
Interdisciplinary Approaches to Deal with Alzheimer’s Disease—From Bench to Bedside: What Feasible Options Do Already Exist Today?
 , ,
, ,
Abstract
:1. Introduction
1.1. Aim of This Review
1.2. Molecular Mechanisms Involved in AD Pathogenesis
2. Individual Approaches
2.1. Nutritional Approaches: Molecular Mechanisms of Dietary Fatty Acids and Vitamins in the Development of Alzheimer’s Disease
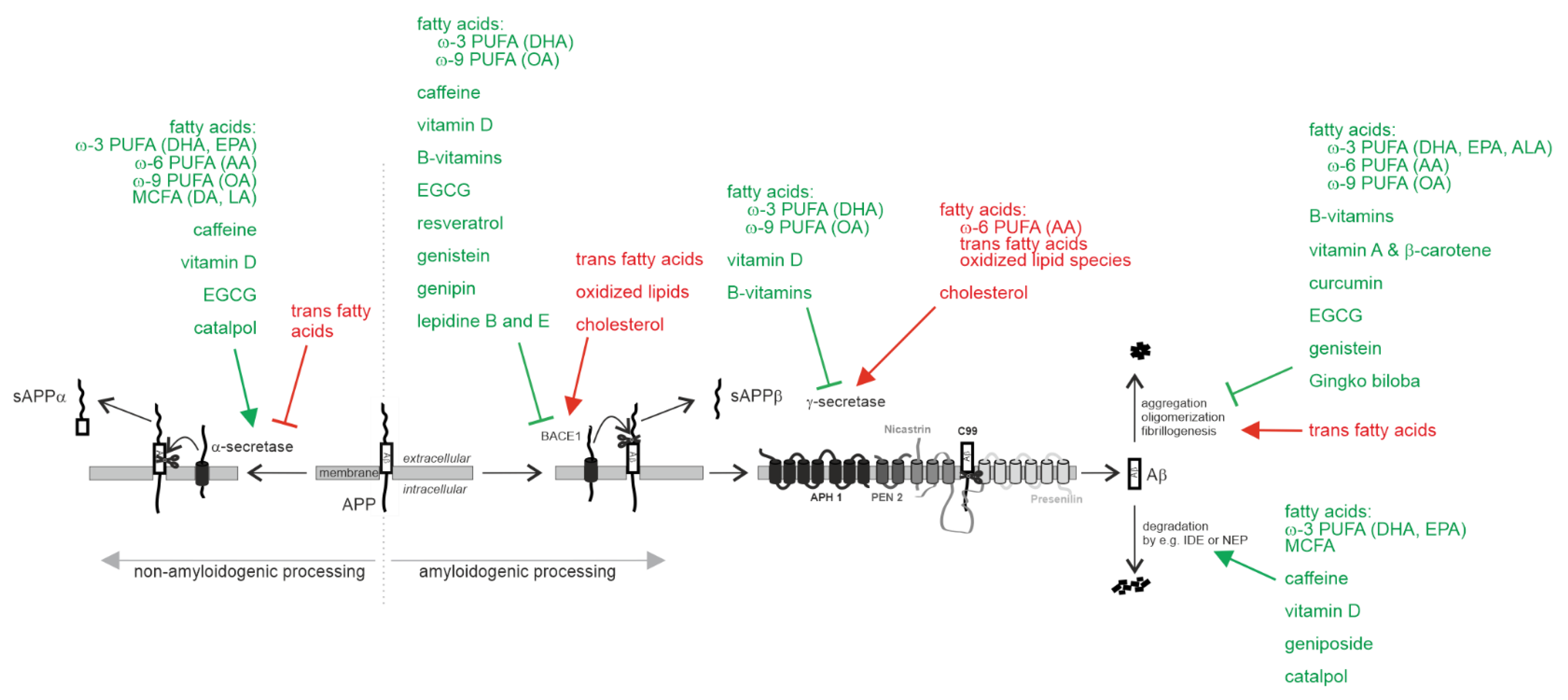
2.1.1. Effect on Aβ Pathology
Non-Amyloidogenic α-Secretase Processing of APP
Amyloidogenic Processing of APP by β-Site Cleaving Enzyme BACE1 and γ-Secretase
Aβ Degradation
Aβ Oligomerization, Aβ Aggregation and Aβ Fibrillogenesis
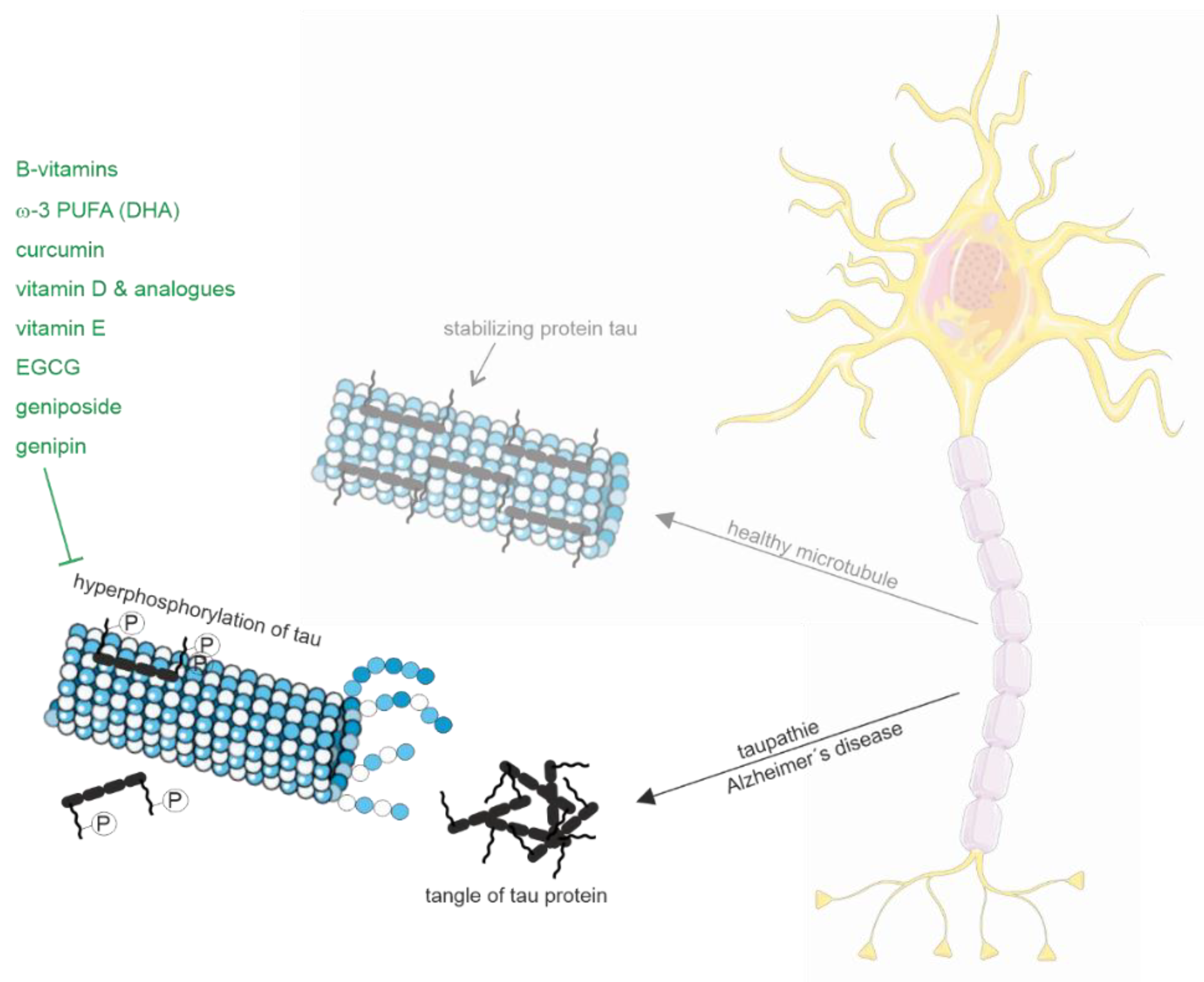
2.1.2. Tau Pathology
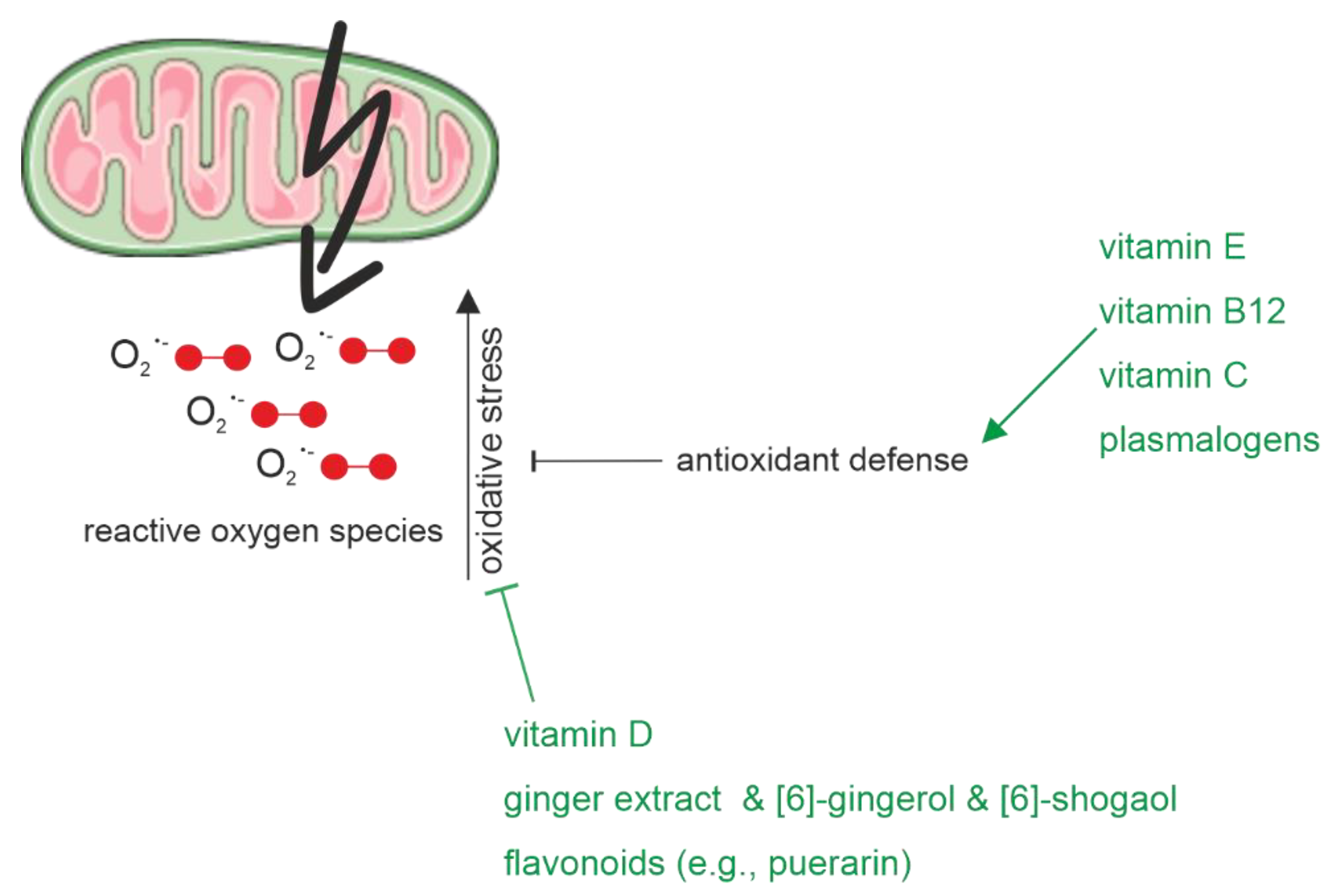
2.1.3. Oxidative Stress
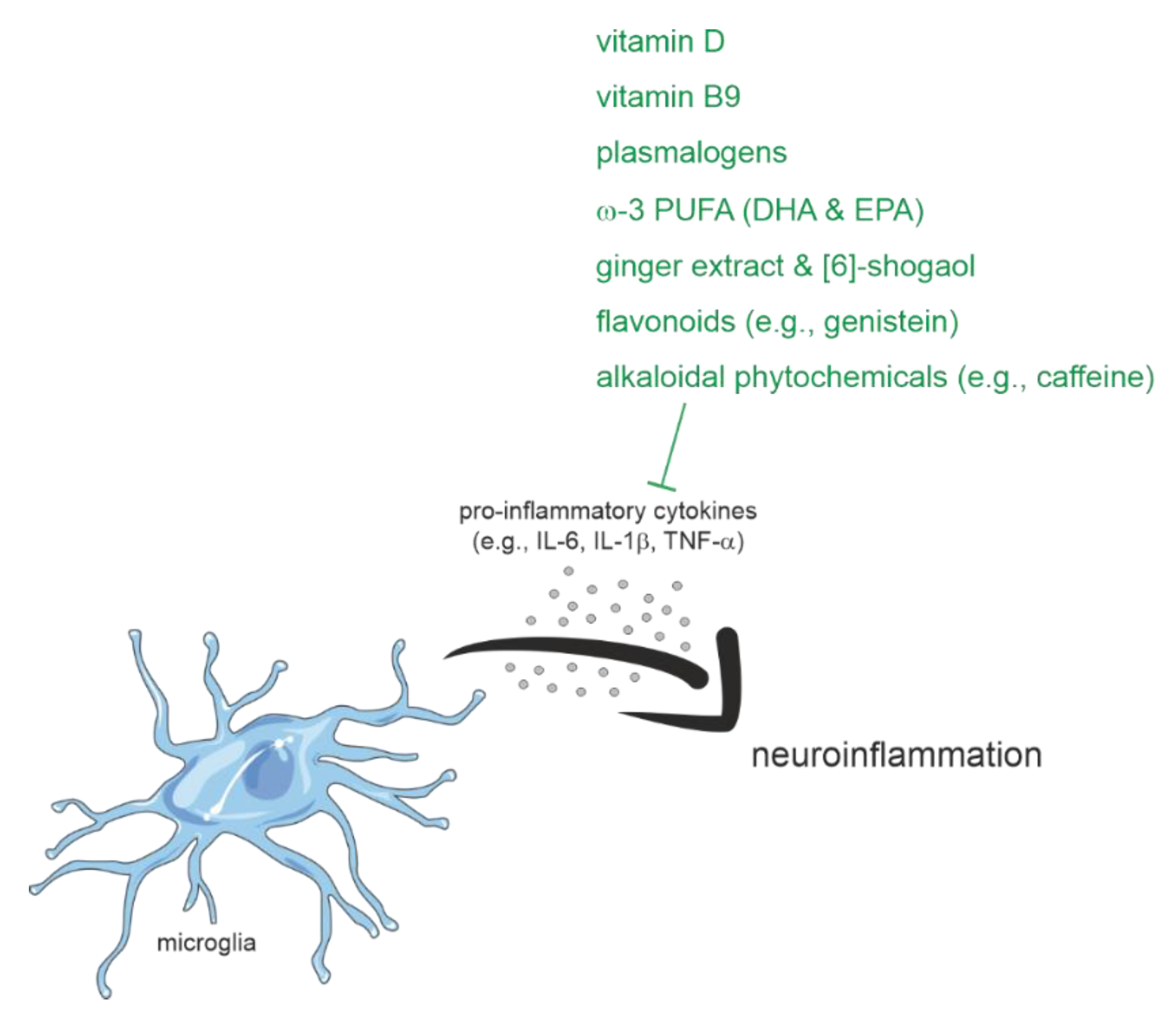
2.1.4. Inflammation
2.1.5. Multicomponent Nutritional Interventions
2.2. Physical (in)Activity and Alzheimer’s Disease
2.3. Cognition-Oriented Treatments
2.3.1. Speech and Language Therapy within the Scope of Cognition-Oriented Treatments
2.3.2. Effects on Language and Communication after Cognitive Stimulation
2.3.3. Effects on Language and Communication after Cognitive Training and Cognitive Rehabilitation
2.3.4. Dyadic Intervention Approaches and Communication Success
2.3.5. Non-Pharmacological Interventions and Functional Outcomes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Language and Communication | Cognition | |||
|---|---|---|---|---|
| Impairments | Resources | Impairments | Resources | |
| Mild Stage | Reception - Comprehension of abstract language/complex conversation | - Comprehension of simple sentences - Reading comprehension | - Declarative/explicit memory - Inconsistent problems with orientation - Visuospatial skills - Divided/selective attention - Inconsistent problems with instrumental activities of daily living (IADL) | - Nondeclarative/sensory memory - Awareness of language and memory lapses - Sustained attention - Concentration |
| Production - Word retrieval for names, objects, locations - Semantic paraphasias - Irrelevant/vague comments - Reduced content/error repairs in discourse | - Grammatical correct sentences - Phonology/articulation - Oral reading/writing | |||
| Moderate Stage | Reception - Comprehending complexinstructions/tasks - Reading comprehension | - Reading comprehension for familiar words/phrases | - Declarative memory - Orientation - Executive functions - Attention in all domains - Visuospatial skills | - Nondeclarative/sensory memory |
| Production - Word retrieval - Increase in circumlocutions/word repetition/paraphasias - Disrupted conversation flow - Decline in sentence length/ grammatical complexity/propositional content - Increase in the use of pronouns/vague terms - Lack of content in conversation - Pragmatic abilities: maintain topics of conversation/knowledge of conversation perspectives/irrelevant content/inaccurate utterances | - Phonology - Syntax - Oral reading of simple texts - Nonverbal conversation | |||
