
Inverse Salt Sensitivity of Blood Pressure Is Associated with an Increased Renin-Angiotensin System Activity



Abstract
:1. Introduction
2. Materials and Methods
2.1. Living Renal Proximal Tubules Obtained from Spot Urines
2.2. uRPTC Culture
2.3. Plasma Membrane D1R and AT2R Expression
2.4. Plasma Membrane D2R and APN Expression
2.5. Total Cellular CTCF, NRF2, and SOD2 Expression
2.6. D2R Binding Assay
2.7. Dopamine D2R siRNA
2.8. D2R Overexpression
2.9. Sodium Influx Assay
2.10. Seminaphtharhodafluor (pH-Sensitive Dye, SNARF), Sodium-Hydrogen Exchanger Type 3 (NHE3), and Sodium-Dependent pH Recovery Assay in Low Sodium Concentrations
2.11. Micro Ribonucleic Acid 485-5p (miR-485-5p) Mimic and Blocker Transfection
2.12. SLC5A11 SNP and qRT-PCR
2.13. Statistical Analysis
3. Results
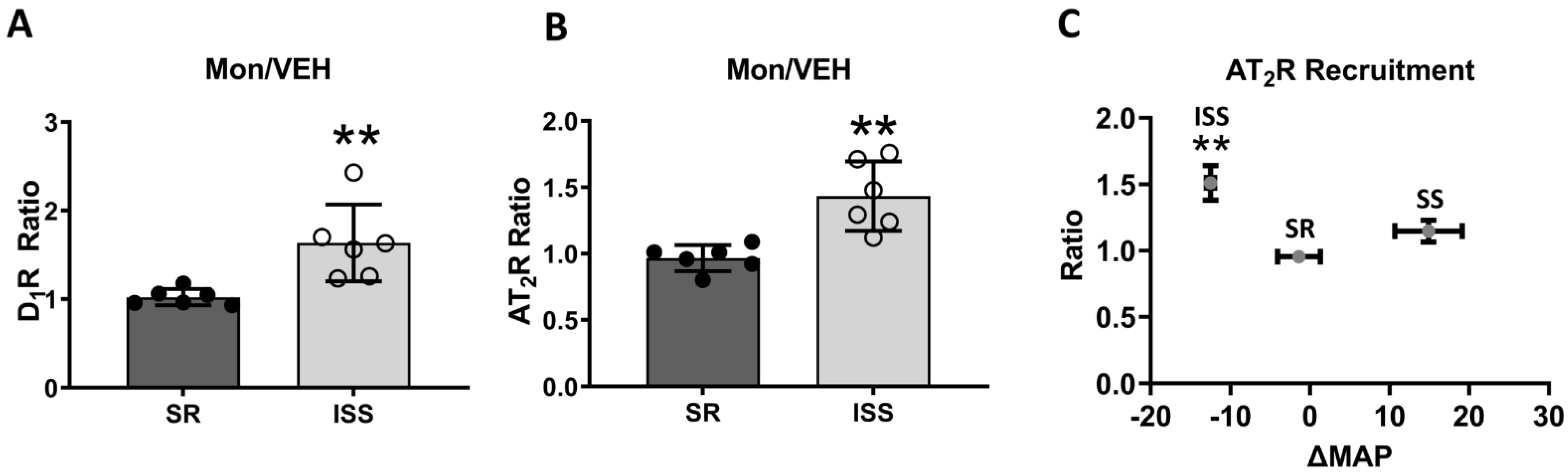
3.1. D1R and AT2R Plasma Membrane Recruitment
3.2. Direct AT2R Plasma Membrane Recruitment in uRPTC
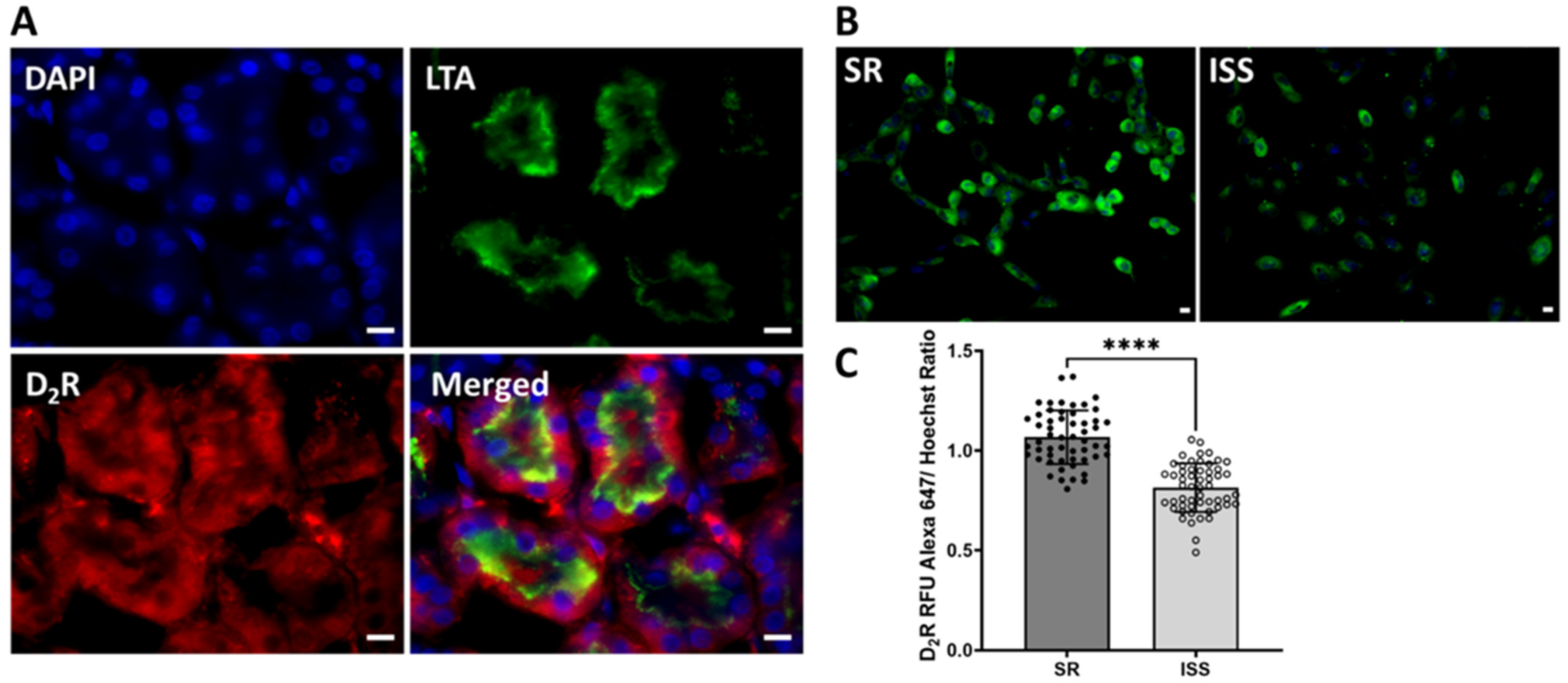
3.3. D2R Binding
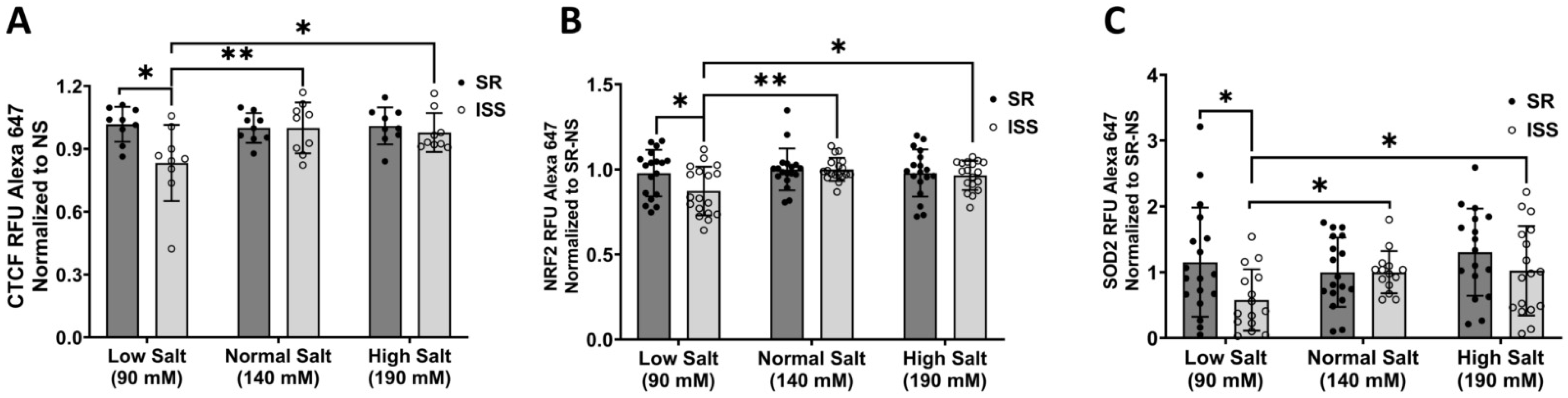
3.4. Total Cellular CTCF, NRF2, and SOD2 Expression
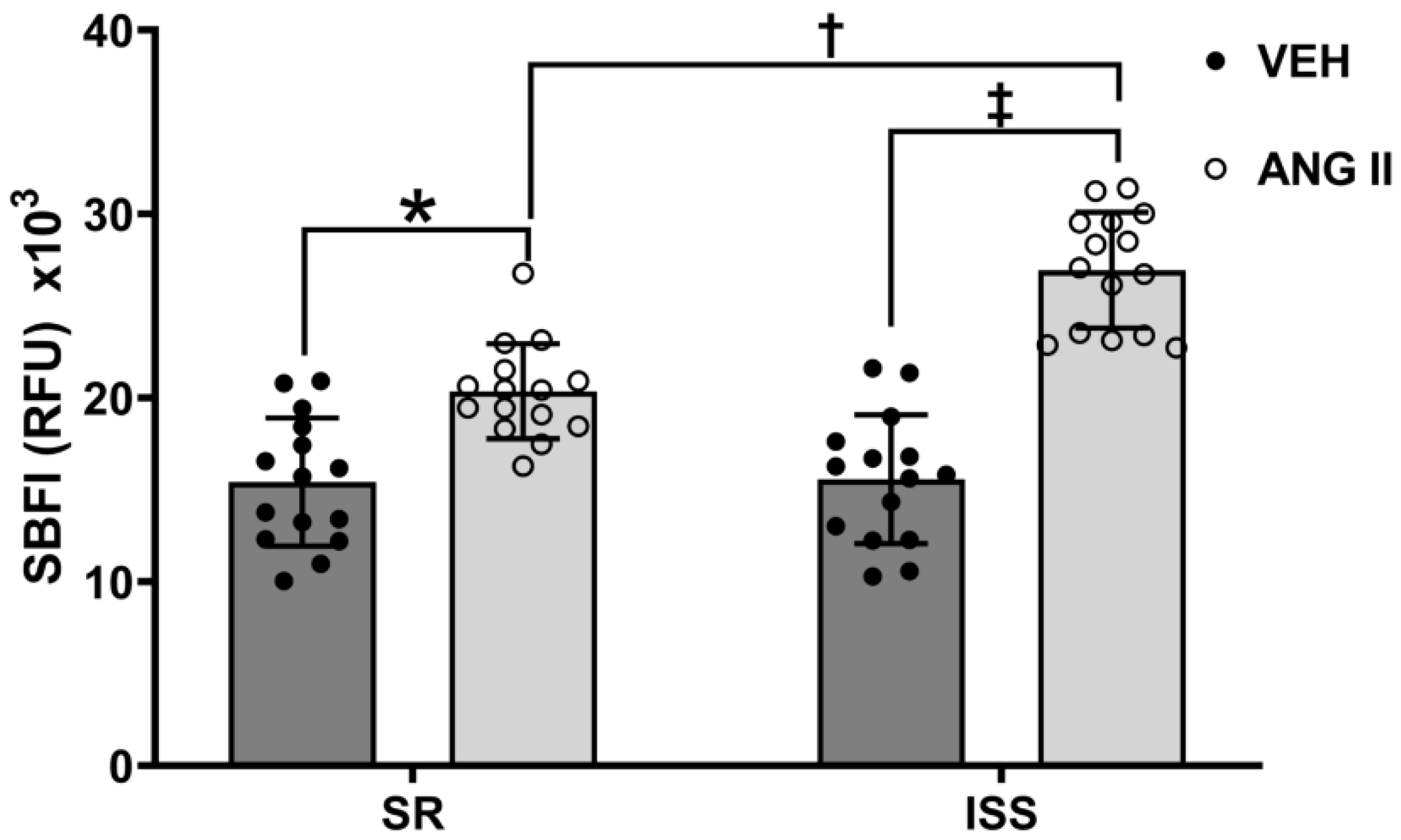
3.5. Ang II-Stimulated Sodium Influx in ISS and SR
3.6. D2R Extracellular Epitope Binding and D2R Overexpression in Living uRPTCs
3.7. Epigenetic Regulation of the D2R by Mir-485-5p
3.8. The Effect of D2R Overexpression on Sodium Transport in uRPTCs
3.9. RPTC Plasma Membrane Amino Peptidase N Expression
3.10. D2 SNPs and SLC5A11 SNP
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Ang I | angiotensin I |
| Ang II | angiotensin II |
| Ang III | angiotensin III |
| Ang IV | angiotensin IV |
| APN | amino peptidase N |
| AT1R | angiotensin II type 1 receptor |
| AT2R | angiotensin II type 2 receptor |
| BP | blood pressure |
| D2R | dopamine type 2 receptor |
| DRD2 | dopamine type 2 receptor gene |
| GRK4 | G protein-coupled receptor kinase type 4 |
| ISS | inverse salt sensitivity |
| MAP | mean arterial pressure |
| RAS | renin-angiotensin system |
| RPTC | renal proximal tubule cell |
| SR | Salt resistance |
| SS | Salt sensitivity |
| uRPTC | urine-derived renal proximal tubule cell |
References
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E.; Collins, K.J.; Dennison Himmelfarb, C.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APHA/ASH/ASPC/ NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: A report of the AMERICAN COLLEGE OF CARDIOLOGY/AMERICAN HEART ASSOCIATION task force on clinical practice guidelines. Hypertension 2018, 71, e13–e115. [Google Scholar] [PubMed]
- He, F.J.; Tan, M.; Ma, Y.; MacGregor, G.A. Salt reduction to prevent hypertension and cardiovascular disease: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2020, 75, 632–647. [Google Scholar] [CrossRef] [PubMed]
- Cappuccio, F.P.; Beer, M.; Strazzullo, P.; Network, E.S.A. Population dietary salt reduction and the risk of cardiovascular disease. A scientific statement from the European Salt Action Network. Nutr. Metab. Cardiovasc. Dis. 2018, 29, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, R.M.; Schoeffel, C.D.; Gildea, J.J.; Jones, J.E.; McGrath, H.E.; Gordon, L.N.; Park, M.J.; Sobota, R.S.; Underwood, P.C.; Williams, J.; et al. Salt sensitivity of blood pressure is associated with polymorphisms in the sodium-bicarbonate cotransporter. Hypertension 2012, 60, 1359–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elijovich, F.; Weinberger, M.H.; Anderson, C.A.; Appel, L.J.; Bursztyn, M.; Cook, N.R.; Dart, R.A.; Newton-Cheh, C.H.; Sacks, F.M.; Laffer, C.L. Hypertension AHAPaPECotCo, Biology CoFGaT, Council aS. Salt sensitivity of blood pressure: A scientific statement from the American Heart Association. Hypertension 2016, 68, e7–e46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rucker, A.J.; Rudemiller, N.P.; Crowley, S.D. Salt, hypertension, and immunity. Annu. Rev. Physiol. 2018, 80, 283–307. [Google Scholar] [CrossRef] [PubMed]
- Lastra, G.; Dhuper, S.; Johnson, M.S.; Sowers, J.R. Salt, aldosterone, and insulin resistance: Impact on the cardiovascular system. Nat. Rev. Cardiol. 2010, 7, 577–584. [Google Scholar] [CrossRef]
