
Current Trends in SPR Biosensing of SARS-CoV-2 Entry Inhibitors



Abstract
:1. Introduction
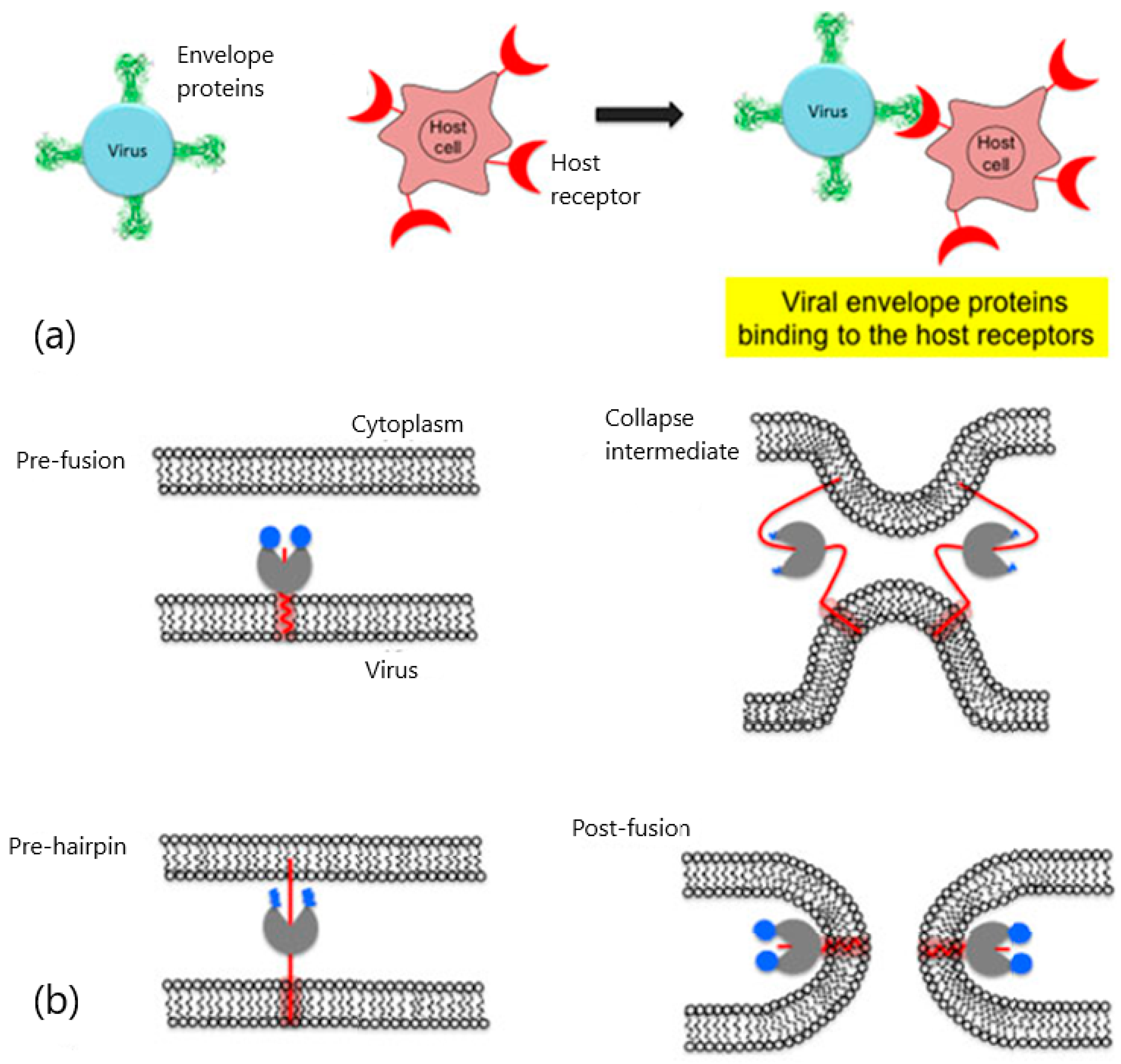
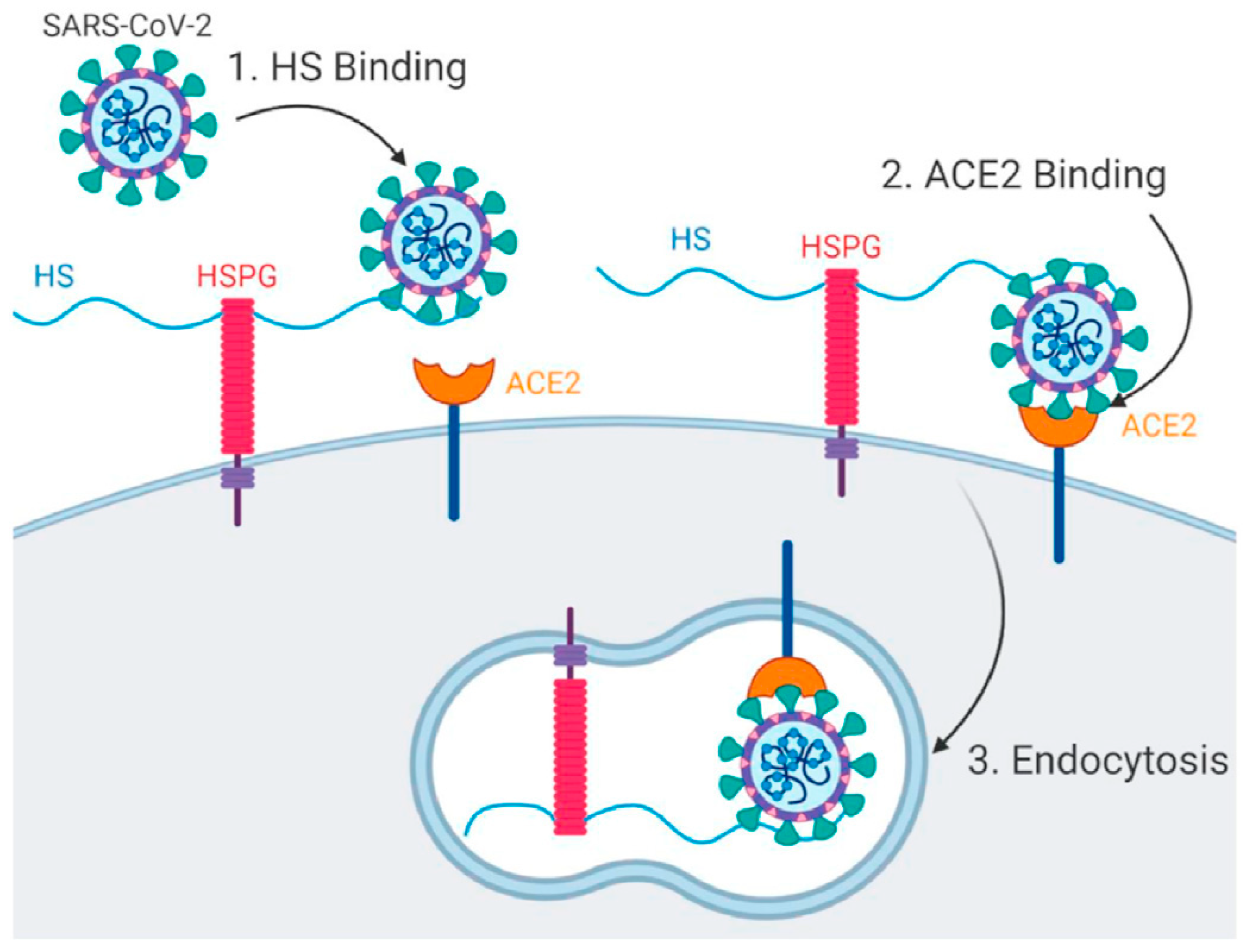
2. Viral Entry and Drug Screening
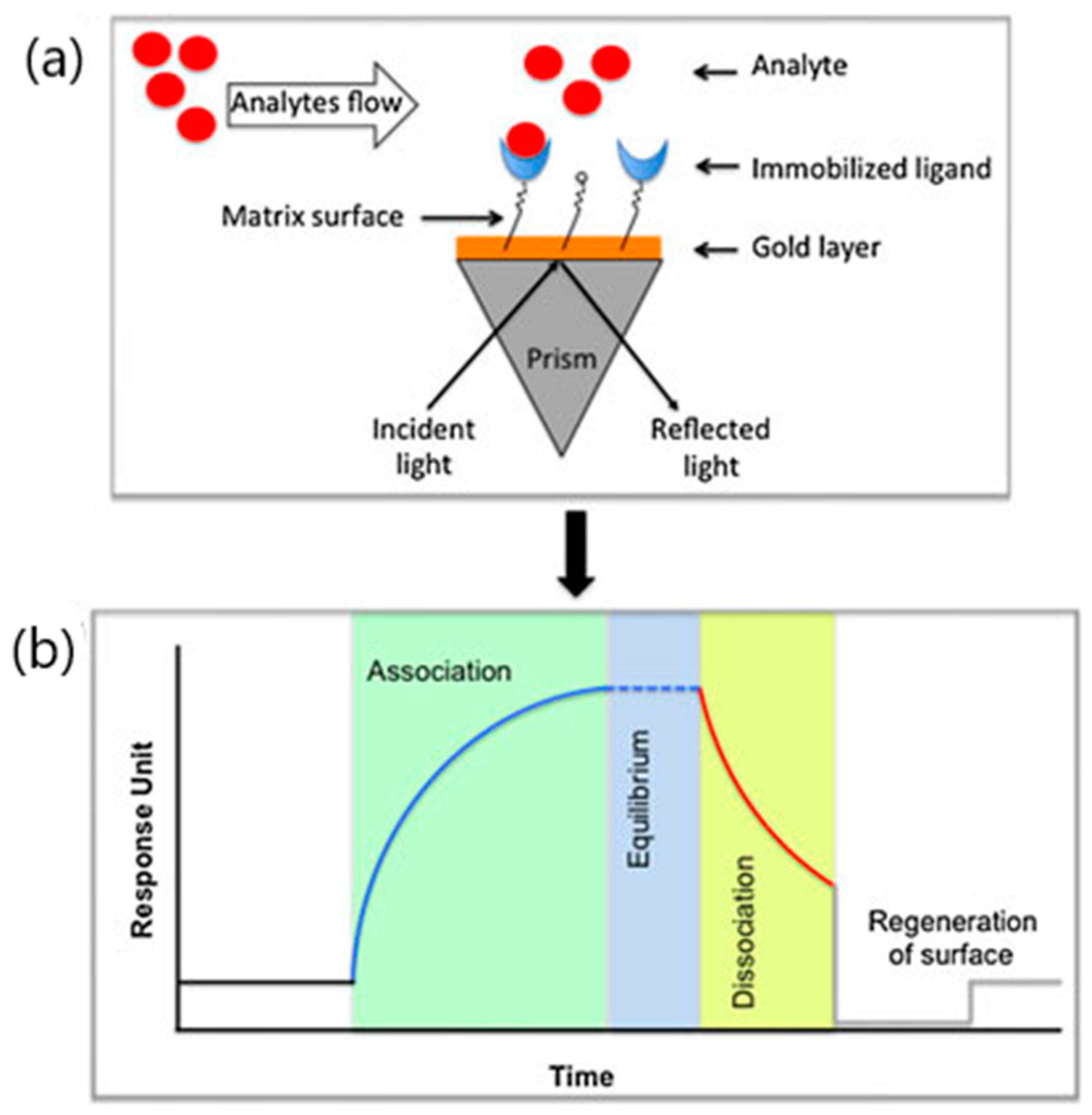
3. SPR Analysis of SARS-CoV-2 Entry Inhibitors
3.1. SPR Strategies for Studying the Blocking of ACE2 Receptors
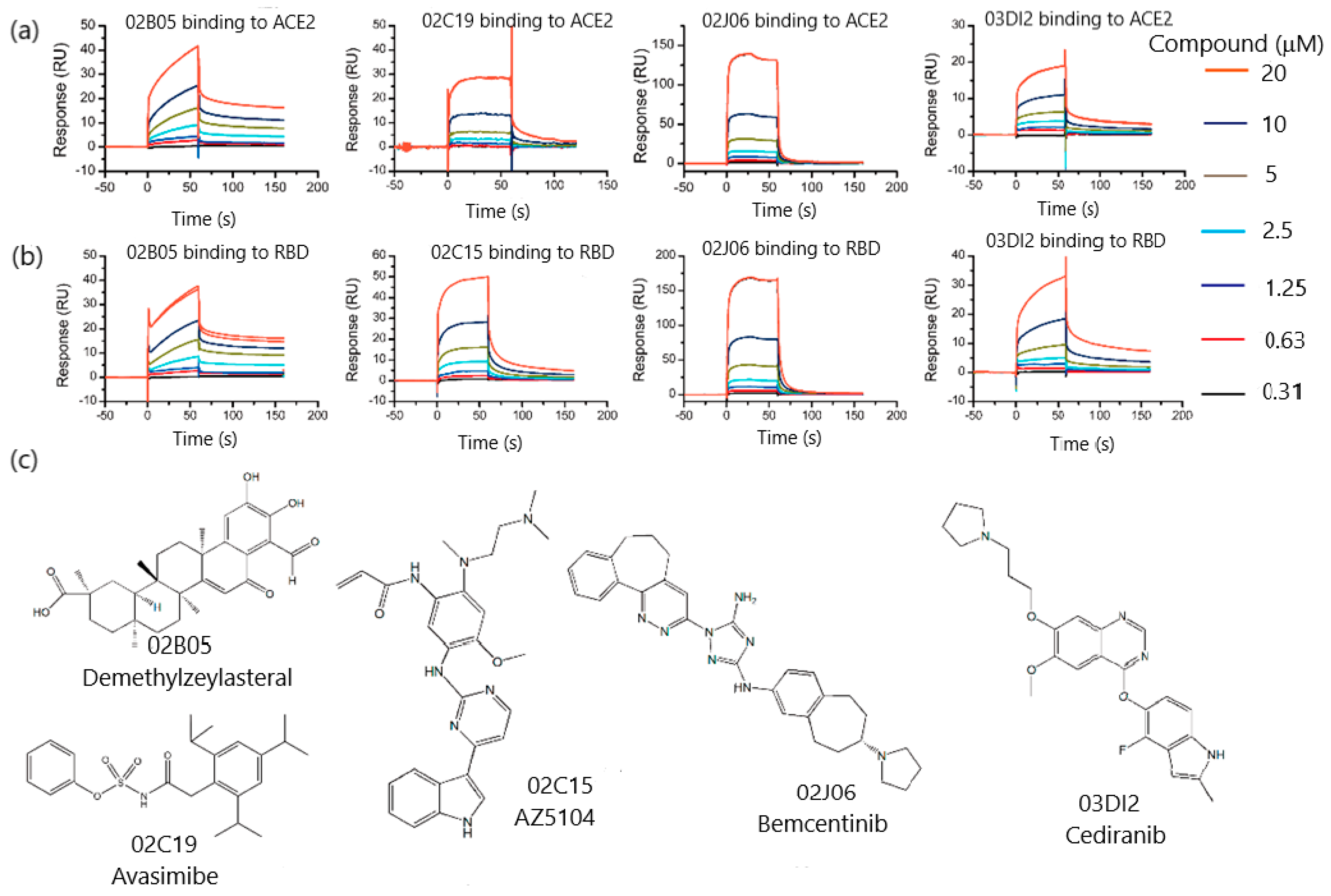
3.1.1. SPR Biosensor as Primary Screening Method
3.1.2. Kinetic Analysis of Natural Products Obtained from Plants
3.1.3. SPR Role in Drug Repurposing
3.1.4. SPR Biosensors as Diagnostic Tools to Detect Neutralizing Antibodies
3.2. SPR Strategies for the Development of SARS-CoV-2 Main Protease Inhibitors
3.2.1. SPR Analysis of Mpro Inhibitors from Plant Origin
3.2.2. Discovery of Mpro Inhibitors via Drug Repurposing
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Pattnaik, G.P.; Chakraborty, H. Entry Inhibitors: Efficient Means to Block Viral Infection. J. Membr. Biol. 2020, 253, 425–444. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.S.; Putilova, V.P.; Yarovaya, O.I.; Zybkina, A.V.; Mordvinova, E.D.; Zaykovskaya, A.V.; Shcherbakov, D.N.; Orshanskaya, I.R.; Sinegubova, E.O.; Esaulkova, I.L.; et al. Synthesis and Antiviral Activity of Camphene Derivatives against Different Types of Viruses. Molecules 2021, 26, 2235. [Google Scholar] [CrossRef]
- Vigant, F.; Santos, N.C.; Lee, B. Broad-Spectrum Antivirals against Viral Fusion. Nat. Rev. Microbiol. 2015, 13, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.K.R. Systematic Screening of Viral Entry Inhibitors Using Surface Plasmon Resonance. Rev. Med. Virol. 2017, 27, e1952. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Z. Recent Progress of Surface Plasmon Resonance in the Development of Coronavirus Disease-2019 Drug Candidates. Eur. J. Med. Chem. Rep. 2021, 1, 100003. [Google Scholar] [CrossRef]
- Brouwer, P.J.M.; Caniels, T.G.; van der Straten, K.; Snitselaar, J.L.; Aldon, Y.; Bangaru, S.; Torres, J.L.; Okba, N.M.A.; Claireaux, M.; Kerster, G.; et al. Potent Neutralizing Antibodies from COVID-19 Patients Define Multiple Targets of Vulnerability. Science 2020, 369, 643–650. [Google Scholar] [CrossRef]
- Shepherd, C.A.; Hopkins, A.L.; Navratilova, I. Fragment Screening by SPR and Advanced Application to GPCRs. Prog. Biophys. Mol. Biol. 2014, 116, 113–123. [Google Scholar] [CrossRef] [Green Version]
- Congreve, M.; Rich, R.L.; Myszka, D.G.; Figaroa, F.; Siegal, G.; Marshall, F.H. Fragment Screening of Stabilized G-Protein-Coupled Receptors Using Biophysical Methods. Methods Enzym. 2011, 493, 115–136. [Google Scholar] [CrossRef]
