
Emerging Role of EGFR Mutations in Creating an Immune Suppressive Tumour Microenvironment


{kind=link}
{kind=link}
Abstract
:1. Introduction
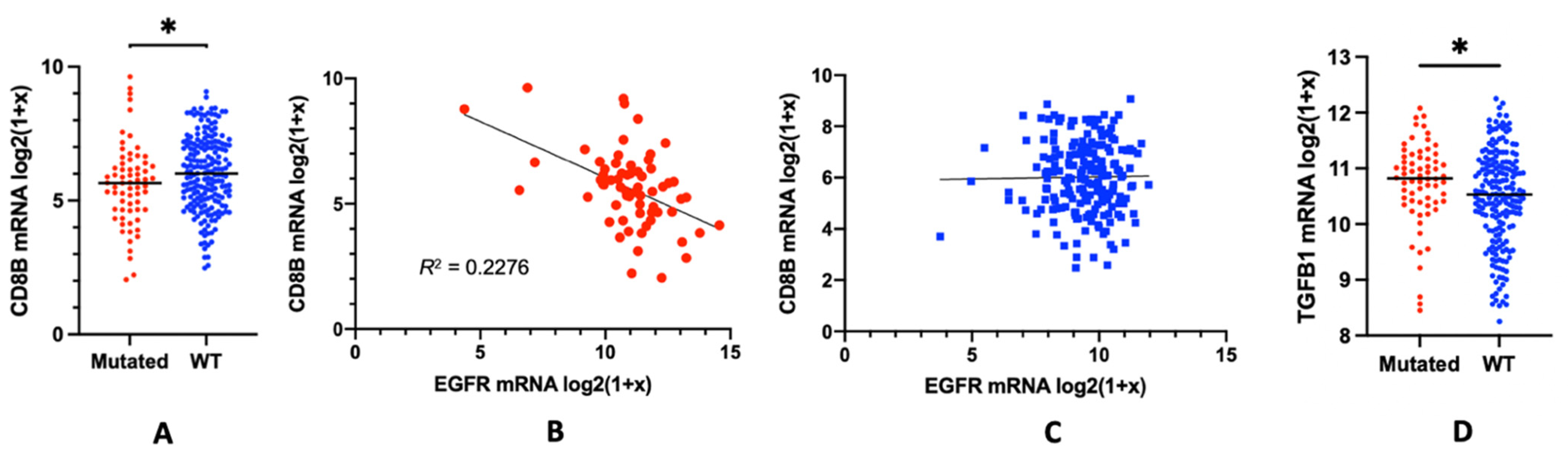
2. The Role of EGFR in Immune Suppression: EGFR Mutations and the Lymphocyte Depletion Phenotype
3. EGFR Mutations and the Lymphocyte Depletion Phenotype: Potential Mechanisms
4. Alternative Model Proposed: Local TGFβ Activation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Curti, B.D.; Faries, M.B. Recent Advances in the Treatment of Melanoma. N. Engl. J. Med. 2021, 384, 2229–2240. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Allison, J.P. Dissecting the mechanisms of immune checkpoint therapy. Nat. Rev. Immunol. 2020, 20, 75–76. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Barrueto, L.; Caminero, F.; Cash, L.; Makris, C.; Lamichhane, P.; Deshmukh, R.R. Resistance to Checkpoint Inhibition in Cancer Immunotherapy. Transl. Oncol. 2020, 13, 100738. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Nicholson, L.T.; Fong, L. Immune Checkpoint Inhibition in Prostate Cancer. Trends Cancer 2020, 6, 174–177. [Google Scholar] [CrossRef]
- Jin, R.; Zhao, J.; Xia, L.; Li, Q.; Li, W.; Peng, L.; Xia, Y. Application of immune checkpoint inhibitors in EGFR-mutant non-small-cell lung cancer: From bed to bench. Ther. Adv. Med. Oncol. 2020, 12, 1758835920930333. [Google Scholar] [CrossRef]
- Liu, F.; Yuan, X.; Jiang, J.; Chu, Q. Immunotherapy in advanced non-small-cell lung cancer with EGFR mutations. Immunotherapy 2020, 12, 1195–1207. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmon, M.A.; Schlessinger, J.; Ferguson, K.M. The EGFR Family: Not So Prototypical Receptor Tyrosine Kinases. Cold Spring Harb. Perspect. Biol. 2014, 6, a020768. [Google Scholar] [CrossRef]
- Koseska, A.; Bastiaens, P.I. Processing Temporal Growth Factor Patterns by an Epidermal Growth Factor Receptor Network Dynamically Established in Space. Annu. Rev. Cell Dev. Biol. 2020, 36, 359–383. [Google Scholar] [CrossRef]
- Weinstein, I.B. Addiction to Oncogenes--the Achilles Heal of Cancer. Science 2002, 297, 63–64. [Google Scholar] [CrossRef]
- Gazdar, A.F.; Shigematsu, H.; Herz, J.; Minna, J.D. Mutations and addiction to EGFR: The Achilles ‘heal’ of lung cancers? Trends Mol. Med. 2004, 10, 481–486. [Google Scholar] [CrossRef]
- Vansteenkiste, J.; Wauters, E. Tyrosine kinase inhibition of EGFR: A successful history of targeted therapy for NSCLC since 20 years. Ann. Oncol. 2018, 29 (Suppl. 1), i1–i2. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, M.G.; Di Noia, V.; D′Argento, E.; Vita, E.; Damiano, P.; Cannella, A.; Ribelli, M.; Pilotto, S.; Milella, M.; Tortora, G.; et al. Oncogene-Addicted Non-Small-Cell Lung Cancer: Treatment Opportunities and Future Perspectives. Cancers 2020, 12, 1196. [Google Scholar] [CrossRef]
- MacDonald, F.; Zaiss, D.M.W. The Immune System’s Contribution to the Clinical Efficacy of EGFR Antagonist Treatment. Front. Pharmacol. 2017, 8, 575. [Google Scholar] [CrossRef] [Green Version]
- Garrido, G.; Lorenzano, P.; Sánchez, B.; Beausoleil, I.; Alonso, D.F.; Pérez, R.; Fernández, L.E. T cells are crucial for the anti-metastatic effect of anti-epidermal growth factor receptor antibodies. Cancer Immunol. Immunother. 2007, 56, 1701–1710. [Google Scholar] [CrossRef]