| Severe Stage | Reception - Auditory and reading comprehension | - Comprehension/interpretation of emotional state via facial expression/gestures/eye contact/prosody/voice tone | - Memory - Attention - Fluctuated alertness | - Affective response to sensory stimuli/music - Basic needs for attention/communication/touch present |
| Production - Production of single words/short phrases - Often inappropriate verbal/vocal production - Repetitive vocal/physical behavior - Mutism in the end stage | - Communication via facial expressions/gestures/eye contact | |||
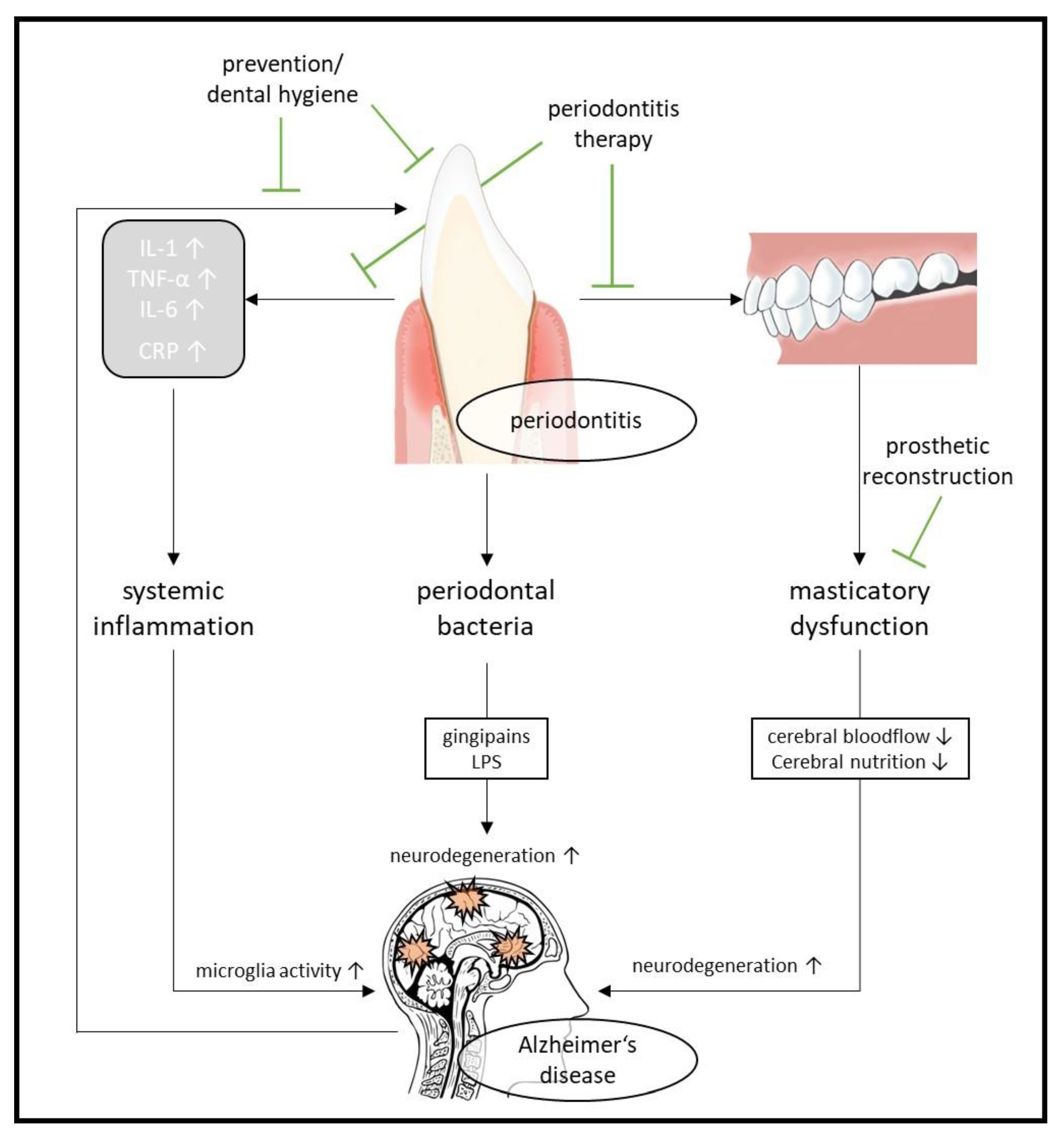
2.4. Oral Health and Alzheimer’s Disease
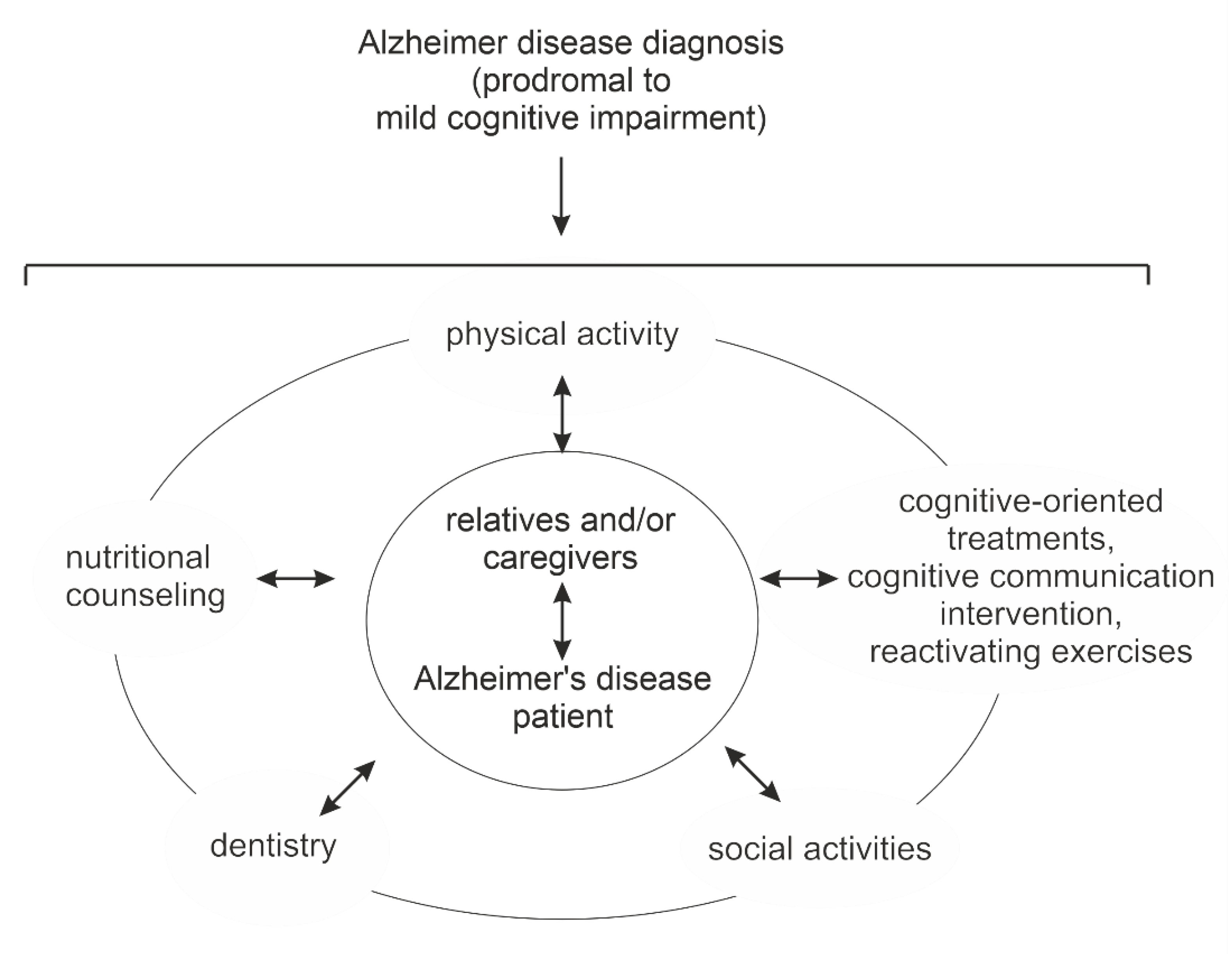
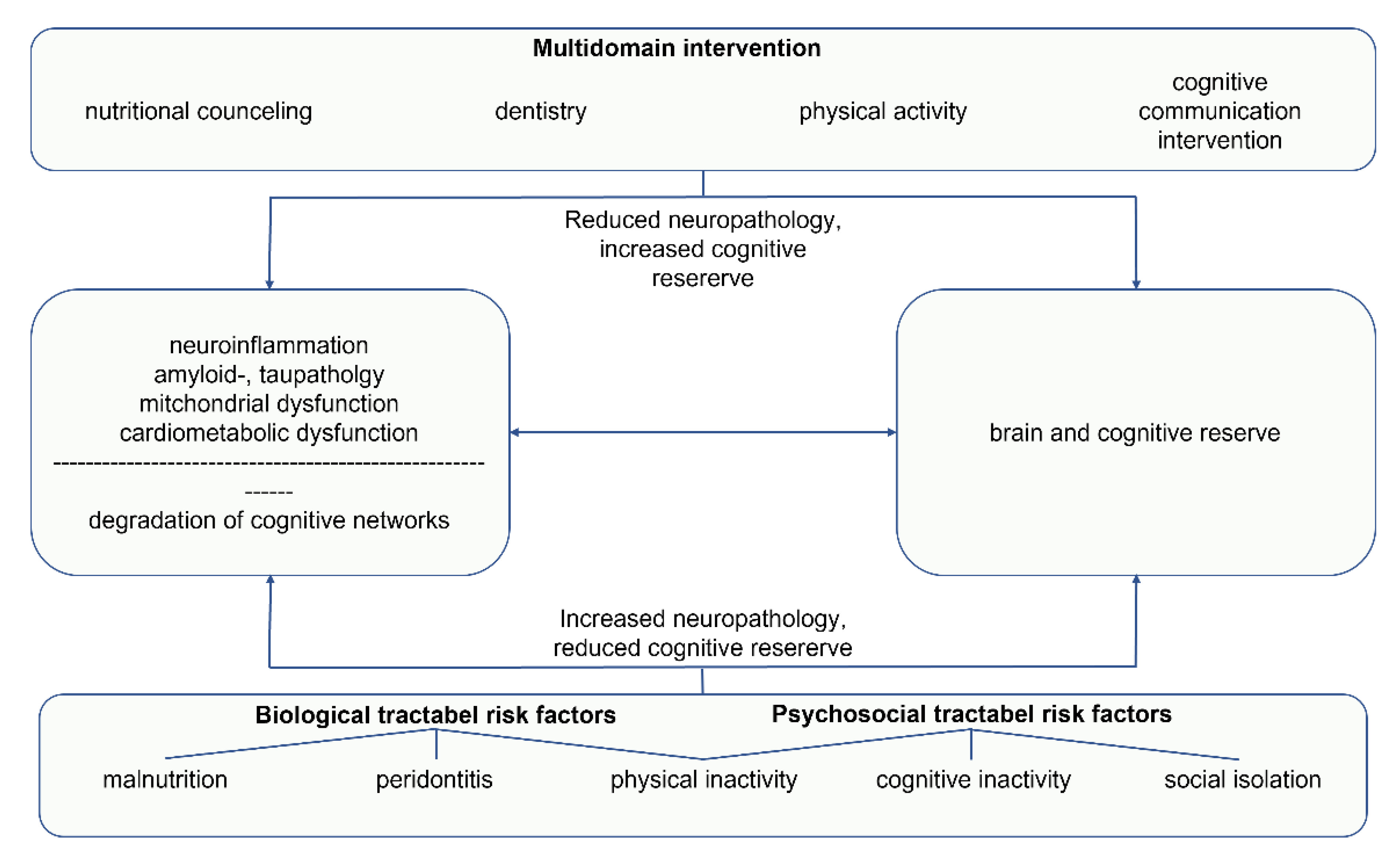
3. Multidomain Interventions
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clare, L.; Teale, J.C.; Toms, G.; Kudlicka, A.; Evans, I.; Abrahams, S.; Goldstein, L.H.; Hindle, J.V.; Ho, A.K.; Jahanshahi, M.; et al. Cognitive rehabilitation, self-management, psychotherapeutic and caregiver support interventions in progressive neurodegenerative conditions: A scoping review. NeuroRehabilitation 2018, 43, 443–471. [Google Scholar] [CrossRef] [PubMed]
- Kudlicka, A.; Martyr, A.; Bahar-Fuchs, A.; Woods, B.; Clare, L. Cognitive rehabilitation for people with mild to moderate dementia. Cochrane Database Syst. Rev. 2019, 28, 707. [Google Scholar] [CrossRef]
- Selkoe, D.J. Cell biology of protein misfolding: The examples of Alzheimer’s and parkinson’s diseases. Nat. Cell Biol. 2004, 6, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimer’s Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef]
- Kumar, A.; Sidhu, J.; Goyal, A.; Tsao, J.W. Alzheimer disease. In Statpearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Breijyeh, Z.; Karaman, R. Comprehensive review on Alzheimer’s disease: Causes and treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Bahar-Fuchs, A.; Martyr, A.; Goh, A.M.; Sabates, J.; Clare, L. Cognitive training for people with mild to moderate dementia. Cochrane Database Syst. Rev. 2019, 3, CD013069. [Google Scholar] [CrossRef]
- Wu, J.; Ma, Y.; Ren, Z. Rehabilitative effects of virtual reality technology for mild cognitive impairment: A systematic review with meta-analysis. Front. Psychol. 2020, 11, 1811. [Google Scholar] [CrossRef]
- Mueller, K.D.; Hermann, B.; Mecollari, J.; Turkstra, L.S. Connected speech and language in mild cognitive impairment and Alzheimer’s disease: A review of picture description tasks. J. Clin. Exp. Neuropsychol. 2018, 40, 917–939. [Google Scholar] [CrossRef]
- Wattmo, C.; Minthon, L.; Wallin, A.K. Mild versus moderate stages of Alzheimer’s disease: Three-year outcomes in a routine clinical setting of cholinesterase inhibitor therapy. Alzheimer’s. Res. 2016, 8, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eldufani, J.; Blaise, G. The role of acetylcholinesterase inhibitors such as neostigmine and rivastigmine on chronic pain and cognitive function in aging: A review of recent clinical applications. Alzheimer’s Dement. 2019, 5, 175–183. [Google Scholar] [CrossRef]