- Vanwetswinkel, S.; Heetebrij, R.J.; van Duynhoven, J.; Hollander, J.G.; Filippov, D.V.; Hajduk, P.J.; Siegal, G. TINS, Target Immobilized NMR Screening: An Efficient and Sensitive Method for Ligand Discovery. Chem. Biol. 2005, 12, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Blundell, T.L.; Jhoti, H.; Abell, C. High-Throughput Crystallography for Lead Discovery in Drug Design. Nat. Rev. Drug. Discov. 2002, 1, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Duong-Thi, M.-D.; Bergström, M.; Fex, T.; Isaksson, R.; Ohlson, S. High-Throughput Fragment Screening by Affinity LC-MS. J. Biomol. Screen. 2013, 18, 160–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladbury, J.E.; Klebe, G.; Freire, E. Adding Calorimetric Data to Decision Making in Lead Discovery: A Hot Tip. Nat. Rev. Drug Discov. 2010, 9, 23–27. [Google Scholar] [CrossRef]
- Kranz, J.K.; Schalk-Hihi, C. Protein Thermal Shifts to Identify Low Molecular Weight Fragments. Methods Enzymol. 2011, 493, 277–298. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.M.; Engle, L.J.; Pierceall, W.E.; Hughes, D.E.; Shaw, K.J. Affinity Capillary Electrophoresis for the Screening of Novel Antimicrobial Targets. J. Biomol. Screen. 2004, 9, 303–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duong-Thi, M.-D.; Meiby, E.; Bergström, M.; Fex, T.; Isaksson, R.; Ohlson, S. Weak Affinity Chromatography as a New Approach for Fragment Screening in Drug Discovery. Anal. Biochem. 2011, 414, 138–146. [Google Scholar] [CrossRef]
- Kaminski, T.; Gunnarsson, A.; Geschwindner, S. Harnessing the Versatility of Optical Biosensors for Target-Based Small-Molecule Drug Discovery. ACS Sens. 2017, 2, 10–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauglitz, G. Critical Assessment of Relevant Methods in the Field of Biosensors with Direct Optical Detection Based on Fibers and Waveguides Using Plasmonic, Resonance, and Interference Effects. Anal. Bioanal. Chem. 2020, 412, 3317–3349. [Google Scholar] [CrossRef] [Green Version]
- Olaru, A.; Bala, C.; Jaffrezic-Renault, N.; Aboul-Enein, H.Y. Surface Plasmon Resonance (SPR) Biosensors in Pharmaceutical Analysis. Crit. Rev. Anal. Chem. 2015, 45, 97–105. [Google Scholar] [CrossRef]
- Shrivastav, A.M.; Cvelbar, U.; Abdulhalim, I. A Comprehensive Review on Plasmonic-Based Biosensors Used in Viral Diagnostics. Commun. Biol. 2021, 4, 70. [Google Scholar] [CrossRef]
- Homola, J. Surface Plasmon Resonance Sensors for Detection of Chemical and Biological Species. Chem. Rev. 2008, 108, 462–493. [Google Scholar] [CrossRef]
- Masson, J.-F. Surface Plasmon Resonance Clinical Biosensors for Medical Diagnostics. ACS Sens. 2017, 2, 16–30. [Google Scholar] [CrossRef]
- Lin, S.; Shih-Yuan Lee, A.; Lin, C.-C.; Lee, C.-K. Determination of Binding Constant and Stoichiometry for Antibody-Antigen Interaction with Surface Plasmon Resonance. Curr. Proteom. 2006, 3, 271–282. [Google Scholar] [CrossRef] [Green Version]
- Chavanieu, A.; Pugnière, M. Developments in SPR Fragment Screening. Expert Opin. Drug Discov. 2016, 11, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Cao, Y.; Shi, Y.; Cai, Y.; Chen, L.; Wang, D.; Liu, Y.; Chen, X.; Zhu, Z.; Hong, Z.; et al. Surface Plasmon Resonance Biosensor Combined with Lentiviral Particle Stabilization Strategy for Rapid and Specific Screening of P-Glycoprotein Ligands. Anal. Bioanal. Chem. 2021, 413, 2021–2031. [Google Scholar] [CrossRef] [PubMed]