- Zaiss, D.M.W.; van Loosdregt, J.; Gorlani, A.; Bekker, C.P.J.; Gröne, A.; Sibilia, M.; van Bergen en Henegouwen, P.M.P.; Roovers, R.C.; Coffer, P.J.; Sijts, A.J.A.M. Amphiregulin Enhances Regulatory T Cell-Suppressive Function via the Epidermal Growth Factor Receptor. Immunity 2013, 38, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Han, C.; Dong, C.; Shen, A.; Hsu, E.; Ren, Z.; Lu, C.; Liu, L.; Zhang, A.; Timmerman, C.; et al. Hypofractionated EGFR tyrosine kinase inhibitor limits tumor relapse through triggering innate and adaptive immunity. Sci. Immunol. 2019, 4, eaav6473. [Google Scholar] [CrossRef]
- Venugopalan, A.; Lee, M.J.; Niu, G.; Medina-Echeverz, J.; Tomita, Y.; Lizak, M.J.; Cultraro, C.M.; Simpson, R.M.; Chen, X.; Trepel, J.B.; et al. EGFR-targeted therapy results in dramatic early lung tumor regression accompanied by imaging response and immune infiltration in EGFR mutant transgenic mouse models. Oncotarget 2016, 7, 54137–54156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Li, X.; Jiang, T.; Zhao, S.; Zhao, C.; Zhang, L.; Liu, X.; Shi, J.; Qiao, M.; Luo, J.; et al. EGFR-targeted therapy alters the tumor microenvironment in EGFR-driven lung tumors: Implications for combination therapies. Int. J. Cancer 2019, 145, 1432–1444. [Google Scholar] [CrossRef] [PubMed]
- Burtness, B.; Goldwasser, M.A.; Flood, W.; Mattar, B.; Forastiere, A.A. Phase III Randomized Trial of Cisplatin Plus Placebo Compared with Cisplatin Plus Cetuximab in Metastatic/Recurrent Head and Neck Cancer: An Eastern Cooperative Oncology Group Study. J. Clin. Oncol. 2005, 23, 8646–8654. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Grandis, J.R.; Rinaldo, A.; Takes, R.P.; Ferlito, A. Emerging perspectives in epidermal growth factor receptor targeting in head and neck cancer. Head Neck 2008, 30, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.Y.; Shia, J.; Kemeny, N.E.; Shah, M.; Schwartz, G.K.; Tse, A.; Hamilton, A.; Pan, D.; Schrag, D.; Schwartz, L.; et al. Cetuximab Shows Activity in Colorectal Cancer Patients With Tumors That Do Not Express the Epidermal Growth Factor Receptor by Immunohistochemistry. J. Clin. Oncol. 2005, 23, 1803–1810. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Gainor, J.F.; Shaw, A.T.; Sequist, L.V.; Fu, X.; Azzoli, C.G.; Piotrowska, Z.; Huynh, T.G.; Zhao, L.; Fulton, L.; Schultz, K.R.; et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non–Small Cell Lung Cancer: A Retrospective Analysis. Clin. Cancer Res. 2016, 22, 4585–4593. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.K.; Man, J.; Lord, S.; Links, M.; Gebski, V.; Mok, T.; Yang, J.C. Checkpoint Inhibitors in Metastatic EGFR-Mutated Non-Small Cell Lung Cancer-A Meta-Analysis. J. Thorac. Oncol. 2017, 12, 403–407. [Google Scholar] [CrossRef] [Green Version]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 Pathway Contributes to Immune Escape in EGFR-Driven Lung Tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Lastwika, K.J.; Wilson, W., 3rd; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Fang, W.; Zhan, J.; Hong, S.; Tang, Y.; Kang, S.; Zhang, Y.; He, X.; Zhou, T.; Qin, T.; et al. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. J. Thorac. Oncol. 2015, 10, 910–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azuma, K.; Ota, K.; Kawahara, A.; Hattori, S.; Iwama, E.; Harada, T.; Matsumoto, K.; Takayama, K.; Takamori, S.; Kage, M.; et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann. Oncol. 2014, 25, 1935–1940. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Nataraj, N.B.; Noronha, A.; Patkar, S.; Sekar, A.; Mukherjee, S.; Winograd-Katz, S.; Kramarski, L.; Verma, A.; Lindzen, M.; et al. PD-L1 recruits phospholipase C and enhances tumorigenicity of lung tumors harboring mutant forms of EGFR. Cell Rep. 2021, 35, 109181. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Yang, T.H.O.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Fang, W.; Zhang, Y.; Hong, S.; Kang, S.; Yan, Y.; Chen, N.; Zhan, J.; He, X.; Qin, T.; et al. The association between PD-L1 and EGFR status and the prognostic value of PD-L1 in advanced non-small cell lung cancer patients treated with EGFR-TKIs. Oncotarget 2015, 6, 14209–14219. [Google Scholar] [CrossRef] [Green Version]
- D′Incecco, A.; Andreozzi, M.; Ludovini, V.; Rossi, E.; Capodanno, A.; Landi, L.; Tibaldi, C.; Minuti, G.; Salvini, J.; Coppi, E.; et al. PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. Br. J. Cancer 2015, 112, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.-Y.; Lin, M.-W.; Chang, Y.-L.; Wu, C.-T.; Yang, P.-C. Programmed cell death-ligand 1 expression in surgically resected stage I pulmonary adenocarcinoma and its correlation with driver mutations and clinical outcomes. Eur. J. Cancer 2014, 50, 1361–1369. [Google Scholar] [CrossRef]
- Cooper, W.A.; Tran, T.; Vilain, R.E.; Madore, J.; Selinger, C.I.; Kohonen-Corish, M.; Yip, P.; Yu, B.; O’Toole, S.A.; McCaughan, B.C.; et al. PD-L1 expression is a favorable prognostic factor in early stage non-small cell carcinoma. Lung Cancer 2015, 89, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.; Jang, J.-Y.; Keam, B.; Kim, S.; Kim, M.-Y.; Go, H.; Kim, T.M.; Kim, D.-W.; Kim, C.-W.; Jeon, Y.K.; et al. EML4-ALK enhances programmed cell death-ligand 1 expression in pulmonary adenocarcinoma via hypoxia-inducible factor (HIF)-1α and STAT3. Oncoimmunology 2016, 5, e1108514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Z.Y.; Zhang, J.T.; Liu, S.Y.; Su, J.; Zhang, C.; Xie, Z.; Zhou, Q.; Tu, H.Y.; Xu, C.R.; Yan, L.X.; et al. EGFR mutation correlates with uninflamed phenotype and weak immunogenicity, causing impaired response to PD-1 blockade in non-small cell lung cancer. Oncoimmunology 2017, 6, e1356145. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, E.; Togashi, Y.; Takeuchi, Y.; Shinya, S.; Tada, Y.; Kataoka, K.; Tane, K.; Sato, E.; Ishii, G.; Goto, K.; et al. Blockade of EGFR improves responsiveness to PD-1 blockade in EGFR -mutated non–small cell lung cancer. Sci. Immunol. 2020, 5, eaav3937. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Chen, P.L.; Roh, W.; Reuben, A.; Cooper, Z.A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Bassett, R.L.; Gopalakrishnan, V.; Wani, K.; et al. Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov. 2016, 6, 827–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambolez, F.; Kronenberg, M.; Cheroutre, H. Thymic differentiation of TCR alpha beta(+) CD8 alpha alpha(+) IELs. Immunol. Rev. 2007, 215, 178–188. [Google Scholar] [CrossRef]