- Singh, R.; Sadiq, N.M. Cholinesterase inhibitors. In Statpearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Chen, J.H. Microscopic features for the identification of chinese drugs in ranunculaceus. Zhong Yao Tong Bao 1985, 10, 15–17. [Google Scholar]
- Khoury, R.; Grysman, N.; Gold, J.; Patel, K.; Grossberg, G.T. The role of 5 ht6-receptor antagonists in Alzheimer’s disease: An update. Expert Opin. Investig. Drugs 2018, 27, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Reddy, P.H. Role of glutamate and nmda receptors in Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 report of the lancet commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Dioguardi, M.; Crincoli, V.; Laino, L.; Alovisi, M.; Sovereto, D.; Mastrangelo, F.; Lo Russo, L.; Lo Muzio, L. The role of periodontitis and periodontal bacteria in the onset and progression of Alzheimer’s disease: A systematic review. J. Clin. Med. 2020, 9, 495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaliamoorthy, S.; Nagarajan, M.; Sethuraman, V.; Jayavel, K.; Lakshmanan, V.; Palla, S. Association of Alzheimer’s disease and periodontitis—A systematic review and meta-analysis of evidence from observational studies. Med. Pharm. Rep. 2022, 95, 144–151. [Google Scholar] [CrossRef]
- Parra-Torres, V.; Melgar-Rodríguez, S.; Muñoz-Manríquez, C.; Sanhueza, B.; Cafferata, E.A.; Paula-Lima, A.C.; Díaz-Zúñiga, J. Periodontal bacteria in the brain-implication for Alzheimer’s disease: A systematic review. Oral Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Spector, A.; Orrell, M. Using a biopsychosocial model of dementia as a tool to guide clinical practice. Int. Psychogeriatr. 2010, 22, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Revolta, C.; Orrell, M.; Spector, A. The biopsychosocial (bps) model of dementia as a tool for clinical practice. A pilot study. Int. Psychogeriatr. 2016, 28, 1079–1089. [Google Scholar] [CrossRef]
- Clare, L.; Linden, D.E.J.; Woods, R.T.; Whitaker, R.; Evans, S.J.; Parkinson, C.H.; van Paasschen, J.; Nelis, S.M.; Hoare, Z.; Yuen, K.S.L.; et al. Goal-oriented cognitive rehabilitation for people with early-stage Alzheimer disease: A single-blind randomized controlled trial of clinical efficacy. Am. J. Geriatr. Psychiatry Off. J. Am. Assoc. Geriatr. Psychiatry 2010, 18, 928–939. [Google Scholar] [CrossRef]
- Clare, L.; Kudlicka, A.; Oyebode, J.R.; Jones, R.W.; Bayer, A.; Leroi, I.; Kopelman, M.; James, I.A.; Culverwell, A.; Pool, J.; et al. Goal-oriented cognitive rehabilitation for early-stage Alzheimer’s and related dementias: The great rct. Health Technol. Assess. 2019, 23, 1–242. [Google Scholar] [CrossRef] [PubMed]
- Ninot, G. Psychosocial interventions and dementia. Pract. Guide Fond. Médéric Alzheimer 2021, 1–100. [Google Scholar] [CrossRef]
