
Impact of COVID-19 and Antibiotic Treatments on Gut Microbiome: A Role for Enterococcus spp.

 , , ,
, , ,
Abstract
:1. Introduction
2. Methods
2.1. Study Population
2.2. Laboratory Analyses
2.3. Data Analysis
3. Results
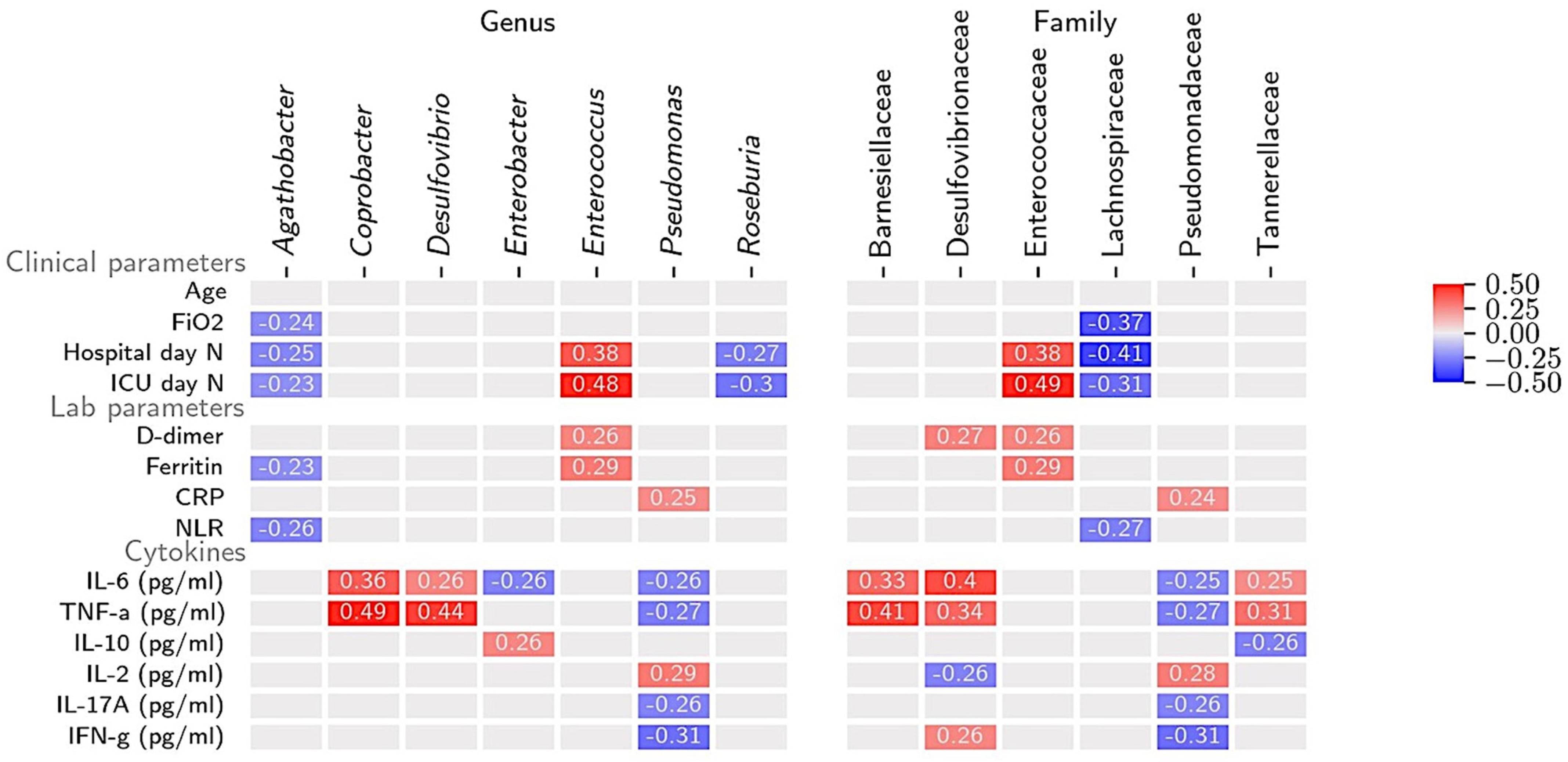
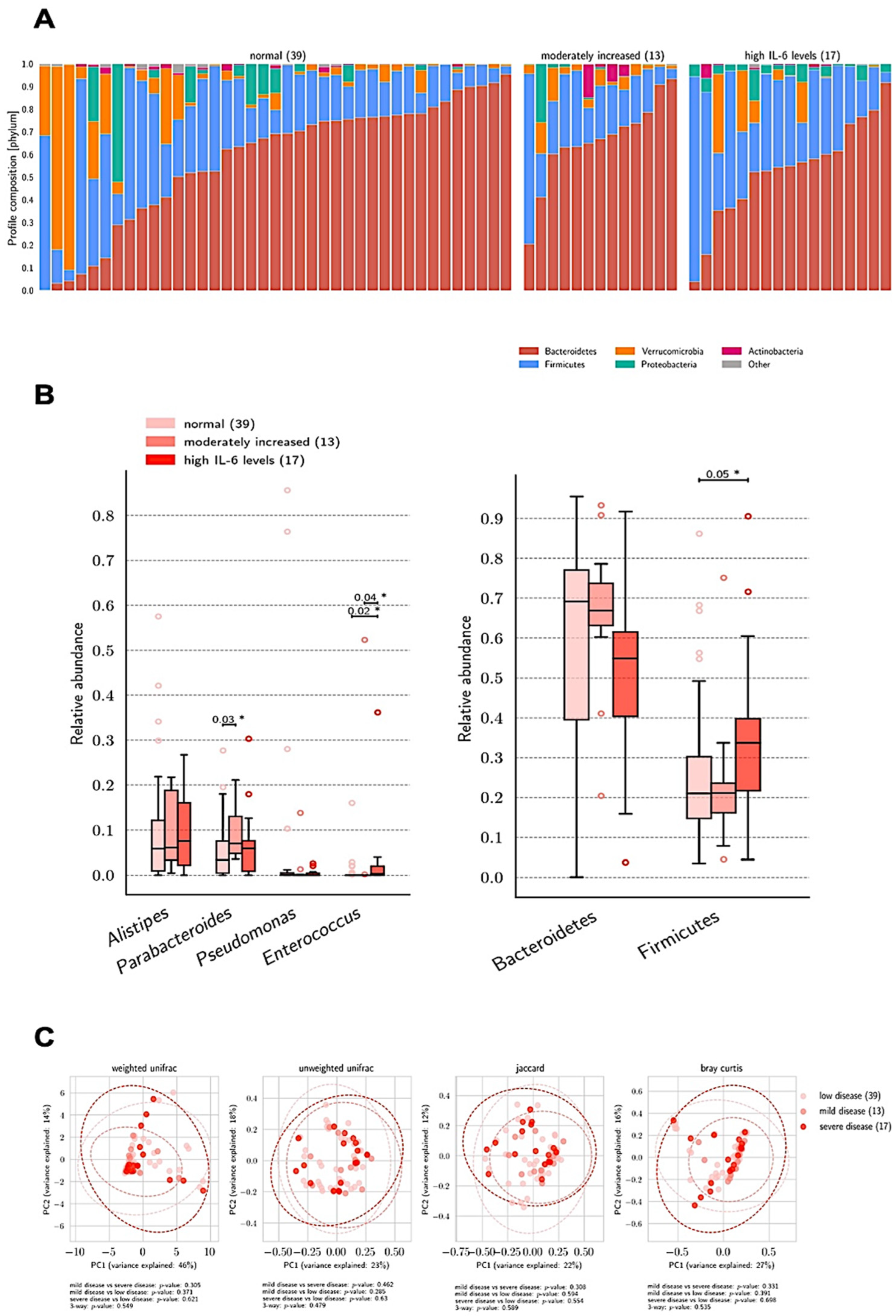
3.1. Gut Microbiome and Clinical Markers
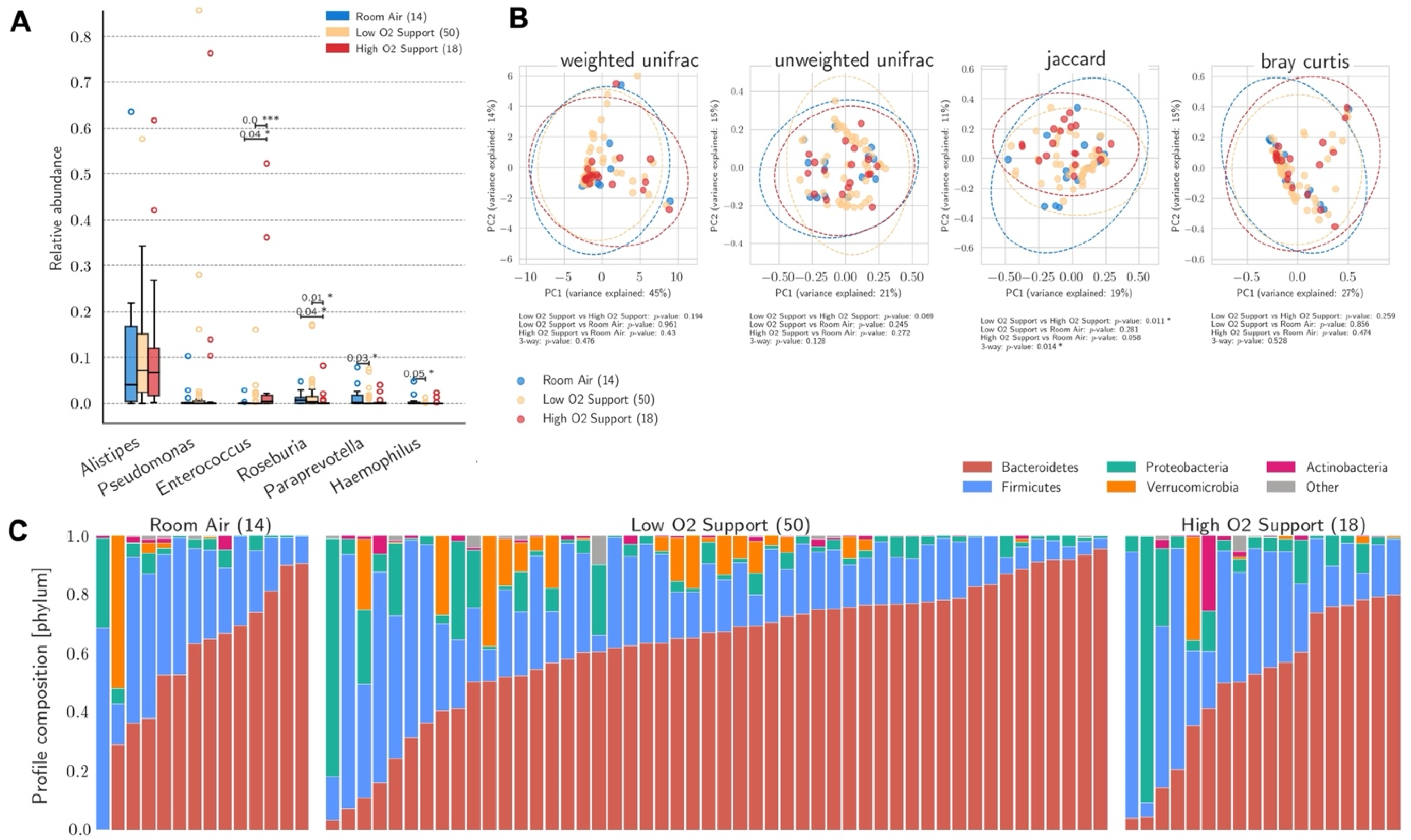
3.2. Gut Microbiome and O2 Therapy
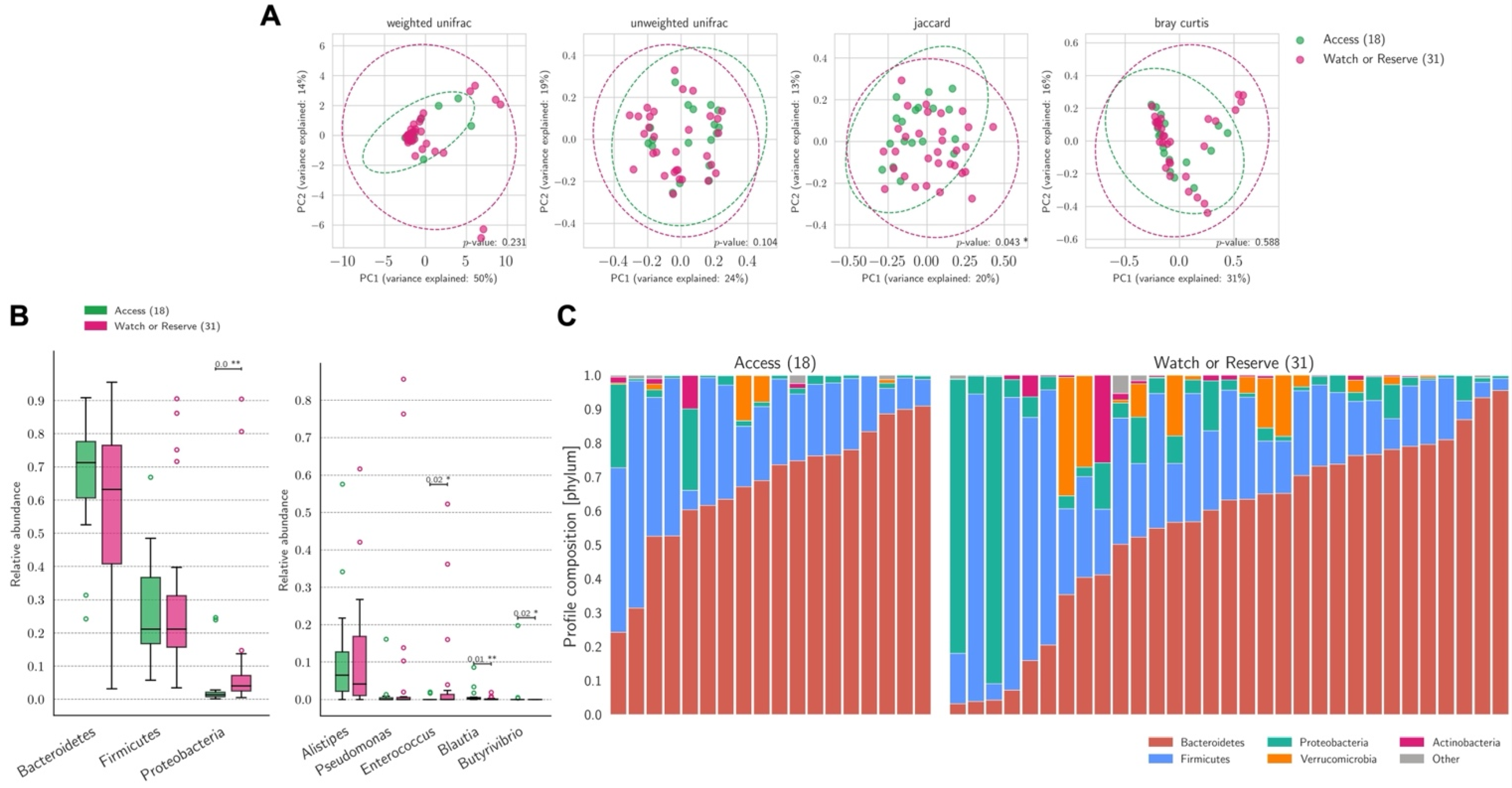
3.3. Gut Microbiome and Therapy
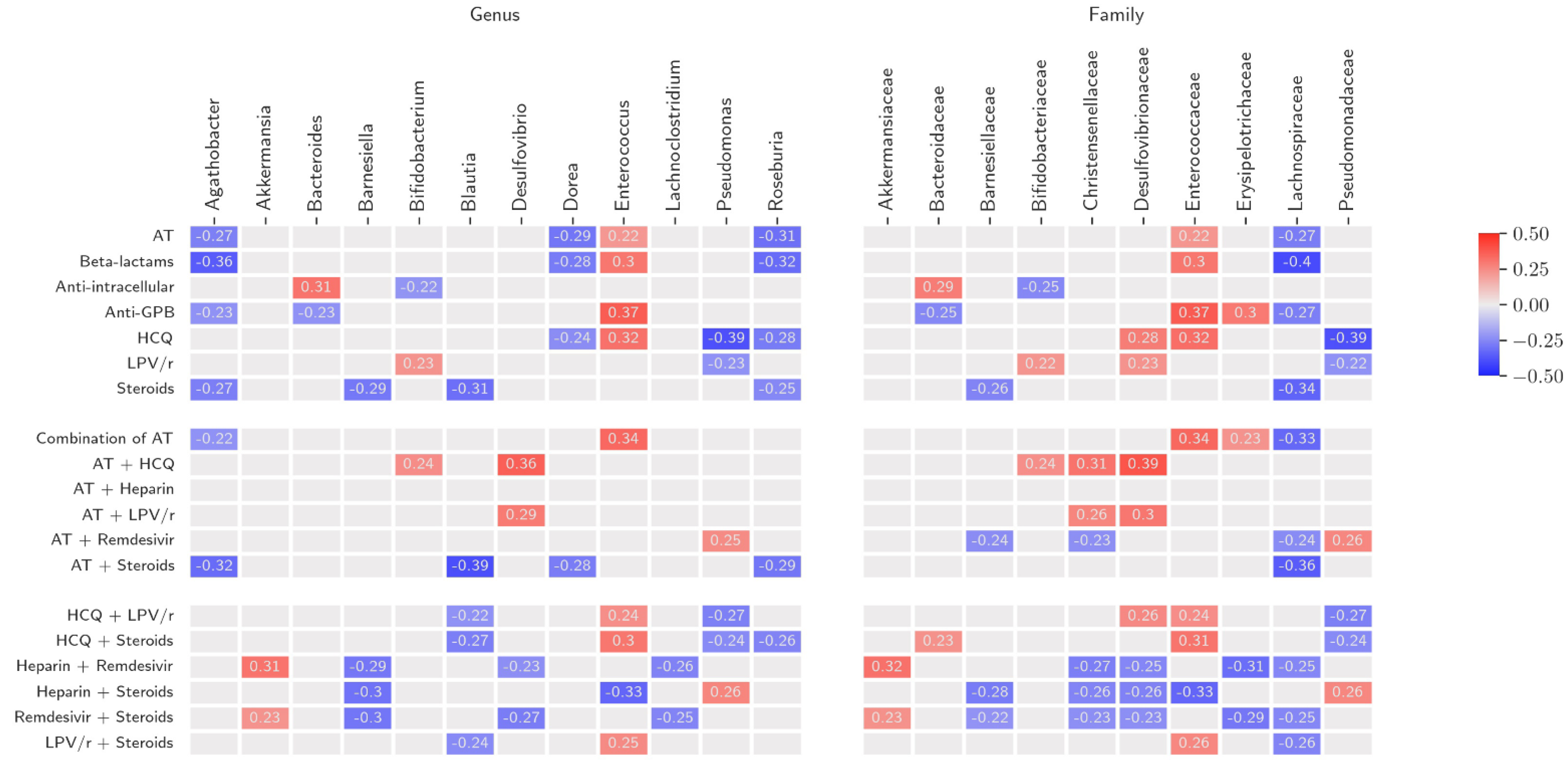
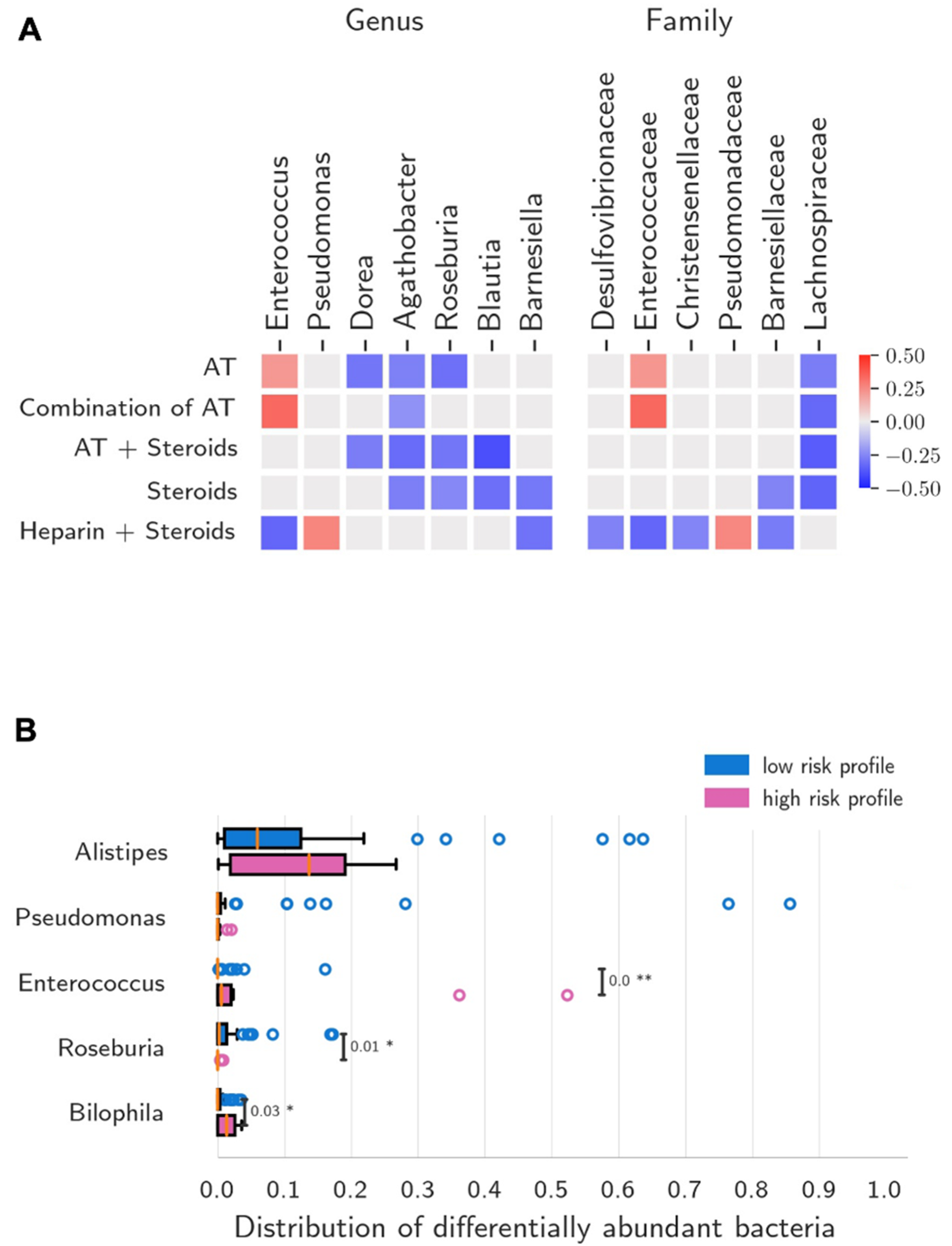
3.4. Antibiotic Therapy (AT)
3.5. Non-Antibiotic COVID-19 Treatment (NACT)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gu, S.; Chen, Y.; Wu, Z.; Chen, Y.; Gao, H.; Lv, L.; Guo, F.; Zhang, X.; Luo, R.; Huang, C.; et