- Kurtz, T.W.; DiCarlo, S.E.; Pravenec, M.; Morris, R.C. The American Heart Association scientific statement on salt sensitivity of blood pressure: Prompting consideration of alternative conceptual frameworks for the pathogenesis of salt sensitivity? J. Hypertens. 2017, 35, 2214–2225. [Google Scholar] [CrossRef]
- Skrabal, F.; Herholz, H.; Neumayr, M.; Hamberger, L.; Ledochowski, M.; Sporer, H.; Hortnagl, H.; Schwarz, S.; Schonitzer, D. Salt sensitivity in humans is linked to enhanced sympathetic responsiveness and to enhanced proximal tubular reabsorption. Hypertension 1984, 6, 152–158. [Google Scholar] [CrossRef] [Green Version]
- Majid, D.S.; Kopkan, L. Nitric oxide and superoxide interactions in the kidney and their implication in the development of salt-sensitive hypertension. Clin. Exp. Pharmacol. Physiol. 2007, 34, 946–952. [Google Scholar] [CrossRef]
- Sigmund, C.D.; Carey, R.M.; Appel, L.J.; Arnett, D.K.; Bosworth, H.B.; Cushman, W.C.; Galis, Z.S.; Green Parker, M.; Hall, J.E.; Harrison, D.G.; et al. Report of the National Heart, Lung, and Blood Institute working group on hypertension: Barriers to translation. Hypertension 2020, 75, 902–917. [Google Scholar] [CrossRef] [PubMed]
- Raizada, M.K.; Joe, B.; Bryan, N.S.; Chang, E.B.; Dewhirst, F.E.; Borisy, G.G.; Galis, Z.S.; Henderson, W.; Jose, P.A.; Ketchum, C.J.; et al. Report of the National Heart, Lung, and Blood Institute working group on the role of microbiota in blood pressure regulation: Current status and future directions. Hypertension 2017, 70, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, M.H.; Fineberg, N.S.; Fineberg, S.E.; Weinberger, M. Salt sensitivity, pulse pressure, and death in normal and hypertensive humans. Hypertension 2001, 37, 429–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alderman, M.H.; Cohen, H.W. Dietary sodium intake and cardiovascular mortality: Controversy resolved? Curr. Hypertens. Rep. 2012, 14, 193–201. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.; Mente, A.; Alderman, M.H.; Brady, A.J.B.; Diaz, R.; Gupta, R.; López-Jaramillo, P.; Luft, F.C.; Lüscher, T.F.; Mancia, G.; et al. Salt and cardiovascular disease: Insufficient evidence to recommend low sodium intake. Eur. Heart. J. 2020, 41, 3363–3373. [Google Scholar] [CrossRef] [PubMed]
- Asayama, K.; Stolarz-Skrzypek, K.; Persu, A.; Staessen, J.A. Systematic review of health outcomes in relation to salt intake highlights the widening divide between guidelines and the evidence. Am. J. Hypertens. 2014, 27, 1138–1142. [Google Scholar] [CrossRef] [Green Version]
- Stolarz-Skrzypek, K.; Kuznetsova, T.; Thijs, L.; Tikhonoff, V.; Seidlerová, J.; Richart, T.; Jin, Y.; Olszanecka, A.; Malyutina, S.; Casiglia, E.; et al. Fatal and nonfatal outcomes, incidence of hypertension, and blood pressure changes in relation to urinary sodium excretion. JAMA 2011, 305, 1777–1785. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, M.; Mente, A.; Rangarajan, S.; McQueen, M.J.; Wang, X.; Liu, L.; Yan, H.; Lee, S.F.; Mony, P.; Devanath, A.; et al. Urinary sodium and potassium excretion, mortality, and cardiovascular events. N. Engl. J. Med. 2014, 371, 612–623. [Google Scholar] [CrossRef] [Green Version]
- Strom, B.L.; Anderson, C.A.; Ix, J.H. Sodium reduction in populations: Insights from the Institute of Medicine committee. JAMA 2013, 310, 31–32. [Google Scholar] [CrossRef]
- Gildea, J.J.; Lahiff, D.T.; Van Sciver, R.E.; Weiss, R.S.; Shah, N.; McGrath, H.E.; Schoeffel, C.D.; Jose, P.A.; Carey, R.M.; Felder, R.A. A linear relationship between the ex-vivo sodium mediated expression of two sodium regulatory pathways as a surrogate marker of salt sensitivity of blood pressure in exfoliated human renal proximal tubule cells: The virtual renal biopsy. Clin. Chim. Acta 2013, 421, 236–242. [Google Scholar] [CrossRef]