- Gavelin, H.M.; Lampit, A.; Hallock, H.; Sabatés, J.; Bahar-Fuchs, A. Cognition-oriented treatments for older adults: A systematic overview of systematic reviews. Neuropsychol. Rev. 2020, 30, 167–193. [Google Scholar] [CrossRef] [Green Version]
- Rody, T.; De Amorim, J.A.; De Felice, F.G. The emerging neuroprotective roles of exerkines in Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 965190. [Google Scholar] [CrossRef] [PubMed]
- Yusufov, M.; Weyandt, L.L.; Piryatinsky, I. Alzheimer’s disease and diet: A systematic review. Int. J. Neurosci. 2017, 127, 161–175. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Nam, E.; Lee, H.J.; Savelieff, M.G.; Lim, M.H. Towards an understanding of amyloid-beta oligomers: Characterization, toxicity mechanisms, and inhibitors. Chem. Soc. Rev. 2017, 46, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Dyrks, T.; Weidemann, A.; Multhaup, G.; Salbaum, J.M.; Lemaire, H.G.; Kang, J.; Muller-Hill, B.; Masters, C.L.; Beyreuther, K. Identification, transmembrane orientation and biogenesis of the amyloid a4 precursor of Alzheimer’s disease. EMBO J. 1988, 7, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Wasco, W.; Bupp, K.; Magendantz, M.; Gusella, J.F.; Tanzi, R.E.; Solomon, F. Identification of a mouse brain cdna that encodes a protein related to the Alzheimer disease-associated amyloid beta protein precursor. Proc. Natl. Acad. Sci. USA 1992, 89, 10758–10762. [Google Scholar] [CrossRef] [Green Version]
- Muller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef]
- Roberts, S.B.; Ripellino, J.A.; Ingalls, K.M.; Robakis, N.K.; Felsenstein, K.M. Non-amyloidogenic cleavage of the beta-amyloid precursor protein by an integral membrane metalloendopeptidase. J. Biol. Chem. 1994, 269, 3111–3116. [Google Scholar] [CrossRef]
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The adam metalloproteinases. Mol. Asp. Med. 2008, 29, 258–289. [Google Scholar] [CrossRef] [PubMed]
- Dulin, F.; Leveille, F.; Ortega, J.B.; Mornon, J.P.; Buisson, A.; Callebaut, I.; Colloc’h, N. P3 peptide, a truncated form of a beta devoid of synaptotoxic effect, does not assemble into soluble oligomers. FEBS Lett. 2008, 582, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Sopher, B.L.; Rydel, R.E.; Begley, J.G.; Pham, D.G.; Martin, G.M.; Fox, M.; Mattson, M.P. Increased activity-regulating and neuroprotective efficacy of alpha-secretase-derived secreted amyloid precursor protein conferred by a c-terminal heparin-binding domain. J. Neurochem. 1996, 67, 1882–1896. [Google Scholar] [CrossRef]
- Meziane, H.; Dodart, J.C.; Mathis, C.; Little, S.; Clemens, J.; Paul, S.M.; Ungerer, A. Memory-enhancing effects of secreted forms of the beta-amyloid precursor protein in normal and amnestic mice. Proc. Natl. Acad. Sci. USA 1998, 95, 12683–12688. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A.; et al. The beta-secretase bace1 in Alzheimer’s disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease bace. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Jing, T.; Wang, X.; Yao, D. Beta-secretase/bace1 promotes app endocytosis and processing in the endosomes and on cell membrane. Neurosci. Lett. 2018, 685, 63–67. [Google Scholar] [CrossRef]