al. Alterations of the Gut Microbiota in Patients With Coronavirus Disease 2019 or H1N1 Influenza. Clin. Infect. Dis. 2020, 71, 2669–2678. [Google Scholar] [CrossRef] [PubMed]
- Yeoh, Y.K.; Zuo, T.; Lui, G.C.Y.; Zhang, F.; Liu, Q.; Li, A.Y.L.; Chung, A.C.; Cheung, C.P.; Tso, E.Y.; Fung, K.S.; et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut 2021, 70, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Gaibani, P.; D’Amico, F.; Bartoletti, M.; Lombardo, D.; Rampelli, S.; Fornaro, G.; Coladonato, S.; Siniscalchi, A.; Re, M.C.; Viale, P.; et al. The Gut Microbiota of Critically Ill Patients with COVID-19. Front. Cell. Infect. Microbiol. 2021, 11, 670424. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Liu, Q.; Zhang, F.; Lui, G.C.Y.; Tso, E.Y.K.; Yeoh, Y.K.; Chen, Z.; Boon, S.S.; Chan, F.K.; Chan, P.K.; et al. Depicting SARS-CoV-2 faecal viral activity in association with gut microbiota composition in patients with COVID-19. Gut 2021, 70, 276–284. [Google Scholar] [CrossRef]
- Reinold, J.; Farahpour, F.; Fehring, C.; Dolff, S.; Konik, M.; Korth, J.; Van Baal, L.; Hoffmann, D.; Buer, J.; Witzke, O.; et al. A Pro-Inflammatory Gut Microbiome Characterizes SARS-CoV-2 Infected Patients and a Reduction in the Connectivity of an Anti-Inflammatory Bacterial Network Associates With Severe COVID-19. Front. Cell. Infect. Microbiol. 2021, 11, 747816. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, J.; Zhang, D.; Ma, W.L.; Wang, X. Linking the gut microbiota to persistent symptoms in survivors of COVID-19 after discharge. J. Microbiol. 2021, 59, 941–948. [Google Scholar] [CrossRef]