- Baume, D.M.; Caligiuri, M.A.; Manley, T.J.; Daley, J.F.; Ritz, J. Differential expression of CD8 alpha and CD8 beta associated with MHC-restricted and non-MHC-restricted cytolytic effector cells. Cell Immunol. 1990, 131, 352–365. [Google Scholar] [CrossRef]
- Mascia, F.; Schloemann, D.T.; Cataisson, C.; McKinnon, K.M.; Krymskaya, L.; Wolcott, K.M.; Yuspa, S.H. Cell autonomous or systemic EGFR blockade alters the immune-environment in squamous cell carcinomas. Int. J. Cancer 2016, 139, 2593–2597. [Google Scholar] [CrossRef] [Green Version]
- Rapp, M.; Wintergerst, M.W.; Kunz, W.G.; Vetter, V.K.; Knott, M.M.; Lisowski, D.; Haubner, S.; Moder, S.; Thaler, R.; Eiber, S.; et al. CCL22 controls immunity by promoting regulatory T cell communication with dendritic cells in lymph nodes. J. Exp. Med. 2019, 216, 1170–1181. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lang, M.; Zhao, T.; Feng, X.; Zheng, C.; Huang, C.; Hao, J.; Dong, J.; Luo, L.; Li, X.; et al. Cancer-FOXP3 directly activated CCL5 to recruit FOXP3+Treg cells in pancreatic ductal adenocarcinoma. Oncogene 2016, 36, 3048–3058. [Google Scholar] [CrossRef]
- Wang, J.; Ioan-Facsinay, A.; van der Voort, E.I.H.; Huizinga, T.W.J.; Toes, R.E.M. Transient expression of FOXP3 in human activated nonregulatory CD4+ T cells. Eur. J. Immunol. 2007, 37, 129–138. [Google Scholar] [CrossRef]
- Dimitrakopoulos, F.-I.; Papadaki, H.; Antonacopoulou, A.G.; Kottorou, A.; Gotsis, A.D.; Scopa, C.; Kalofonos, H.; Mouzaki, A. Association of FOXP3 expression with non-small cell lung cancer. Anticancer. Res. 2011, 31, 1677–1683. [Google Scholar]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Massague, J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Gunderson, A.J.; Yamazaki, T.; McCarty, K.; Fox, N.; Phillips, M.; Alice, A.; Blair, T.; Whiteford, M.; O′Brien, D.; Ahmad, R.; et al. TGFbeta suppresses CD8(+) T cell expression of CXCR3 and tumor trafficking. Nat. Commun. 2020, 11, 1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-beta family signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef] [Green Version]
- Gleizes, P.E.; Munger, J.S.; Nunes, I.; Harpel, J.G.; Mazzieri, R.; Noguera, I.; Rifkin, D.B. TGF-beta latency: Biological significance and mechanisms of activation. Stem Cells 1997, 15, 190–197. [Google Scholar] [CrossRef]
- Khan, Z.; Marshall, J.F. The role of integrins in TGFbeta activation in the tumour stroma. Cell Tissue Res. 2016, 365, 657–673. [Google Scholar] [CrossRef] [Green Version]
- Minutti, C.M.; Modak, R.V.; Macdonald, F.; Li, F.; Smyth, D.J.; Dorward, D.A.; Blair, N.; Husovsky, C.; Muir, A.; Giampazolias, E.; et al. A Macrophage-Pericyte Axis Directs Tissue Restoration via Amphiregulin-Induced Transforming Growth Factor Beta Activation. Immunity 2019, 50, 645–654.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freed, D.; Bessman, N.J.; Kiyatkin, A.; Salazar-Cavazos, E.; Byrne, P.O.; Moore, J.O.; Valley, C.C.; Ferguson, K.M.; Leahy, D.J.; Lidke, D.S.; et al. EGFR Ligands Differentially Stabilize Receptor Dimers to Specify Signaling Kinetics. Cell 2017, 171, 683–695.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besson, A.; Wilson, T.L.; Yong, V.W. The anchoring protein RACK1 links protein kinase Cepsilon to integrin beta chains. Requirements for adhesion and motility. J. Biol. Chem. 2002, 277, 22073–22084. [Google Scholar] [CrossRef] [Green Version]