- Gouras, G.K.; Xu, H.; Jovanovic, J.N.; Buxbaum, J.D.; Wang, R.; Greengard, P.; Relkin, N.R.; Gandy, S. Generation and regulation of beta-amyloid peptide variants by neurons. J. Neurochem. 1998, 71, 1920–1925. [Google Scholar] [CrossRef]
- Wang, R.; Sweeney, D.; Gandy, S.E.; Sisodia, S.S. The profile of soluble amyloid beta protein in cultured cell media. Detection and quantification of amyloid beta protein and variants by immunoprecipitation-mass spectrometry. J. Biol. Chem. 1996, 271, 31894–31902. [Google Scholar] [CrossRef] [Green Version]
- Schieb, H.; Kratzin, H.; Jahn, O.; Mobius, W.; Rabe, S.; Staufenbiel, M.; Wiltfang, J.; Klafki, H.W. Beta-amyloid peptide variants in brains and cerebrospinal fluid from amyloid precursor protein (app) transgenic mice: Comparison with human Alzheimer amyloid. J. Biol. Chem. 2011, 286, 33747–33758. [Google Scholar] [CrossRef]
- Haass, C. Take five--bace and the gamma-secretase quartet conduct Alzheimer’s amyloid beta-peptide generation. EMBO J. 2004, 23, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Riddell, D.R.; Christie, G.; Hussain, I.; Dingwall, C. Compartmentalization of beta-secretase (asp2) into low-buoyant density, noncaveolar lipid rafts. Curr. Biol. 2001, 11, 1288–1293. [Google Scholar] [CrossRef] [Green Version]
- Vetrivel, K.S.; Cheng, H.; Kim, S.H.; Chen, Y.; Barnes, N.Y.; Parent, A.T.; Sisodia, S.S.; Thinakaran, G. Spatial segregation of gamma-secretase and substrates in distinct membrane domains. J. Biol. Chem. 2005, 280, 25892–25900. [Google Scholar] [CrossRef] [Green Version]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundke-Iqbal, I.; Iqbal, K.; Quinlan, M.; Tung, Y.C.; Zaidi, M.S.; Wisniewski, H.M. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 1986, 261, 6084–6089. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Cairns, N.J.; Crowther, R.A. Tau proteins of Alzheimer paired helical filaments: Abnormal phosphorylation of all six brain isoforms. Neuron 1992, 8, 159–168. [Google Scholar] [CrossRef]
- Binder, L.I.; Frankfurter, A.; Rebhun, L.I. The distribution of tau in the mammalian central nervous system. J. Cell Biol. 1985, 101, 1371–1378. [Google Scholar] [CrossRef] [Green Version]
- Butner, K.A.; Kirschner, M.W. Tau protein binds to microtubules through a flexible array of distributed weak sites. J. Cell Biol. 1991, 115, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Lindwall, G.; Cole, R.D. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J. Biol. Chem. 1984, 259, 5301–5305. [Google Scholar] [CrossRef]
- Alonso, A.C.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 5562–5566. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.L.; Yardin, C.; Terro, F. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res. Rev. 2013, 12, 289–309. [Google Scholar] [CrossRef]
- Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Contributions of protein phosphatases pp1, pp2a, pp2b and pp5 to the regulation of tau phosphorylation. Eur. J. Neurosci. 2005, 22, 1942–1950. [Google Scholar] [CrossRef]
- Kins, S.; Kurosinski, P.; Nitsch, R.M.; Gotz, J. Activation of the erk and jnk signaling pathways caused by neuron-specific inhibition of pp2a in transgenic mice. Am. J. Pathol. 2003, 163, 833–843. [Google Scholar] [CrossRef] [Green Version]
- Vogelsberg-Ragaglia, V.; Schuck, T.; Trojanowski, J.Q.; Lee, V.M. Pp2a mrna expression is quantitatively decreased in Alzheimer’s disease hippocampus. Exp. Neurol. 2001, 168, 402–412. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, P.; Jansen-West, K.; Beccard, A.; Ceballos-Diaz, C.; Levites, Y.; Verbeeck, C.; Zubair, A.C.; Dickson, D.; Golde, T.E.; Das, P. Massive gliosis induced by interleukin-6 suppresses abeta deposition in vivo: Evidence against inflammation as a driving force for amyloid deposition. FASEB J. 2010, 24, 548–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaftel, S.S.; Kyrkanides, S.; Olschowka, J.A.; Miller, J.N.; Johnson, R.E.; O’Banion, M.K. Sustained hippocampal il-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J. Clin. Investig. 2007, 117, 1595–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grammas, P. Neurovascular dysfunction, inflammation and endothelial activation: Implications for the pathogenesis of Alzheimer’s disease. J. Neuroinflammation 2011, 8, 26. [Google Scholar] [CrossRef] [Green Version]
- Meraz-Rios, M.A.; Toral-Rios, D.; Franco-Bocanegra, D.; Villeda-Hernandez, J.; Campos-Pena, V. Inflammatory process in Alzheimer’s disease. Front. Integr. Neurosci. 2013, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- Goldgaber, D.; Harris, H.W.; Hla, T.; Maciag, T.; Donnelly, R.J.; Jacobsen, J.S.; Vitek, M.P.; Gajdusek, D.C. Interleukin 1 regulates synthesis of amyloid beta-protein precursor mrna in human endothelial cells. Proc. Natl. Acad. Sci. USA 1989, 86, 7606–7610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintanilla, R.A.; Orellana, D.I.; Gonzalez-Billault, C.; Maccioni, R.B. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp. Cell Res. 2004, 295, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Redox proteomics identification of oxidatively modified proteins in Alzheimer’s disease brain and in vivo and in vitro models of ad centered around abeta(1-42). J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2006, 833, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Pratico, D.; Clark, C.M.; Liun, F.; Rokach, J.; Lee, V.Y.; Trojanowski, J.Q. Increase of brain oxidative stress in mild cognitive impairment: A possible predictor of Alzheimer disease. Arch. Neurol. 2002, 59, 972–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, J.N.; Schmitt, F.A.; Scheff, S.W.; Ding, Q.; Chen, Q.; Butterfield, D.A.; Markesbery, W.R. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology 2005, 64, 1152–1156. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Sheu, K.F.; Blass, J.P. Abnormalities of mitochondrial enzymes in Alzheimer disease. J. Neural Transm. 1998, 105, 855–870. [Google Scholar] [CrossRef]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharm. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Bartochowski, Z.; Conway, J.; Wallach, Y.; Chakkamparambil, B.; Alakkassery, S.; Grossberg, G.T. Dietary interventions to prevent or delay Alzheimer’s disease: What the evidence shows. Curr. Nutr. Rep. 2020, 9, 210–225. [Google Scholar] [CrossRef]