- Giannetti, A.M. From Experimental Design to Validated Hits a Comprehensive Walk-through of Fragment Lead Identification Using Surface Plasmon Resonance. Methods Enzymol. 2011, 493, 169–218. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, M.; Marsh, M. Targeting Viral Entry as a Strategy for Broad-Spectrum Antivirals. F1000Research 2019, 8. F1000 Faculty Rev-1628. [Google Scholar] [CrossRef] [Green Version]
- Hajduk, P.J.; Greer, J. A Decade of Fragment-Based Drug Design: Strategic Advances and Lessons Learned. Nat. Rev. Drug Discov. 2007, 6, 211–219. [Google Scholar] [CrossRef]
- Pollack, S.J.; Beyer, K.S.; Lock, C.; Müller, I.; Sheppard, D.; Lipkin, M.; Hardick, D.; Blurton, P.; Leonard, P.M.; Hubbard, P.A.; et al. A Comparative Study of Fragment Screening Methods on the P38α Kinase: New Methods, New Insights. J. Comput. Aided Mol. Des. 2011, 25, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Boettcher, A.; Ruedisser, S.; Erbel, P.; Vinzenz, D.; Schiering, N.; Hassiepen, U.; Rigollier, P.; Mayr, L.M.; Woelcke, J. Fragment-Based Screening by Biochemical Assays: Systematic Feasibility Studies with Trypsin and MMP12. J. Biomol. Screen. 2010, 15, 1029–1041. [Google Scholar] [CrossRef] [Green Version]
- Rouhana, J.; Hoh, F.; Estaran, S.; Henriquet, C.; Boublik, Y.; Kerkour, A.; Trouillard, R.; Martinez, J.; Pugnière, M.; Padilla, A.; et al. Fragment-Based Identification of a Locus in the Sec7 Domain of Arno for the Design of Protein-Protein Interaction Inhibitors. J. Med. Chem. 2013, 56, 8497–8511. [Google Scholar] [CrossRef] [PubMed]
- Hulswit, R.J.G.; de Haan, C.A.M.; Bosch, B.-J. Coronavirus Spike Protein and Tropism Changes. Adv. Virus Res. 2016, 96, 29–57. [Google Scholar] [CrossRef] [PubMed]
- Pellett, P.E.; Mitra, S.; Holland, T.C. Basics of Virology. Handb. Clin. Neurol. 2014, 123, 45–66. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks, M.; O’Hara, G.; Houlihan, C.; Bell, L.; Heightman, M.; Hart, N. Severe Acute Respiratory Syndrome Coronavirus 2. Ref. Modul. Biomed. Sci. 2021. [Google Scholar] [CrossRef]
- Hikmet, F.; Méar, L.; Edvinsson, Å.; Micke, P.; Uhlén, M.; Lindskog, C. The Protein Expression Profile of ACE2 in Human Tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Pogue, A.; Hill, J.M. SARS-CoV-2 Infectivity and Neurological Targets in the Brain. Cell Mol. Neurobiol. 2020, 1–8. [Google Scholar] [CrossRef]
- Zhu, Z.-L.; Qiu, X.-D.; Wu, S.; Liu, Y.-T.; Zhao, T.; Sun, Z.-H.; Li, Z.-R.; Shan, G.-Z. Blocking Effect of Demethylzeylasteral on the Interaction between Human ACE2 Protein and SARS-CoV-2 RBD Protein Discovered Using SPR Technology. Molecules 2020, 26, 57. [Google Scholar] [CrossRef]
- Day, C.J.; Bailly, B.; Guillon, P.; Dirr, L.; Jen, F.E.-C.; Spillings, B.L.; Mak, J.; von Itzstein, M.; Haselhorst, T.; Jennings, M.P. Multidisciplinary Approaches Identify Compounds That Bind to Human ACE2 or SARS-CoV-2 Spike Protein as Candidates to Block SARS-CoV-2–ACE2 Receptor Interactions. mBio 2021, 12, e03681-20. [Google Scholar] [CrossRef]