- Graudal, N.; Jürgens, G. Conflicting evidence on health effects associated with salt reduction calls for a redesign of the salt dietary guidelines. Prog. Cardiovasc. Dis. 2018, 61, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Alderman, M.H.; Blumenfeld, J.D. Hypertension: Evolving from standardized to individualized care. J. Hypertens. 2020, 38, 1251–1254. [Google Scholar] [CrossRef] [PubMed]
- Montasser, M.E.; Douglas, J.A.; Roy-Gagnon, M.H.; Van Hout, C.V.; Weir, M.R.; Vogel, R.; Parsa, A.; Steinle, N.I.; Snitker, S.; Brereton, N.H.; et al. Determinants of blood pressure response to low-salt intake in a healthy adult population. J. Clin. Hypertens. 2011, 13, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Overlack, A.; Ruppert, M.; Kolloch, R.; Göbel, B.; Kraft, K.; Diehl, J.; Schmitt, W.; Stumpe, K.O. Divergent hemodynamic and hormonal responses to varying salt intake in normotensive subjects. Hypertension 1993, 22, 331–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overlack, A.; Ruppert, M.; Kolloch, R.; Kraft, K.; Stumpe, K.O. Age is a major determinant of the divergent blood pressure responses to varying salt intake in essential hypertension. Am. J. Hypertens. 1995, 8, 829–836. [Google Scholar] [CrossRef]
- Cuka, E.; Simonini, M.; Lanzani, C.; Zagato, L.; Citterio, L.; Messaggio, E.; Faienza, S.; Brioni, E.; Hamlyn, J.M.; Manunta, P. Inverse salt sensitivity: An independent risk factor for cardiovascular damage in essential hypertension. J. Hypertens. 2022, 40, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Bihorac, A.; Tezcan, H.; Ozener, C.; Oktay, A.; Akoglu, E. Association between salt sensitivity and target organ damage in essential hypertension. Am. J. Hypertens. 2000, 13, 864–872. [Google Scholar] [CrossRef] [Green Version]
- Felder, R.A.; White, M.J.; Williams, S.M.; Jose, P.A. Diagnostic tools for hypertension and salt sensitivity testing. Curr. Opin. Nephrol. Hypertens. 2013, 22, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Iatrino, R.; Manunta, P.; Zagato, L. Salt Sensitivity: Challenging and Controversial Phenotype of Primary Hypertension. Curr. Hypertens. Rep. 2016, 18, 70. [Google Scholar] [CrossRef]
- Institute of Medicine (US) Committee on Strategies to Reduce Sodium Intake. Strategies to Reduce Sodium Intake in the United States; Henney, J.E., Taylor, C.L., Boon, C.S., Eds.; National Academies Press: Washington, DC, USA, 2010.
- O’Donnell, M.; Mente, A.; Rangarajan, S.; McQueen, M.J.; O’Leary, N.; Yin, L.; Liu, X.; Swaminathan, S.; Khatib, R.; Rosengren, A.; et al. Joint association of urinary sodium and potassium excretion with cardiovascular events and mortality: Prospective cohort study. BMJ 2019, 364, l772. [Google Scholar] [CrossRef]
- Castiglioni, P.; Parati, G.; Lazzeroni, D.; Bini, M.; Faini, A.; Brambilla, L.; Brambilla, V.; Coruzzi, P. Hemodynamic and autonomic response to different salt intakes in normotensive individuals. J. Am. Heart. Assoc. 2016, 5, e003736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gildea, J.J.; Carlson, J.M.; Schoeffel, C.D.; Carey, R.M.; Felder, R.A. Urinary exosome miRNome analysis and its applications to salt sensitivity of blood pressure. Clin. Biochem. 2013, 46, 1131–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barros, E.R.; Carvajal, C.A. Urinary Exosomes and Their Cargo: Potential Biomarkers for Mineralocorticoid Arterial Hypertension? Front. Endocrinol. 2017, 8, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, P.P.S.; Hall, D.; Chilian, W.M.; Chia, Y.C.; Mohd Zain, S.; Lim, H.M.; Kumar, D.N.; Ching, S.M.; Low, T.Y.; Md Noh, M.F.; et al. Exosomal microRNAs in the development of essential hypertension and its potential as biomarkers. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H1486–H1497. [Google Scholar] [CrossRef] [PubMed]