- Shigematsu, H.; Lin, L.; Takahashi, T.; Nomura, M.; Suzuki, M.; Wistuba, I.I.; Fong, K.M.; Lee, H.; Toyooka, S.; Shimizu, N.; et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J. Natl. Cancer Inst. 2005, 97, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.-H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of Lung Cancer-Derived EGFR Mutants and Inhibitor Complexes: Mechanism of Activation and Insights into Differential Inhibitor Sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Shan, Y.; Eastwood, M.P.; Zhang, X.; Kim, E.T.; Arkhipov, A.; Dror, R.O.; Jumper, J.; Kuriyan, J.; Shaw, D.E. Oncogenic Mutations Counteract Intrinsic Disorder in the EGFR Kinase and Promote Receptor Dimerization. Cell 2012, 149, 860–870. [Google Scholar] [CrossRef] [Green Version]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-Sensitizing EGFR Mutations in Lung Cancer Activate Anti-Apoptotic Pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef]
- Huang, P.H.; Mukasa, A.; Bonavia, R.; Flynn, R.A.; Brewer, Z.E.; Cavenee, W.K.; Furnari, F.B.; White, F.M. Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc. Natl. Acad. Sci. USA 2007, 104, 12867–12872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albeck, J.G.; Mills, G.B.; Brugge, J.S. Frequency-Modulated Pulses of ERK Activity Transmit Quantitative Proliferation Signals. Mol. Cell 2013, 49, 249–261. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.; Fenoglio, S.; Gao, D.C.; Camiolo, M.; Stiles, B.; Lindsted, T.; Schlederer, M.; Johns, C.; Altorki, N.; Mittal, V.; et al. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 15535–15540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, K.B.; Spranger, S. Modulation of the immune microenvironment by tumor-intrinsic oncogenic signaling. J. Cell Biol. 2019, 219, e201908224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blakely, C.M.; Watkins, T.B.K.; Wu, W.; Gini, B.; Chabon, J.J.; McCoach, C.E.; McGranahan, N.; Wilson, G.A.; Birkbak, N.; Olivas, V.R.; et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat. Genet. 2017, 49, 1693–1704. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapoor, S.S.; Zaiss, D.M.W. Emerging Role of EGFR Mutations in Creating an Immune Suppressive Tumour Microenvironment. Biomedicines 2022, 10, 52. https://doi.org/10.3390/biomedicines10010052
Kapoor SS, Zaiss DMW. Emerging Role of EGFR Mutations in Creating an Immune Suppressive Tumour Microenvironment. Biomedicines. 2022; 10(1):52. https://doi.org/10.3390/biomedicines10010052
Chicago/Turabian StyleKapoor, Simran S., and Dietmar M. W. Zaiss. 2022. "Emerging Role of EGFR Mutations in Creating an Immune Suppressive Tumour Microenvironment" Biomedicines 10, no. 1: 52. https://doi.org/10.3390/biomedicines10010052
APA StyleKapoor, S. S., & Zaiss, D. M. W. (2022). Emerging Role of EGFR Mutations in Creating an Immune Suppressive Tumour Microenvironment. Biomedicines, 10(1), 52. https://doi.org/10.3390/biomedicines10010052