- Xu, H.; Liu, B.; Xiao, Z.; Zhou, M.; Ge, L.; Jia, F.; Liu, Y.; Jin, H.; Zhu, X.; Gao, J.; et al. Computational and Experimental Studies Reveal That Thymoquinone Blocks the Entry of Coronaviruses Into In Vitro Cells. Infect Dis. Ther. 2021, 10, 483–494. [Google Scholar] [CrossRef]
- Yu, S.; Zhu, Y.; Xu, J.; Yao, G.; Zhang, P.; Wang, M.; Zhao, Y.; Lin, G.; Chen, H.; Chen, L.; et al. Glycyrrhizic Acid Exerts Inhibitory Activity against the Spike Protein of SARS-CoV-2. Phytomedicine 2021, 85, 153364. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.; Zhou, Y.; Yang, X.; Zhang, F.; Liu, X.; Yu, B. Active Components in Ephedra Sinica Stapf Disrupt the Interaction between ACE2 and SARS-CoV-2 RBD: Potent COVID-19 Therapeutic Agents. J. Ethnopharmacol. 2021, 278, 114303. [Google Scholar] [CrossRef]
- Gao, J.; Ding, Y.; Wang, Y.; Liang, P.; Zhang, L.; Liu, R. Oroxylin A Is a Severe Acute Respiratory Syndrome Coronavirus 2-spiked Pseudotyped Virus Blocker Obtained from Radix Scutellariae Using Angiotensin-converting Enzyme II /Cell Membrane Chromatography. Phytother. Res. 2021, 35, 3194–3204. [Google Scholar] [CrossRef] [PubMed]
- Binette, V.; Côté, S.; Haddad, M.; Nguyen, P.T.; Bélanger, S.; Bourgault, S.; Ramassamy, C.; Gaudreault, R.; Mousseau, N. Corilagin and 1,3,6-Tri- O -Galloy-β- D -Glucose: Potential Inhibitors of SARS-CoV-2 Variants. Phys. Chem. Chem. Phys. 2021, 23, 14873–14888. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Singh, S.; Singh, R. Targeting Novel Coronavirus SARS-CoV-2 Spike Protein with Phytoconstituents of Momordica Charantia. J. Ovarian Res. 2021, 14, 126. [Google Scholar] [CrossRef]
- Ge, S.; Wang, X.; Hou, Y.; Lv, Y.; Wang, C.; He, H. Repositioning of Histamine H1 Receptor Antagonist: Doxepin Inhibits Viropexis of SARS-CoV-2 Spike Pseudovirus by Blocking ACE2. Eur. J. Pharmacol. 2021, 896, 173897. [Google Scholar] [CrossRef]
- Ge, S.; Lu, J.; Hou, Y.; Lv, Y.; Wang, C.; He, H. Azelastine Inhibits Viropexis of SARS-CoV-2 Spike Pseudovirus by Binding to SARS-CoV-2 Entry Receptor ACE2. Virology 2021, 560, 110–115. [Google Scholar] [CrossRef]
- Hou, Y.; Ge, S.; Li, X.; Wang, C.; He, H.; He, L. Testing of the Inhibitory Effects of Loratadine and Desloratadine on SARS-CoV-2 Spike Pseudotyped Virus Viropexis. Chem.-Biol. Interact. 2021, 338, 109420. [Google Scholar] [CrossRef]
- Wang, N.; Han, S.; Liu, R.; Meng, L.; He, H.; Zhang, Y.; Wang, C.; Lv, Y.; Wang, J.; Li, X.; et al. Chloroquine and Hydroxychloroquine as ACE2 Blockers to Inhibit Viropexis of 2019-NCoV Spike Pseudotyped Virus. Phytomedicine 2020, 79, 153333. [Google Scholar] [CrossRef]
- Lu, J.; Hou, Y.; Ge, S.; Wang, X.; Wang, J.; Hu, T.; Lv, Y.; He, H.; Wang, C. Screened Antipsychotic Drugs Inhibit SARS-CoV-2 Binding with ACE2 in Vitro. Life Sci. 2021, 266, 118889. [Google Scholar] [CrossRef]
- Cheng, H.; Lear-Rooney, C.M.; Johansen, L.; Varhegyi, E.; Chen, Z.W.; Olinger, G.G.; Rong, L. Inhibition of Ebola and Marburg Virus Entry by G Protein-Coupled Receptor Antagonists. J. Virol. 2015, 89, 9932–9938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Xia, S.; Pu, J.; Wang, Q.; Li, P.; Lu, L.; Jiang, S. The Antihistamine Drugs Carbinoxamine Maleate and Chlorpheniramine Maleate Exhibit Potent Antiviral Activity Against a Broad Spectrum of Influenza Viruses. Front. Microbiol. 2018, 9, 2643. [Google Scholar] [CrossRef] [PubMed]