- Wu, Y.; Cheng, X.; Jiang, G.; Tang, H.; Ming, S.; Tang, L.; Lu, J.; Guo, C.; Shan, H.; Huang, X. Altered oral and gut microbiota and its association with SARS-CoV-2 viral load in COVID-19 patients during hospitalization. NPJ Biofilms Microbiomes 2021, 7, 61. [Google Scholar] [CrossRef]
- Tao, W.; Zhang, G.; Wang, X.; Guo, M.; Zeng, W.; Xu, Z.; Cao, D.; Pan, A.; Wang, Y.; Zhang, K.; et al. Analysis of the intestinal microbiota in COVID-19 patients and its correlation with the inflammatory factor IL-18. Med. Microecol. 2020, 5, 100023. [Google Scholar] [CrossRef]
- Zuo, T.; Zhang, F.; Lui, G.C.Y.; Yeoh, Y.K.; Li, A.Y.L.; Zhan, H.; Wan, Y.; Chung, A.C.; Cheung, C.P.; Chen, N.; et al. Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology 2020, 159, 944–955. [Google Scholar] [CrossRef]
- He, F.; Zhang, T.; Xue, K.; Fang, Z.; Jiang, G.; Huang, S.; Li, K.; Gu, Z.; Shi, H.; Zhang, Z.; et al. Fecal multi-omics analysis reveals diverse molecular alterations of gut ecosystem in COVID-19 patients. Anal. Chim. Acta 2021, 1180, 338881. [Google Scholar] [CrossRef]
- Langford, B.J.; So, M.; Raybardhan, S.; Leung, V.; Soucy, J.R.; Westwood, D.; Daneman, N.; MacFadden, D.R. Antibiotic prescribing in patients with COVID-19: Rapid review and meta-analysis. Clin. Microbiol. Infect. 2021, 27, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Rawson, T.M.; Ming, D.; Ahmad, R.; Moore, L.S.P.; Holmes, A.H. Antimicrobial use, drug-resistant infections and COVID-19. Nat. Rev. Microbiol. 2020, 18, 409–410. [Google Scholar] [CrossRef] [PubMed]
- Vegivinti, C.T.R.; Evanson, K.W.; Lyons, H.; Akosman, I.; Barrett, A.; Hardy, N.; Kane, B.; Keesari, P.R.; Pulakurthi, Y.S.; Sheffels, E.; et al. Efficacy of antiviral therapies for COVID-19: A systematic review of randomized controlled trials. BMC Infect. Dis. 2022, 22, 107. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (CDC). Risk for COVID-19 Infection, Hospitalization, and Death by Age Group. 2022. Available online: www.cdc.gov/coronavirus/2019-ncov/covid-data/investigations-discovery/hospitalization-death-by-age.htlm (accessed on 12 October 2022).
- 2021 AWaRe Classification. WHO Access, Watch, Reserve, Classification of Antibiotics for Evaluation and Monitoring of Use. Available online: https://www.who.int/publications/i/item/2021-aware-classification (accessed on 16 May 2022).