- Manosroi, W.; Williams, G.H. Genetics of human primary hypertension: Focus on hormonal mechanisms. Endocr. Rev. 2019, 40, 825–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayner, B.; Ramesar, R. The importance of G protein-coupled receptor kinase 4 (GRK4) in pathogenesis of salt sensitivity, salt sensitive hypertension and response to antihypertensive treatment. Int. J. Mol. Sci. 2015, 16, 5741–5749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, S.; Kim, J.Y.; Cho, Y.; Koh, S.B.; Kim, N.; Choi, J.R. Genetically, dietary sodium intake is causally associated with salt-sensitive hypertension risk in a community-based cohort study: A mendelian randomization approach. Curr. Hypertens. Rep. 2020, 22, 45. [Google Scholar] [CrossRef]
- Lee, M.; Kim, M.K.; Kim, S.M.; Park, H.; Park, C.G.; Park, H.K. Gender-based differences on the association between salt-sensitive genes and obesity in Korean children aged between 8 and 9 years. PLoS ONE 2015, 10, e0120111. [Google Scholar] [CrossRef] [Green Version]
- Sanada, H.; Yatabe, J.; Midorikawa, S.; Hashimoto, S.; Watanabe, T.; Moore, J.H.; Ritchie, M.D.; Williams, S.M.; Pezzullo, J.C.; Sasaki, M.; et al. Single nucleotide polymorphisms for diagnosis of salt-sensitive hypertension. Clin. Chem. 2006, 52, 352–360. [Google Scholar] [CrossRef] [Green Version]
- Bengra, C.; Mifflin, T.E.; Khripin, Y.; Manunta, P.; Williams, S.M.; Jose, P.A.; Felder, R.A. Genotyping of essential hypertension single-nucleotide polymorphisms by a homogeneous PCR method with universal energy transfer primers. Clin. Chem. 2002, 48, 2131–2140. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zeng, C.; Villar, V.A.; Chen, S.Y.; Konkalmatt, P.; Wang, X.; Asico, L.D.; Jones, J.E.; Yang, Y.; Sanada, H.; et al. Human GRK4γ142V Variant Promotes Angiotensin II Type I Receptor-Mediated Hypertension via Renal Histone Deacetylase Type 1 Inhibition. Hypertension 2016, 67, 325–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diao, Z.; Asico, L.D.; Villar, V.A.M.; Zheng, X.; Cuevas, S.; Armando, I.; Jose, P.A.; Wang, X. Increased renal oxidative stress in salt-sensitive human GRK4gamma486V transgenic mice. Free Radic. Biol. Med. 2017, 106, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Armando, I.; Konkalmatt, P.; Felder, R.A.; Jose, P.A. The renal dopaminergic system: Novel diagnostic and therapeutic approaches in hypertension and kidney disease. Transl. Res. 2015, 165, 505–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, R.C.; Zhang, M.Z. Dopamine, the kidney, and hypertension. Curr. Hypertens. Rep. 2012, 14, 138–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padia, S.H.; Kemp, B.A.; Howell, N.L.; Fournie-Zaluski, M.C.; Roques, B.P.; Carey, R.M. Conversion of renal angiotensin II to angiotensin III is critical for AT2 receptor-mediated natriuresis in rats. Hypertension 2008, 51, 460–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felder, R.A.; Gildea, J.J.; Xu, P.; Yue, W.; Armando, I.; Carey, R.M.; Jose, P.A. Inverse Salt Sensitivity of Blood Pressure: Mechanisms and Potential Relevance for Prevention of Cardiovascular Disease. Curr. Hypertens. Rep. 2022, 9, 361–374. [Google Scholar] [CrossRef]
- Li, X.; Campbell, D.; Ohishi, M.; Yuan, S.; Zhuo, J. AT1 receptor-activated signaling mediates angiotensin IV-induced renal cortical vasoconstriction in rats. Am. J. Physiol. Renal. Physiol. 2006, 290, F1024–F1033. [Google Scholar] [CrossRef] [Green Version]
- Danziger, R.S. Aminopeptidase N in arterial hypertension. Heart Fail. Rev. 2008, 13, 293–298. [Google Scholar] [CrossRef]
- Padia, S.H.; Howell, N.L.; Kemp, B.A.; Fournie-Zaluski, M.C.; Roques, B.P.; Carey, R.M. Intrarenal aminopeptidase N inhibition restores defective angiontesin II type 2-mediated natriuresis in spontaneously hypertensive rats. Hypertension 2010, 55, 474–480. [Google Scholar] [CrossRef] [Green Version]
- Kemp, B.A.; Howell, N.L.; Gildea, J.J.; Keller, S.R.; Carey, R.M. Identification of a Primary Renal AT2 Receptor Defect in Spontaneously Hypertensive Rats. Circ. Res. 2020, 126, 644–659. [Google Scholar] [CrossRef]