- Mercuro, N.J.; Yen, C.F.; Shim, D.J.; Maher, T.R.; McCoy, C.M.; Zimetbaum, P.J.; Gold, H.S. Risk of QT Interval Prolongation Associated With Use of Hydroxychloroquine with or without Concomitant Azithromycin Among Hospitalized Patients Testing Positive for Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, e201834. [Google Scholar] [CrossRef] [PubMed]
- Hewlett, I.; Lee, S.; Molnar, J.; Foldeak, S.; Pine, P.S.; Weaver, J.L.; Aszalos, A. Inhibition of HIV Infection of H9 Cells by Chlorpromazine Derivatives. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1997, 15, 16–20. [Google Scholar] [CrossRef]
- Walker, S.N.; Chokkalingam, N.; Reuschel, E.L.; Purwar, M.; Xu, Z.; Gary, E.N.; Kim, K.Y.; Helble, M.; Schultheis, K.; Walters, J.; et al. SARS-CoV-2 Assays To Detect Functional Antibody Responses That Block ACE2 Recognition in Vaccinated Animals and Infected Patients. J. Clin. Microbiol. 2020, 58, 13. [Google Scholar] [CrossRef]
- Ebihara, T.; Masuda, A.; Takahashi, D.; Hino, M.; Mon, H.; Kakino, K.; Fujii, T.; Fujita, R.; Ueda, T.; Lee, J.M.; et al. Production of ScFv, Fab, and IgG of CR3022 Antibodies Against SARS-CoV-2 Using Silkworm-Baculovirus Expression System. Mol. Biotechnol. 2021, 63, 1223–1234. [Google Scholar] [CrossRef]
- Ravichandran, S.; Coyle, E.M.; Klenow, L.; Tang, J.; Grubbs, G.; Liu, S.; Wang, T.; Golding, H.; Khurana, S. Antibody Signature Induced by SARS-CoV-2 Spike Protein Immunogens in Rabbits. Sci. Transl. Med. 2020, 12, eabc3539. [Google Scholar] [CrossRef]
- Ye, G.; Gallant, J.; Zheng, J.; Massey, C.; Shi, K.; Tai, W.; Odle, A.; Vickers, M.; Shang, J.; Wan, Y.; et al. The Development of Nanosota-1 as Anti-SARS-CoV-2 Nanobody Drug Candidates. eLife 2021, 10, e64815. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-Based Design of Antiviral Drug Candidates Targeting the SARS-CoV-2 Main Protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-Ketoamide Inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Hilgenfeld, R. From SARS to MERS: Crystallographic Studies on Coronaviral Proteases Enable Antiviral Drug Design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Lin, X.; Xing, N.; Zhang, Z.; Zhang, H.; Wu, H.; Xue, W. Structure-Based Discovery of Novel Nonpeptide Inhibitors Targeting SARS-CoV-2 M pro. J. Chem. Inf. Model. 2021, 61, 3917–3926. [Google Scholar] [CrossRef] [PubMed]
- Du, A.; Zheng, R.; Disoma, C.; Li, S.; Chen, Z.; Li, S.; Liu, P.; Zhou, Y.; Shen, Y.; Liu, S.; et al. Epigallocatechin-3-Gallate, an Active Ingredient of Traditional Chinese Medicines, Inhibits the 3CLpro Activity of SARS-CoV-2. Int. J. Biol. Macromol. 2021, 176, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xu, F.; Liu, C.; Cai, A.; Dain, J.A.; Li, D.; Seeram, N.P.; Cho, B.P.; Ma, H. Inhibitory Effects and Surface Plasmon Resonance-Based Binding Affinities of Dietary Hydrolyzable Tannins and Their Gut Microbial Metabolites on SARS-CoV-2 Main Protease. J. Agric. Food Chem. 2021, 69, 12197–12208. [Google Scholar] [CrossRef]