- Liu, J.; Liu, Y.; Xiang, P.; Pu, L.; Xiong, H.; Li, C.; Zhang, M.; Tan, J.; Xu, Y.; Song, R.; et al. Neutrophil-to-lymphocyte ratio predicts critical illness patients with 2019 coronavirus disease in the early stage. J. Transl. Med. 2020, 18, 206. [Google Scholar] [CrossRef]
- Santa Cruz, A.; Mendes-Frias, A.; Oliveira, A.I.; Dias, L.; Matos, A.R.; Carvalho, A.; Capela, C.; Pedrosa, J.; Castro, A.G.; Silvestre, R. Interleukin-6 Is a Biomarker for the Development of Fatal Severe Acute Respiratory Syndrome Coronavirus 2 Pneumonia. Front. Immunol. 2021, 12, 613422. [Google Scholar] [CrossRef]
- Takahashi, S.; Tomita, J.; Nishioka, K.; Hisada, T.; Nishijima, M. Development of a prokaryotic universal primer for simultaneous analysis of Bacteria and Archaea using next-generation sequencing. PLoS ONE 2014, 9, e105592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. MEGAN Community Edition —Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef]
- Hunter, J.D. MATPLOTLIB: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Jones, E.; Oliphant, T.; Peterson, P. SciPy: Open Source Scientific Tools for Python. 2001. Available online: http://www.scipy.org/ (accessed on 10 March 2022).
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Scikit-Bio Development Team. Scikit-Bio: A Bioinformatics Library for Data Scientists, Students, and Developers. Version 0.5.5. 2020. Available online: http://scikit-bio.org (accessed on 10 March 2022).
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, E.; Crass, R.L.; Jorgensen, S.C.J.; Raybardhan, S.; Langford, B.J.; Moore, W.J.; Rhodes, N.J. Pharmacokinetic/Pharmacodynamic Considerations of Alternate Dosing Strategies of Tocilizumab in COVID-19. Clin. Pharm. 2022, 61, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.Y.; Yin, C.H.; Yao, Y.M. Update Advances on C-Reactive Protein in COVID-19 and Other Viral Infections. Front. Immunol. 2021, 12, 720363. [Google Scholar] [CrossRef]
- Dhar, S.K.; Damodar, S.; Gujar, S.; Das, M. IL-6 and IL-10 as predictors of disease severity in COVID-19 patients: Results from meta-analysis and regression. Heliyon 2021, 7, e06155. [Google Scholar] [CrossRef]
- Hu, Z.J.; Xu, J.; Yin, J.M.; Li, L.; Hou, W.; Zhang, L.L.; Zhou, Z.; Yu, Y.Z.; Li, H.J.; Feng, Y.M.; et al. Lower Circulating Interferon-Gamma Is a Risk Factor for Lung Fibrosis in COVID-19 Patients. Front. Immunol. 2020, 11, 585647. [Google Scholar] [CrossRef]
- Francino, M.P. Antibiotics and the Human Gut Microbiome: Dysbioses and Accumulation of Resistances. Front. Microbiol. 2016, 6, 1543. [Google Scholar] [CrossRef] [Green Version]
- Gu, S.L.; Gong, Y.; Zhang, J.; Chen, Y.; Wu, Z.; Xu, Q.; Fang, Y.; Wang, J.; Tang, L.L. Effect of the Short-Term Use of Fluoroquinolone and β-Lactam Antibiotics on Mouse Gut Microbiota. Infect. Drug Resist. 2020, 13, 4547–4558. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef]
- Brandl, K.; Plitas, G.; Mihu, C.N.; Ubeda, C.; Jia, T.; Fleisher, M.; Schnabl, B.; DeMatteo, R.P.; Pamer, E.G. Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature 2008, 455, 804–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoletti, M.; Azap, O.; Barac, A.; Bussini, L.; Ergonul, O.; Krause, R.; Paño-Pardo, J.R.; Power, N.R.; Sibani, M.; Szabo, B.G.; et al. ESCMID COVID-19 living guidelines: Drug treatment and clinical management. Clin. Microbiol. Infect. 2022, 28, 222–238. [Google Scholar] [CrossRef] [PubMed]
- Saag, M.S.; Gandhi, R.T.; Hoy, J.F.; Landovitz, R.J.; Thompson, M.A.; Sax, P.E.; Smith, D.M.; Benson, C.A.; Buchbinder, S.P.; Del Rio, C.; et al. Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults: 2020 Recommendations of the International Antiviral Society-USA Panel. JAMA 2020, 324, 1651–1669. [Google Scholar] [CrossRef]