- Monacelli, F.; Acquarone, E.; Giannotti, C.; Borghi, R.; Nencioni, A. Vitamin c, aging and Alzheimer’s disease. Nutrients 2017, 9, 670. [Google Scholar] [CrossRef] [PubMed]
- Roman, G.C.; Jackson, R.E.; Gadhia, R.; Roman, A.N.; Reis, J. Mediterranean diet: The role of long-chain omega-3 fatty acids in fish; polyphenols in fruits, vegetables, cereals, coffee, tea, cacao and wine; probiotics and vitamins in prevention of stroke, age-related cognitive decline, and Alzheimer disease. Rev. Neurol. 2019, 175, 724–741. [Google Scholar] [CrossRef] [PubMed]
- McGrattan, A.M.; McGuinness, B.; McKinley, M.C.; Kee, F.; Passmore, P.; Woodside, J.V.; McEvoy, C.T. Diet and inflammation in cognitive ageing and Alzheimer’s disease. Curr. Nutr. Rep. 2019, 8, 53–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastry, P.S. Lipids of nervous tissue: Composition and metabolism. Prog. Lipid Res. 1985, 24, 69–176. [Google Scholar] [CrossRef]
- Naudi, A.; Cabre, R.; Jove, M.; Ayala, V.; Gonzalo, H.; Portero-Otin, M.; Ferrer, I.; Pamplona, R. Lipidomics of human brain aging and Alzheimer’s disease pathology. Int. Rev. Neurobiol. 2015, 122, 133–189. [Google Scholar]
- Pararasa, C.; Ikwuobe, J.; Shigdar, S.; Boukouvalas, A.; Nabney, I.T.; Brown, J.E.; Devitt, A.; Bailey, C.J.; Bennett, S.J.; Griffiths, H.R. Age-associated changes in long-chain fatty acid profile during healthy aging promote pro-inflammatory monocyte polarization via ppargamma. Aging Cell 2016, 15, 128–139. [Google Scholar] [CrossRef]
- Schonfeld, P.; Reiser, G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow Metab. 2013, 33, 1493–1499. [Google Scholar] [CrossRef] [Green Version]
- Grimm, M.O.; Mett, J.; Hartmann, T. The impact of vitamin e and other fat-soluble vitamins on Alzheimer s disease. Int. J. Mol. Sci. 2016, 17, 1785. [Google Scholar] [CrossRef] [Green Version]
- Lauer, A.A.; Grimm, H.S.; Apel, B.; Golobrodska, N.; Kruse, L.; Ratanski, E.; Schulten, N.; Schwarze, L.; Slawik, T.; Sperlich, S.; et al. Mechanistic link between vitamin b12 and Alzheimer’s disease. Biomolecules 2022, 12, 129. [Google Scholar] [CrossRef]
- Naudi, A.; Cabre, R.; Dominguez-Gonzalez, M.; Ayala, V.; Jove, M.; Mota-Martorell, N.; Pinol-Ripoll, G.; Gil-Villar, M.P.; Rue, M.; Portero-Otin, M.; et al. Region-specific vulnerability to lipid peroxidation and evidence of neuronal mechanisms for polyunsaturated fatty acid biosynthesis in the healthy adult human central nervous system. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 485–495. [Google Scholar] [CrossRef]