- Lew, R.A.; Mustafa, T.; Ye, S.; McDowall, S.G.; Chai, S.Y.; Albiston, A.L. Angiotensin AT4 ligands are potent, competitive inhibitors of insulin regulated aminopeptidase (IRAP). J. Neurochem. 2003, 86, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Walton, S.L.; Mirabito Colafella, K.M.; Ansari, A.; Chai, S.Y.; Denton, K.M. Insulin-regulated aminopeptidase deficiency impairs cardiovascular adaptations and placental development during pregnancy. Clin. Sci. 2020, 134, 3213–3228. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.; Abdul-Wahab, R.; Kotak, D.K.; Lokhandwala, M.F. Bromocriptine regulates angiotensin II response on sodium pump in proximal tubules. Hypertension 1998, 32, 1054–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, C.; Ganju, R.K.; Pompili, V.J.; Chakroborty, D. Enhanced peripheral dopamine impairs post-ischemic healing by suppressing angiotensin receptor type 1 expression in endothelial cells and inhibiting angiogenesis. Angiogenesis 2017, 20, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.R.; Kouyoumdzian, N.M.; Rukavina Mikusic, N.L.; Kravetz, M.C.; Rosón, M.I.; Rodríguez Fermepin, M.; Fernández, B.E. Renal dopaminergic system: Pathophysiological implications and clinical perspectives. World J. Nephrol. 2015, 4, 196–212. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Konkalmatt, P.; Yang, Y.; Gildea, J.J.; Jones, J.E.; Cuevas, S.; Felder, R.A.; Jose, P.A.; Armando, I. Single-nucleotide polymorphisms of the dopamine D2 receptor increase inflammation and fibrosis in human renal proximal tubule cells. Hypertension 2014, 63, e74–e80. [Google Scholar] [CrossRef] [PubMed]
- Konkalmatt, P.R.; Asico, L.D.; Zhang, Y.; Yang, Y.; Drachenberg, C.; Zheng, X.; Han, F.; Jose, P.A.; Armando, I. Renal rescue of dopamine D2 receptor function reverses renal injury and high blood pressure. JCI Insight 2016, 1, e85888. [Google Scholar] [CrossRef]
- Zhang, Y.; Cuevas, S.; Asico, L.D.; Escano, C.; Yang, Y.; Pascua, A.M.; Wang, X.; Jones, J.E.; Grandy, D.; Eisner, G.; et al. Deficient dopamine D2 receptor function causes renal inflammation independently of high blood pressure. PLoS ONE 2012, 7, e38745. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Jiang, X.; Qin, C.; Cuevas, S.; Jose, P.A.; Armando, I. Dopamine D2 receptors’ effects on renal inflammation are mediated by regulation of PP2A function. Am. J. Physiol. Renal. Physiol. 2016, 310, F128–F134. [Google Scholar] [CrossRef] [Green Version]
- Barrese, V.; Stott, J.B.; Baldwin, S.N.; Mondejar-Parreño, G.; Greenwood, I.A. SMIT (Sodium-Myo-Inositol Transporter) 1 Regulates Arterial Contractility Through the Modulation of Vascular Kv7 Channels. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2468–2480. [Google Scholar] [CrossRef]
- Lahjouji, K.; Aouameur, R.; Bissonnette, P.; Coady, M.J.; Bichet, D.G.; Lapointe, J.Y. Expression and functionality of the Na+/myo-inositol cotransporter SMIT2 in rabbit kidney. Biochim. Biophys. Acta 2007, 1768, 1154–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gildea, J.J.; McGrath, H.E.; Van Sciver, R.E.; Wang, D.B.; Felder, R.A. Isolation, growth, and characterization of human renal epithelial cells using traditional and 3D methods. Methods Mol. Biol. 2013, 945, 329–345. [Google Scholar] [PubMed]
- Gildea, J.J.; Xu, P.; Kemp, B.A.; Carey, R.M.; Jose, P.A.; Felder, R.A. The dopamine D1 receptor and angiotensin II type-2 receptor are required for inhibition of sodium transport through a protein phosphatase 2A pathway. Hypertension 2019, 73, 1258–1265. [Google Scholar] [CrossRef]
- Rosić, N.K.; Standaert, M.L.; Pollet, R.J. The mechanism of insulin stimulation of (Na+,K+)-ATPase transport activity in muscle. J. Biol. Chem. 1985, 260, 6206–6212. [Google Scholar] [CrossRef]
- Stojanovic, T.; Orlova, M.; Sialana, F.J.; Höger, H.; Stuchlik, S.; Milenkovic, I.; Aradska, J.; Lubec, G. Validation of dopamine receptor Drd1 and Drd2 antibodies using receptor deficient mice. Amino Acids 2017, 49, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Newton, C.L.; Wood, M.D.; Strange, P.G. Examining the Effects of Sodium Ions on the Binding of Antagonists to Dopamine D2 and D3 Receptors. PLoS ONE 2016, 11, e0158808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeman, P.; Van Tol, H.H. Dopamine receptor pharmacology. Curr. Opin. Neurol. Neurosurg. 1993, 6, 602–608. [Google Scholar] [CrossRef]