- Eberle, R.J.; Olivier, D.S.; Amaral, M.S.; Gering, I.; Willbold, D.; Arni, R.K.; Coronado, M.A. The Repurposed Drugs Suramin and Quinacrine Cooperatively Inhibit SARS-CoV-2 3CLpro In Vitro. Viruses 2021, 13, 873. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.K.; Upadhyay, S.; Singh, M.; Raghavendhar, S.; Bhardwaj, M.; Sharma, P.; Patel, A.K. Screening and Evaluation of Approved Drugs as Inhibitors of Main Protease of SARS-CoV-2. Int. J. Biol. Macromol. 2020, 164, 2622–2631. [Google Scholar] [CrossRef]
- Gupta, A.; Rani, C.; Pant, P.; Vijayan, V.; Vikram, N.; Kaur, P.; Singh, T.P.; Sharma, S.; Sharma, P. Structure-Based Virtual Screening and Biochemical Validation to Discover a Potential Inhibitor of the SARS-CoV-2 Main Protease. ACS Omega 2020, 5, 33151–33161. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Analyte (MW, Common Uses) | Analytical Approach | SPR Instrument | Binding Affinities (KD: Equilibrium Dissociation Constant) | Reference |
|---|---|---|---|---|
| Demethylzeylasteral (480.59 (g/mol); immunosuppressor, anti-inflammatory, anti-tumoral) | Protein–protein interaction testing Screening and kinetic analysis (binding to ACE2 and S-RBD) Competition assay (blocking RBD-ACE2 interactions) | Biacore T200 (Washington, DC, USA) or S200 instrument (GE Healthcare Life Sciences). | KD = 1.736 mM; kon (1/Ms) = 1989; koff (×10−3 1/s) = 3.345 (ACE2) KD = 1.039 mM (S-RBD) | [38] |
| Sodium lifitegrast (among 21 screened compounds) (637.5 g/mol; keratoconjunctivitis) | SPR screening combined approach Kinetic analysis (binding to S-RBD) Competition assay (blocking RBD-ACE2 interactions) | Biacore S200 system (GE Healthcare Life Sciences) | KD = 1.92 nM (sodium lifitegrast) (KD < 3 mM: rest of compounds) (kon and koff values N/A) | [39] |
| Thymoquinone (164,201 g/mol; antioxidant, anti-inflammatory, chemotherapic) | Binding affinity to ACE2 receptors | Biacore T200 System (GE Healthcare, Uppsala, Sweden) | KD = 32.140 mM (kon and koff values N/A) | [40] |
| Ginsenoside Ra2, ginsenoside Rb1, ginsenoside Rb3, glycyrrhizic acid and berberine chloride (1211.38, 1109.29, 1079.27, 822.94 and 371.81 g/mol respectively; herbs within clinically effective TCM schemes) | Binding activity of TCM-derived components with SARS-CoV-2 S1 subunit | BIAcore T200 instrument (BIAcore T200, GE Healthcare, Chicago, IL, USA) | KD = 55.6 mM (ginsenoside Ra2); KD = 29.7 mM (ginsenoside Rb3); KD = 2.0 mM (ginsenoside Rb1); KD = 66.8 mM (glycyrrhizic acid); KD = 23.9 mM (berberine chloride) (kon and koff values N/A) | [41] |
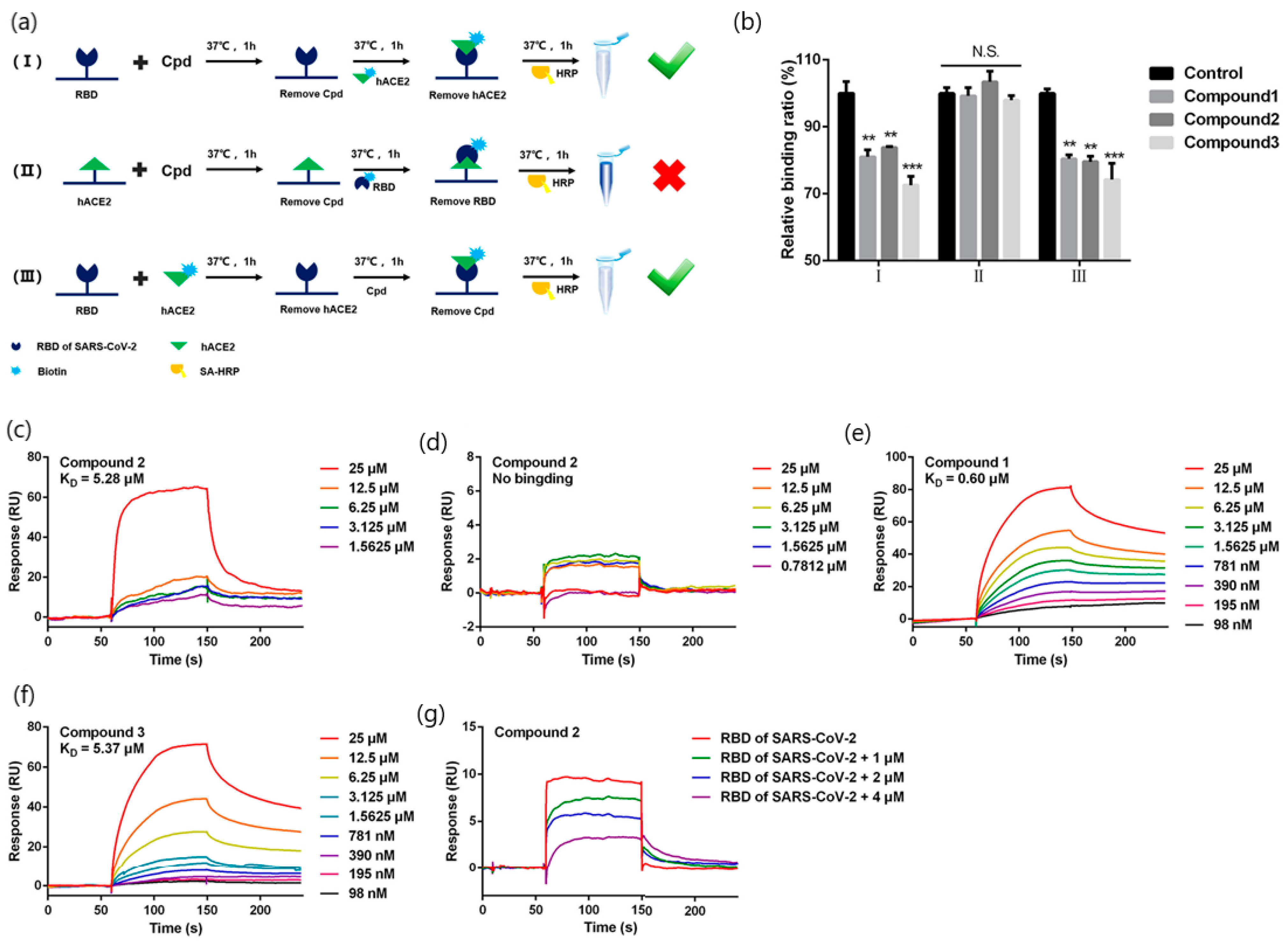
| Quinoline-2-carboxylic acids (3 compounds) (173.168 g/mol, Ephedra sinica extracts; lung diseases treatment) | Binding affinity constants (to S-RBD) Competition assay (blocking RBD-ACE2 interactions) | BIAcore T200 instrument (GE Healthcare Life Sciences) | KD = 0.60–5.37 mM (kon and koff values N/A) | [42] |