- Pinto-Cardoso, S.; Klatt, N.R.; Reyes-Terán, G. Impact of antiretroviral drugs on the microbiome: Unknown answers to important questions. Curr. Opin. HIV AIDS 2018, 13, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Z.Y.; Chang, Y.X.; Han, N.; Hou, F.Y.; Lee, B.Y.; Zhi, F.C.; Yang, R.F.; Bi, Y.J. Short-term high-dose gavage of hydroxychloroquine changes gut microbiota but not the intestinal integrity and immunological responses in mice. Life Sci. 2021, 264, 118450. [Google Scholar] [CrossRef] [PubMed]
- Balmant, B.D.; Torrinhas, R.S.; Rocha, I.M.; Fonseca, D.C.; Formiga, F.F.; Bonfá, E.S.; Borba, E.F.; Waitzberg, D.L. SARS-CoV-2 infection, gut dysbiosis, and heterogeneous clinical results of hydroxychloroquine on COVID-19 therapy-Is there a link? Nutrition 2021, 85, 111115. [Google Scholar] [CrossRef]
- Huang, E.Y.; Inoue, T.; Leone, V.A.; Dalal, S.; Touw, K.; Wang, Y.; Musch, M.W.; Theriault, B.; Higuchi, K.; Donovan, S.; et al. Using corticosteroids to reshape the gut microbiome: Implications for inflammatory bowel diseases. Inflamm. Bowel Dis. 2015, 21, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, J.E.; Albrich, W.C.; Dmitrijeva, M.; Kahlert, C.R. The Effects of Corticosteroids on the Respiratory Microbiome: A Systematic Review. Front. Med. 2016, 8, 1543. [Google Scholar] [CrossRef]
- Macchione, I.G.; Lopetuso, L.R.; Ianiro, G.; Napoli, M.; Gibiino, G.; Rizzatti, G.; Petito, V.; Gasbarrini, A.; Scaldaferri, F. Akkermansia muciniphila: Key player in metabolic and gastrointestinal disorders. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8075–8083. [Google Scholar]
- Toc, D.A.; Mihaila, R.M.; Botan, A.; Bobohalma, C.N.; Risteiu, G.A.; Simut-Cacuci, B.N.; Steorobelea, B.; Troanca, S.; Junie, L.M. Enterococcus and COVID-19: The Emergence of a Perfect Storm? Int. J. Transl. Med. 2022, 2, 220–229. [Google Scholar] [CrossRef]
- Segal, J.P.; Mak, J.W.Y.; Mullish, B.H.; Alexander, J.L.; Ng, S.C.; Marchesi, J.R. The gut microbiome: An under-recognised contributor to the COVID-19 pandemic? Therap. Adv. Gastroenterol. 2020, 13, 1756284820974914. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | COVID-19 Hospitalized Patients (n = 82) |
|---|---|
| Male gender (%) | 57 (70) |
| Median age, years (Q1–Q3) | 66 (57–77) |
| Median hospitalization (H), days (Q1–Q3) | 13 (7–22) |
| Comorbidities (%) | |
| - No comorbidities | 27 (33) |
| - Hypertension | 48 (59) |
| - Diabetes mellitus | 13 (16) |
| - Heart disease | 19 (23) |
| - Two or more comorbidities | 21 (26) |
| O2 Support (%) during H | |
| - None | 14 (17) |
| - Nasal cannulae or face mask | 50 (61) |
| - Non-invasive or mechanical ventilation | 18 (22) |
| ICU admission (%) during H | 18 (22) |
| Length of ICU stay, days (Q1–Q3) | 9 (5–12) |
| Laboratory parameters (mean ± SE) | |
| - Ferritin µg/L | 944 ± 71 |
| - D-dimer ng/ml | 1994 ± 239 |
| - CRP mg/L | 47 ± 6.1 |
| - NLR | 7.13 ± 0.71 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Righi, E.; Lambertenghi, L.; Gorska, A.; Sciammarella, C.; Ivaldi, F.; Mirandola, M.; Sartor, A.; Tacconelli, E. Impact of COVID-19 and Antibiotic Treatments on Gut Microbiome: A Role for Enterococcus spp. Biomedicines 2022, 10, 2786. https://doi.org/10.3390/biomedicines10112786
Righi E, Lambertenghi L, Gorska A, Sciammarella C, Ivaldi F, Mirandola M, Sartor A, Tacconelli E. Impact of COVID-19 and Antibiotic Treatments on Gut Microbiome: A Role for Enterococcus spp. Biomedicines. 2022; 10(11):2786. https://doi.org/10.3390/biomedicines10112786
Chicago/Turabian StyleRighi, Elda, Lorenza Lambertenghi, Anna Gorska, Concetta Sciammarella, Federico Ivaldi, Massimo Mirandola, Assunta Sartor, and Evelina Tacconelli. 2022. "Impact of COVID-19 and Antibiotic Treatments on Gut Microbiome: A Role for Enterococcus spp." Biomedicines 10, no. 11: 2786. https://doi.org/10.3390/biomedicines10112786
APA StyleRighi, E., Lambertenghi, L., Gorska, A., Sciammarella, C., Ivaldi, F., Mirandola, M., Sartor, A., & Tacconelli, E. (2022). Impact of COVID-19 and Antibiotic Treatments on Gut Microbiome: A Role for Enterococcus spp. Biomedicines, 10(11), 2786. https://doi.org/10.3390/biomedicines10112786