- 741742 L. Guide to Pharmacology. 2021. Available online: https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3302 (accessed on 13 October 2021).
- Kling-Petersen, T.; Ljung, E.; Svensson, K. Effects on locomotor activity after local application of D3 preferring compounds in discrete areas of the rat brain. J. Neural. Transm. Gen. Sect. 1995, 102, 209–220. [Google Scholar] [CrossRef]
- Gao, D.Q.; Canessa, L.M.; Mouradian, M.M.; Jose, P.A. Expression of the D2 subfamily of dopamine receptor genes in kidney. Am. J. Physiol. 1994, 266, F646–F650. [Google Scholar] [CrossRef]
- Gildea, J.J.; Xu, P.; Kemp, B.A.; Carlson, J.M.; Tran, H.T.; Bigler Wang, D.; Langouët-Astrié, C.J.; McGrath, H.E.; Carey, R.M.; Jose, P.A.; et al. Sodium bicarbonate cotransporter NBCe2 gene variants increase sodium and bicarbonate transport in human renal proximal tubule cells. PLoS ONE 2018, 13, e0189464. [Google Scholar] [CrossRef]
- Diarra, A.; Sheldon, C.; Church, J. In situ calibration and [H+] sensitivity of the fluorescent Na+ indicator SBFI. Am. J. Physiol. Cell Physiol. 2001, 280, C1623–C1633. [Google Scholar] [CrossRef] [PubMed]
- Crocco, P.; Montesanto, A.; Passarino, G.; Rose, G. Polymorphisms falling within putative miRNA target sites in the 3’UTR region of SIRT2 and DRD2 genes are correlated with human longevity. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 586–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauberg, M.E.; Roussos, P.; Grove, J.; Børglum, A.D.; Mattheisen, M.; Schizophrenia Working Group of the Psychiatric Genomics Consortium. Analyzing the role of microRNAs in schizophrenia in the context of common genetic risk variants. JAMA Psychiatry 2016, 73, 369–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maffioletti, E.; Tardito, D.; Gennarelli, M.; Bocchio-Chiavetto, L. Micro spies from the brain to the periphery: New clues from studies on micrornas in neuropsychiatric disorders. Front. Cell. Neurosci. 2014, 8, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Gildea, J.J.; Zhang, C.; Konkalmatt, P.; Cuevas, S.; Bigler Wang, D.; Tran, H.T.; Jose, P.A.; Felder, R.A. Stomach gastrin is regulated by sodium via PPAR-α and dopamine D1 receptor. J. Mol. Endocrinol. 2020, 64, 53–65. [Google Scholar] [CrossRef]
- Luo, Y.; Hitz, B.C.; Gabdank, I.; Hilton, J.A.; Kagda, M.S.; Lam, B.; Myers, Z.; Sud, P.; Jou, J.; Lin, K.; et al. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res. 2020, 48, D882–D889. [Google Scholar] [CrossRef]
- Yang, Y.; Cuevas, S.; Yang, S.; Villar, V.A.; Escano, C.; Asico, L.; Yu, P.; Jiang, X.; Weinman, E.J.; Armando, I.; et al. Sestrin2 decreases renal oxidative stress, lowers blood pressure, and mediates dopamine D2 receptor-induced inhibition of reactive oxygen species production. Hypertension 2014, 64, 825–832. [Google Scholar] [CrossRef] [Green Version]
- Cuevas, S.; Yang, Y.; Konkalmatt, P.; Asico, L.D.; Feranil, J.; Jones, J.; Villar, V.A.; Armando, I.; Jose, P.A. Role of nuclear factor erythroid 2-related factor 2 in the oxidative stress-dependent hypertension associated with the depletion of DJ-1. Hypertension 2015, 65, 1251–1257. [Google Scholar] [CrossRef]
- Ryoo, I.G.; Kwak, M.K. Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol. Appl. Pharmacol. 2018, 359, 24–33. [Google Scholar] [CrossRef]
- Padia, S.H.; Kemp, B.A.; Howell, N.L.; Gildea, J.J.; Keller, S.R.; Carey, R.M. Intrarenal angiotensin III infusion induces natriuresis and angiotensin type 2 receptor translocation in Wistar-Kyoto but not in spontaneously hypertensive rats. Hypertension 2009, 53, 338–343. [Google Scholar] [CrossRef]
- Guirguis-Blake, J.M.; Evans, C.V.; Webber, E.M.; Coppola, E.L.; Perdue, L.A.; Weyrich, M.S. Screening for Hypertension in adults: Updated Evidence Report and Systematic Review for the US Preventive Services Task Force; Report No.: 20–05265-EF-1; Agency for Healthcare Research and Quality: Rockville, MD, USA, 2021.