| Radix Scutellariae extract: Oroxylin A (284.26 g/mol; respiratory diseases, diabetes, diarrhea treatment) | Binding affinity constants (to ACE2) | Open SPR™ (Nicoya Lifesciences, Waterloo, Canada) | KD = 9.72 × 10−6 M (kon and koff values N/A) | [43] |
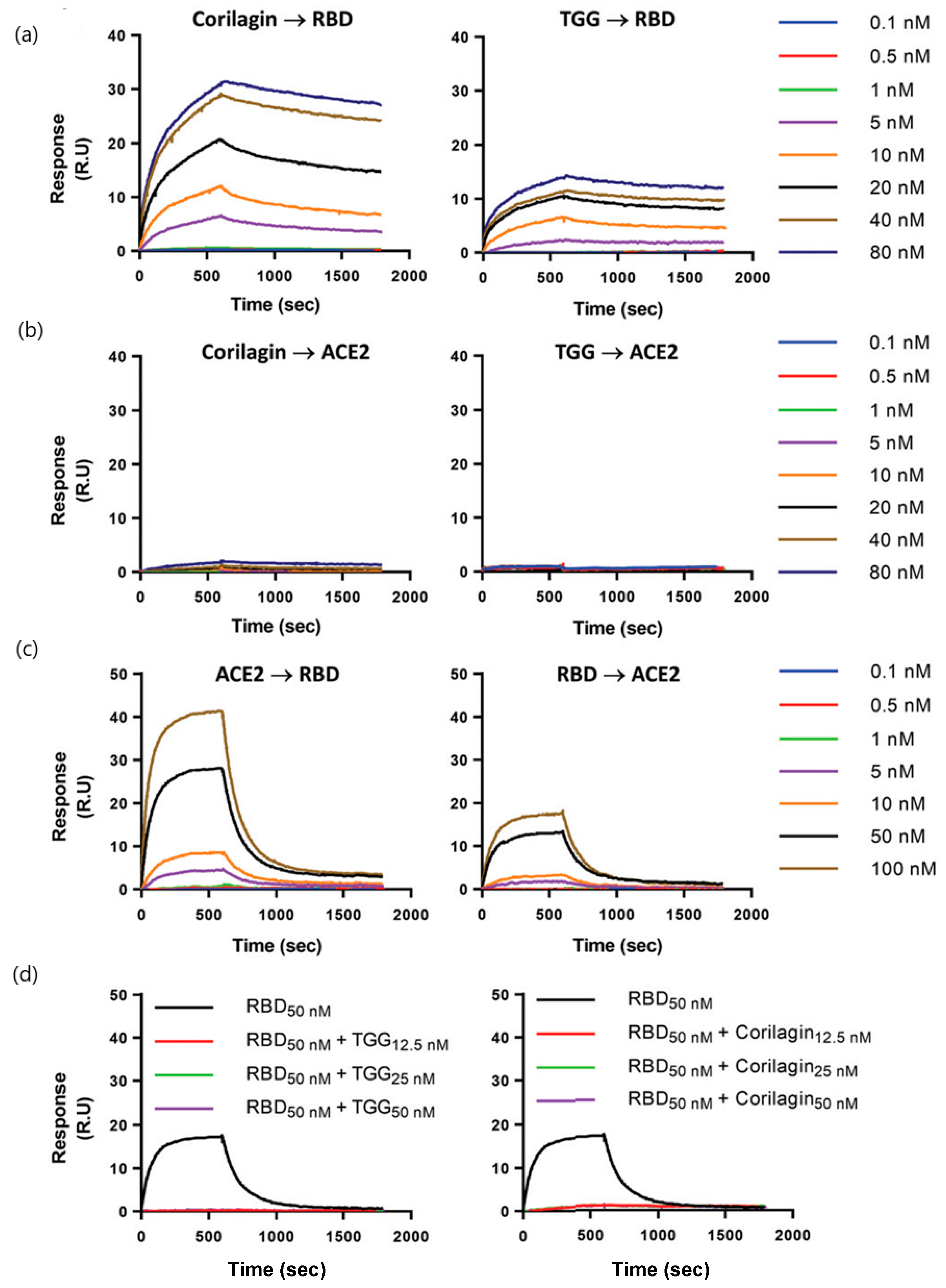
| Polyphenols: corilagin and TGG (636.46 and 423.40 g/mol, respectively; antioxidant, anti-inflammatory, and antidiabetic treatments) | Binding affinity constants (to S-RBD) Binding affinity of TGG and corilagin with RBD mutations of three main SARS-CoV-2 variants Competition assay (blocking RBD–ACE2 interactions) | Biacore T200 instrument (GE Healthcare) | KD = 1.8 nM (corilagin) KD = 1.3 nM (TGG) (kon and koff values N/A) | [44] |
| Erythrodiol (442.7 g/mol; Momordica charantia component: treatment of nephropathy, neuropathy, gastroparesis, cataracts and atherosclerosis) | Binding affinity constants (to SARS-CoV-2 S1 subunit) | Open SPR instrument (Nicoya Lifescience, ON, Canada) | KD = 1.15 μM (kon and koff values N/A) | [45] |
| Histamine H1 receptor antagonists: doxepin, chlorpheniramine, and doxylamine (279.376, 274.788 and 270.369 g/mol respectively; allergic rhinitis, allergic conjunctivitis and allergic dermatitis) | Analysis of bimolecular interactions with ACE2 receptor | Open SPRTM (Nicoya, Waterloo, Canada) | KD = 9.54 mM (doxepin); KD = 0.30 mM (chlorpheniramine); KD = 47.3 mM (doxylamine) (kon and koff values N/A) | [46] |
| Azelastine (381,898 g/mol; allergic conjunctivitis) | Kinetic analysis (binding to ACE2 receptor) | Open SPRTM (Nicoya, Waterloo, Canada) | KD = 0.258 μM (kon and koff values N/A) | [47] |
| Desloratadine and loratadine (310.82 and 382.88 g/mol; treatment of allergic disease) | Kinetic analysis (binding to ACE2 receptor) | Open SPRTM (Nicoya, Waterloo, Canada) | KD = 9.13 μM (desloratadine); KD = 0.1.02 μM (loratadine) (kon and koff values N/A) | [48] |
| Chloroquine (CQ) and hydroxychloroquine (HCQ) (319.872 and 335.872 g/mol; antimalarial drugs) | Kinetic analysis (binding to ACE2 receptor) | Open SPRTM (Nicoya, Waterloo, Canada) | KD = (7.31 ± 0.62) × 10−7 M (CQ) KD = (4.82 ± 0.87) × 10−7 M (HCQ) (kon and koff values N/A) | [49] |
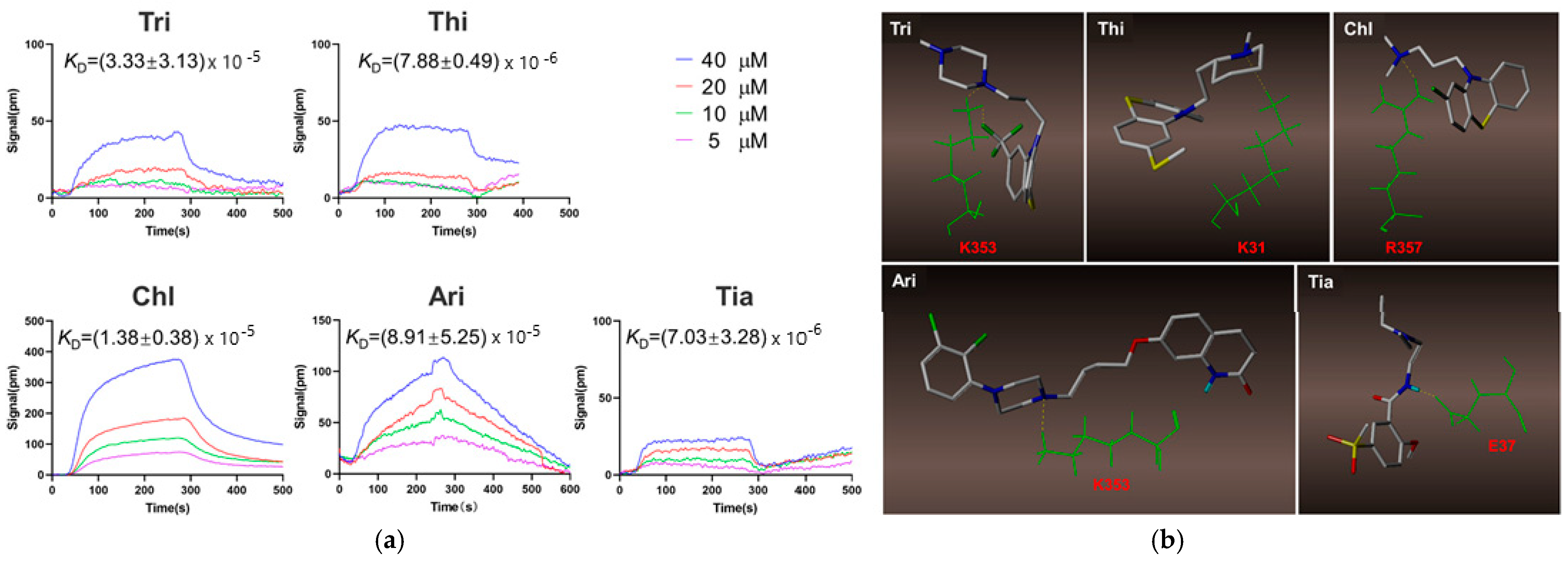
| Trifluoperazine (Tri); thioridazine (Thi); chlorpromazine (Chl), aripiprazole (Ari), tiapride (Tia) (407.497, 370.6, 318.86, 448.385 and 328,427 g/mol, respectively; antipsychotic drugs) | Kinetic analysis (binding to ACE2 receptor) | Open SPRTM (Nicoya, Waterloo, Canada) | KD = (7.03 ± 3.28) × 10−6 M (Tri); KD = (8.91 ± 5.25) × 10−5 M (Thi); KD = (1.38 ± 0.38) × 10−5 M (Chl); KD = (7.88 ± 0.49) × 10−6 M (Ari), and (3.33 ± 3.13) × 10−5 M (Tia) (kon and koff values N/A) | [50] |
| Target Analyte (MW; Common Uses) | Analytical Approach | SPR Instrument | Binding Affinities (KD = Equilibrium Dissociation Constant) | Reference |
|---|---|---|---|---|
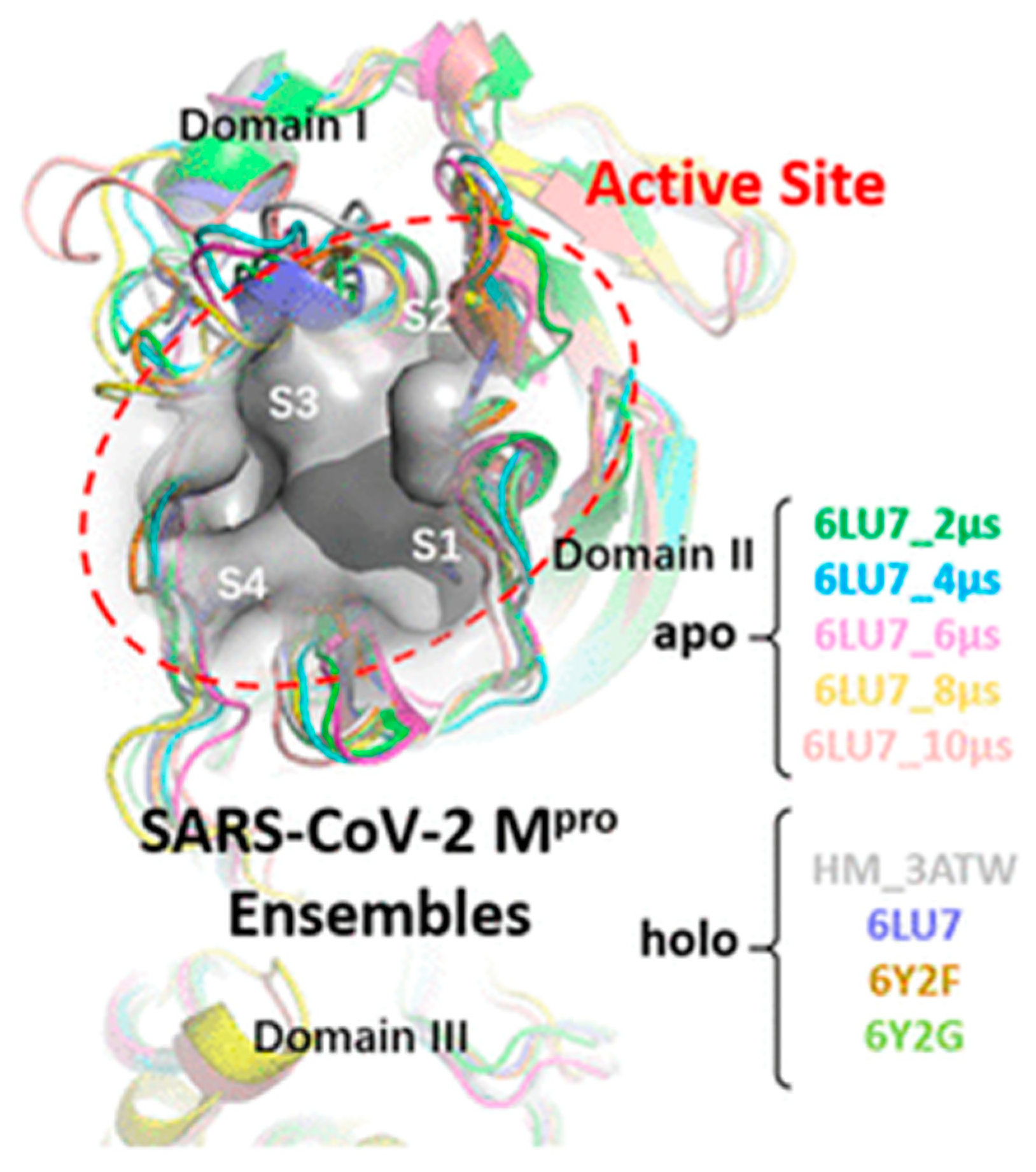
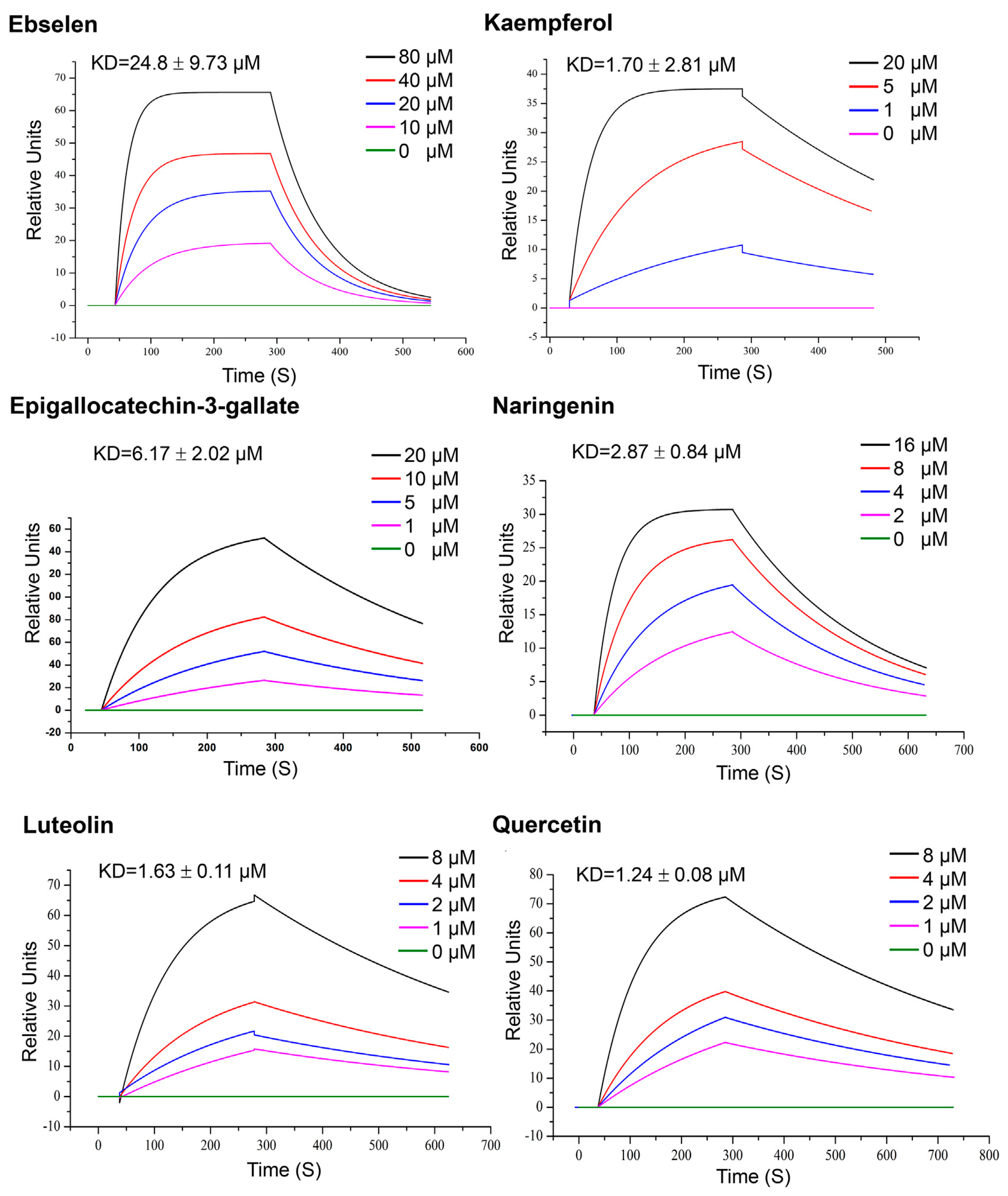
