with Copper and Nickel Centres for Electrochemical In Vitro Sensing of Nitric Oxide in an Aqueous Solution. Chemosensors 2022, 10, 175. https://doi.org/10.3390/chemosensors10050175
Moeketse TN, Baker PG, Farao AC, Iwuoha EI. Microwave-Assisted Synthesis of Schiff Base Metal–Ligand Complexes with Copper and Nickel Centres for Electrochemical In Vitro Sensing of Nitric Oxide in an Aqueous Solution. Chemosensors. 2022; 10(5):175. https://doi.org/10.3390/chemosensors10050175
Chicago/Turabian StyleMoeketse, Teboho N., Priscilla G. Baker, Al. C. Farao, and Emmanuel I. Iwuoha. 2022. "Microwave-Assisted Synthesis of Schiff Base Metal–Ligand Complexes with Copper and Nickel Centres for Electrochemical In Vitro Sensing of Nitric Oxide in an Aqueous Solution" Chemosensors 10, no. 5: 175. https://doi.org/10.3390/chemosensors10050175
APA StyleMoeketse, T. N., Baker, P. G., Farao, A. C., & Iwuoha, E. I. (2022). Microwave-Assisted Synthesis of Schiff Base Metal–Ligand Complexes with Copper and Nickel Centres for Electrochemical In Vitro Sensing of Nitric Oxide in an Aqueous Solution. Chemosensors, 10(5), 175. https://doi.org/10.3390/chemosensors10050175