- MacFadyen, R.J.; Lees, K.R.; Reid, J.L. Responses to low dose intravenous perindoprilat infusion in salt deplete/salt replete normotensive volunteers. Br. J. Clin. Pharmacol. 1994, 38, 329–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimsdale, J.E.; Ziegler, M.; Mills, P.; Berry, C. Prediction of salt sensitivity. Am. J. Hypertens. 1990, 3, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Kumar, U.; Patel, Y.; Patel, S.C.; Sidhu, A. Differential expression of D2-like dopamine receptors in the kidney of the spontaneously hypertensive rat. J. Hypertens. 2003, 21, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.J.; Thomas, G.N.; Xu, Z.L.; Fang, J.Q.; Critchley, J.A.; Tomlinson, B. An affected pedigree member analysis of linkage between the dopamine D2 receptor gene Taq1 polymorphism and obesity and hypertension. Int. J. Cardiol. 2005, 102, 111–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, J.; Thomas, N.; Singleton, A.; Piggott, M.; Lloyd, S.; Perry, E.K.; Morris, C.M.; Perry, R.H.; Ferrier, I.N.; Court, J.A. D2 dopamine receptor gene (DRD2) Taq1 a polymorphism: Reduced dopamine D2 receptor binding in the human striatum associated with the A1 allele. Pharmacogenetics 1997, 7, 479–484. [Google Scholar] [CrossRef]
- Qaddumi, W.N.; Jose, P.A. The role of the renal dopaminergic system and oxidative stress in the pathogenesis of hypertension. Biomedicines 2021, 9, 139. [Google Scholar] [CrossRef]
- Armando, I.; Asico, L.D.; Wang, X.; Jones, J.E.; Serrão, M.P.; Cuevas, S.; Grandy, D.K.; Soares-da-Silva, P.; Jose, P.A. Antihypertensive effect of etamicastat in dopamine D2 receptor-deficient mice. Hypertens. Res. 2018, 41, 489–498. [Google Scholar] [CrossRef]
- Han, F.; Konkalmatt, P.; Mokashi, C.; Kumar, M.; Zhang, Y.; Ko, A.; Farino, Z.J.; Asico, L.D.; Xu, G.; Gildea, J.; et al. Dopamine D2 receptor modulates Wnt expression and control of cell proliferation. Sci. Rep. 2019, 9, 16861. [Google Scholar] [CrossRef] [Green Version]
- Han, F.; Konkalmatt, P.; Chen, J.; Gildea, J.; Felder, R.A.; Jose, P.A.; Armando, I. miR-217 mediates the protective effects of the dopamine D2 receptor on fibrosis in human renal proximal tubule cells. Hypertension 2015, 65, 1118–1125. [Google Scholar] [CrossRef] [Green Version]
- Armando, I.; Villar, V.A.; Jose, P.A. Dopamine and renal function and blood pressure regulation. Compr. Physiol. 2011, 1, 1075–1117. [Google Scholar]
- Zhang, M.Z.; Yao, B.; Fang, X.; Wang, S.; Smith, J.P.; Harris, R.C. Intrarenal dopaminergic system regulates renin expression. Hypertension 2009, 53, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Bromocriptine. Guide to Pharmacology. 2021. Available online: https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=35 (accessed on 13 October 2021).
- Kemp, B.A.; Howell, N.L.; Keller, S.R.; Gildea, J.J.; Shao, W.; Navar, L.G.; Carey, R.M. Defective renal angiotensin III and AT receptor signaling in prehypertensive spontaneously hypertensive rats. J. Am. Heart Assoc. 2019, 8, e012016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotlo, K.; Shukla, S.; Tawar, U.; Skidgel, R.A.; Danziger, R.S. Aminopeptidase N reduces basolateral Na+ -K+ -ATPase in proximal tubule cells. Am. J. Physiol. Renal. Physiol. 2007, 293, F1047–F1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Smolders, I.; Dupont, A.G. Blood pressure and renal hemodynamic effects of angiotensin fragments. Hypertens. Res. 2011, 34, 674–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handa, R.K. Biphasic actions of angiotensin IV on renal blood flow in the rat. Regul. Pept. 2006, 136, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, S.; Villar, V.A.; Jose, P.A.; Armando, I. Renal dopamine receptors, oxidative stress, and hypertension. Int. J. Mol. Sci. 2013, 14, 17553–17572. [Google Scholar] [CrossRef] [Green Version]
- Rukavina Mikusic, N.L.; Silva, M.G.; Mazzitelli, L.R.; Santos, R.A.S.; Gómez, K.A.; Grecco, H.E.; Gironacci, M.M. Interaction between the angiotensin-(1–7) mas receptor and the dopamine D2 receptor: Implications in inflammation. Hypertension 2021, 77, 1659–1669. [Google Scholar] [CrossRef]
- Han, X.; Li, B.; Ye, X.; Mulatibieke, T.; Wu, J.; Dai, J.; Wu, D.; Ni, J.; Zhang, R.; Xue, J.; et al. Dopamine D2 receptor signalling controls inflammation in acute pancreatitis via a PP2A-dependent Akt/NF-κB signalling pathway. Br. J. Pharmacol. 2017, 174, 4751–4770. [Google Scholar] [CrossRef] [Green Version]
- Udali, S.; Guarini, P.; Moruzzi, S.; Choi, S.W.; Friso, S. Cardiovascular epigenetics: From DNA methylation to microRNAs. Mol. Asp. Med. 2013, 34, 883–901. [Google Scholar] [CrossRef]
- Chen, L.J.; Wei, S.Y.; Chiu, J.J. Mechanical regulation of epigenetics in vascular biology and pathobiology. J. Cell Mol. Med. 2013, 17, 437–448. [Google Scholar] [CrossRef]
- Findeisen, H.M.; Kahles, F.K.; Bruemmer, D. Epigenetic regulation of vascular smooth muscle cell function in atherosclerosis. Curr. Atheroscler. Rep. 2013, 15, 319. [Google Scholar] [CrossRef]