| Quercetin, Luteolin, kaempferol, naringenin and epigallocatechin-3-gallate (302.236, 286.24, 286.23, 272.257 and 458,372 g/mol, respectively; antioxidant, anti-inflammatory) | Kinetic analysis (binding to 3CLpro) | Open SPR instrument (Nicoya Life Science, Inc., Kitchener, Canada) | KD = 1.24 μM; KD = 1.63 μM (luteolin); KD = 1.70 μM (kaempferol); KD = 2.87 μM (naringenin); KD = 6.17 μM (epigallocatechin-3-gallate). (kon and koff values N/A) | [63] |
| Punicalagin: PA; ellagic acid: EA; tannic acid: TA, pentagalloyl glucose: PGG; ginnalin A, and gallic acid: GA; urolithins: UB and pyrogallol: PYG (1.084.71, 302.297, 1701.19, 940.67, 468.4, 170.12, 212.20 and 126.11 g/mol respectively; antioxidant, anti-inflammatory) | Kinetic analysis and binding affinities (binding to 3CLpro) | Biacore T200 instrument (GE Healthcare; Marlborough, MA, USA) | KD = 6.8 × 10−6; kon (1/Ms) = 697.3; koff (1/s) = 0.0047 (PA) KD = 2.7 × 10−6 M kon (1/Ms) = 4755.2; koff (1/s) = 0.0130 (EA); KD = 1.13 × 10−6 M kon (1/Ms) = 6309.0; koff (1/s) = 0.0071 (TA); KD = 4.33 × 10−6 M kon (1/Ms) = 3991.0; koff (1/s) = 0.0173 (PGG); KD = 1.18 × 10−6 M kon (1/Ms) = 2657; koff (1/s) = 0.0031 (GA); KD = 5.27 × 10−5 M kon (1/Ms) = 1874.0; koff (1/s) = 0.0988 (UB); KD = 3.59 × 10−6 M kon (1/Ms) = 661.8; koff (1/s) = 0.0024 (PYG) | [64] |
| Suramin and quinacrine (1.297.29 and 399.957 g/mol, respectively; treatment of protozoal infection) | Binding affinity to 3CLpro | Biacore T200 instrument (GE Healthcare, Uppsala, Sweden) | KD = 59.7 μM (suramin) and KD = 227.9 μM (quinacrine) (kon and koff values N/A) | [65] |
| Teicoplanin (1709.4 g/mol; glycopeptide antibiotic) | Binding affinity to 3CLpro | Biacore 3000 (GE Healthcare.) | KD = 1.6 mM kon (1/Ms) = 7.8 × 103; koff (1/s) = 0.012 | [66] |
| Cobicistat and cangrelor and denufosol (776.023, 776.35 and 773.323 g/mol, respectively; pulmonary diseases) | Binding affinity to 3CLpro | BIAcore-3000 (Biacore Inc., Uppsala, Sweden) | KD = 2.1 × 10−6 (cobicistat); KD = 6.4 × 10−4 M (cangrelor); KD = 1.4 × 10−3 M (denufosol) (kon and koff values N/A) | [67] |
| Nonpeptide inhibitors (compounds Z1244904919 and Z1759961356, MW and common uses N/A) | Binding affinity to 3CLpro | Biacore 8K device (Cytiva, Previously GE Healthcare Life Sciences) | KD = 465 μM (Z1244904919); KD = 133 μM (Z1244904919) (kon and koff values N/A) | [62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mauriz, E.; Lechuga, L.M. Current Trends in SPR Biosensing of SARS-CoV-2 Entry Inhibitors. Chemosensors 2021, 9, 330. https://doi.org/10.3390/chemosensors9120330
Mauriz E, Lechuga LM. Current Trends in SPR Biosensing of SARS-CoV-2 Entry Inhibitors. Chemosensors. 2021; 9(12):330. https://doi.org/10.3390/chemosensors9120330
Chicago/Turabian StyleMauriz, Elba, and Laura M. Lechuga. 2021. "Current Trends in SPR Biosensing of SARS-CoV-2 Entry Inhibitors" Chemosensors 9, no. 12: 330. https://doi.org/10.3390/chemosensors9120330
APA StyleMauriz, E., & Lechuga, L. M. (2021). Current Trends in SPR Biosensing of SARS-CoV-2 Entry Inhibitors. Chemosensors, 9(12), 330. https://doi.org/10.3390/chemosensors9120330