- Wang, J.; Yin, N.; Deng, Y.; Wei, Y.; Huang, Y.; Pu, X.; Li, L.; Zheng, Y.; Guo, J.; Yu, J.; et al. Ascorbic acid protects against hypertension through downregulation of ACE1 gene expression mediated by histone deacetylation in prenatal inflammation-induced offspring. Sci. Rep. 2016, 6, 39469. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, R.W.; Vogel, H.; Schürmann, A. Genetic and epigenetic control of metabolic health. Mol. Metab. 2013, 2, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Tammen, S.A.; Friso, S.; Choi, S.W. Epigenetics: The link between nature and nurture. Mol. Asp. Med. 2013, 34, 753–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banday, A.A.; Fazili, F.R.; Lokhandwala, M.F. Oxidative stress causes renal dopamine D1 receptor dysfunction and hypertension via mechanisms that involve nuclear factor-kappab and protein kinase C. J. Am. Soc. Nephrol. 2007, 18, 1446–1457. [Google Scholar] [CrossRef] [Green Version]
- Kawarazaki, W.; Fujita, T. Kidney and epigenetic mechanisms of salt-sensitive hypertension. Nat. Rev. Nephrol. 2021, 17, 350–363. [Google Scholar] [CrossRef]
- Cuevas, S.; Villar, V.A.M.; Jose, P.A. Genetic polymorphisms associated with reactive oxygen species and blood pressure regulation. Pharm. J. 2019, 19, 315–336. [Google Scholar] [CrossRef]
- Lee, H.; Jiang, X.; Perwaiz, I.; Yu, P.; Wang, J.; Wang, Y.; Hüttemann, M.; Felder, R.A.; Sibley, D.R.; Polster, B.M.; et al. Dopamine D(5) receptor-mediated decreases in mitochondrial reactive oxygen species production are cAMP and autophagy dependent. Hypertens. Res. 2021, 44, 628–641. [Google Scholar] [CrossRef]
- Gildea, J.J.; Tran, H.T.; Van Sciver, R.E.; Bigler Wang, D.; Carlson, J.M.; Felder, R.A. A novel role for c-Myc in G protein-coupled receptor kinase 4 (GRK4) transcriptional regulation in human kidney proximal tubule cells. Hypertension 2013, 61, 1021–1027. [Google Scholar] [CrossRef] [Green Version]
- Filippova, G.N.; Fagerlie, S.; Klenova, E.M.; Myers, C.; Dehner, Y.; Goodwin, G.; Neiman, P.E.; Collins, S.J.; Lobanenkov, V.V. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol. Cell. Biol. 1996, 16, 2802–2813. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Liang, G.; Duan, H.; Yang, X.; Qin, G.; Sang, N. Synergistic effects of sulfur dioxide and polycyclic aromatic hydrocarbons on pulmonary pro-fibrosis via mir-30c-1–3p/ transforming growth factor β type ii receptor axis. Chemosphere 2019, 219, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Foo, J.N.; O’Roak, B.J.; Zhao, H.; Larson, M.G.; Simon, D.B.; Newton-Cheh, C.; State, M.W.; Levy, D.; Lifton, R.P. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 2008, 40, 592–599. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Sudarikova, A.V.; Ilatovskaya, D.V.; Gildea, J.J.; Akhter, M.; Carey, R.M.; Yue, W.; Jose, P.A.; Felder, R.A. Epithelial Sodium Channel Alpha Subunit (αENaC) Is Associated with Inverse Salt Sensitivity of Blood Pressure. Biomedicines 2022, 10, 981. [Google Scholar] [CrossRef] [PubMed]
- Horniblow, R.D.; Pathak, P.; Balacco, D.L.; Acharjee, A.; Lles, E.; Gkoutos, G.; Beggs, A.D.; Tselepis, C. Iron-mediated epigenetic activation of NRF2 targets. J. Nutr. Biochem. 2022, 101, 108929. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, P.; Park, J.; Hurtado Del Pozo, C.; Li, L.; Doke, T.; Huang, S.; Zhao, J.; Kang, H.M.; Shrestra, R.; Balzer, M.S.; et al. The Nuclear Receptor ESRRA Protects from Kidney Disease by Coupling Metabolism and Differentiation. Cell Metab. 2021, 33, 379–394.e8. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Salt Sensitivity | WT | Heterozygous | SNP |
|---|---|---|---|
| ISS | 9 (29%) | 19 (61%) | 3 (10%) |
| SR | 111 (51%) | 83 (38%) | 24 (11%) |
| SS | 20 (38%) | 23 (43%) | 10 (19%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gildea, J.J.; Xu, P.; Schiermeyer, K.A.; Yue, W.; Carey, R.M.; Jose, P.A.; Felder, R.A. Inverse Salt Sensitivity of Blood Pressure Is Associated with an Increased Renin-Angiotensin System Activity. Biomedicines 2022, 10, 2811. https://doi.org/10.3390/biomedicines10112811
Gildea JJ, Xu P, Schiermeyer KA, Yue W, Carey RM, Jose PA, Felder RA. Inverse Salt Sensitivity of Blood Pressure Is Associated with an Increased Renin-Angiotensin System Activity. Biomedicines. 2022; 10(11):2811. https://doi.org/10.3390/biomedicines10112811
Chicago/Turabian StyleGildea, John J., Peng Xu, Katie A. Schiermeyer, Wei Yue, Robert M. Carey, Pedro A. Jose, and Robin A. Felder. 2022. "Inverse Salt Sensitivity of Blood Pressure Is Associated with an Increased Renin-Angiotensin System Activity" Biomedicines 10, no. 11: 2811. https://doi.org/10.3390/biomedicines10112811
APA StyleGildea, J. J., Xu, P., Schiermeyer, K. A., Yue, W., Carey, R. M., Jose, P. A., & Felder, R. A. (2022). Inverse Salt Sensitivity of Blood Pressure Is Associated with an Increased Renin-Angiotensin System Activity. Biomedicines, 10(11), 2811. https://doi.org/10.3390/biomedicines10112811