- Sahlin, C.; Pettersson, F.E.; Nilsson, L.N.; Lannfelt, L.; Johansson, A.S. Docosahexaenoic acid stimulates non-amyloidogenic app processing resulting in reduced abeta levels in cellular models of Alzheimer’s disease. Eur. J. Neurosci. 2007, 26, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Kuchenbecker, J.; Grosgen, S.; Burg, V.K.; Hundsdorfer, B.; Rothhaar, T.L.; Friess, P.; de Wilde, M.C.; Broersen, L.M.; Penke, B.; et al. Docosahexaenoic acid reduces amyloid beta production via multiple pleiotropic mechanisms. J. Biol. Chem. 2011, 286, 14028–14039. [Google Scholar] [CrossRef] [PubMed]
- Eckert, G.P.; Chang, S.; Eckmann, J.; Copanaki, E.; Hagl, S.; Hener, U.; Muller, W.E.; Kogel, D. Liposome-incorporated dha increases neuronal survival by enhancing non-amyloidogenic app processing. Biochim. Biophys. Acta 2011, 1808, 236–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, M.O.; Haupenthal, V.J.; Mett, J.; Stahlmann, C.P.; Blumel, T.; Mylonas, N.T.; Endres, K.; Grimm, H.S.; Hartmann, T. Oxidized docosahexaenoic acid species and lipid peroxidation products increase amyloidogenic amyloid precursor protein processing. Neurodegener. Dis. 2016, 16, 44–54. [Google Scholar] [CrossRef]
- Yang, X.; Sheng, W.; Sun, G.Y.; Lee, J.C. Effects of fatty acid unsaturation numbers on membrane fluidity and alpha-secretase-dependent amyloid precursor protein processing. Neurochem. Int. 2011, 58, 321–329. [Google Scholar] [CrossRef] [Green Version]
- Amtul, Z.; Westaway, D.; Cechetto, D.F.; Rozmahel, R.F. Oleic acid ameliorates amyloidosis in cellular and mouse models of Alzheimer’s disease. Brain Pathol. 2011, 21, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Haupenthal, V.J.; Rothhaar, T.L.; Zimmer, V.C.; Grosgen, S.; Hundsdorfer, B.; Lehmann, J.; Grimm, H.S.; Hartmann, T. Effect of different phospholipids on alpha-secretase activity in the non-amyloidogenic pathway of Alzheimer’s disease. Int. J. Mol. Sci. 2013, 14, 5879–5898. [Google Scholar] [CrossRef] [Green Version]
- Grimm, M.O.W.; Thiel, A.; Lauer, A.A.; Winkler, J.; Lehmann, J.; Regner, L.; Nelke, C.; Janitschke, D.; Benoist, C.; Streidenberger, O.; et al. Vitamin d and its analogues decrease amyloid-beta (abeta) formation and increase abeta-degradation. Int. J. Mol. Sci. 2017, 18, 2764. [Google Scholar] [CrossRef] [Green Version]
- Emken, E.A. Nutrition and biochemistry of trans and positional fatty acid isomers in hydrogenated oils. Annu. Rev. Nutr. 1984, 4, 339–376. [Google Scholar] [CrossRef]
- Grimm, M.O.; Rothhaar, T.L.; Grosgen, S.; Burg, V.K.; Hundsdorfer, B.; Haupenthal, V.J.; Friess, P.; Kins, S.; Grimm, H.S.; Hartmann, T. Trans fatty acids enhance amyloidogenic processing of the Alzheimer amyloid precursor protein (app). J. Nutr. Biochem. 2012, 23, 1214–1223. [Google Scholar] [CrossRef]
- Phivilay, A.; Julien, C.; Tremblay, C.; Berthiaume, L.; Julien, P.; Giguere, Y.; Calon, F. High dietary consumption of trans fatty acids decreases brain docosahexaenoic acid but does not alter amyloid-beta and tau pathologies in the 3xTg-ad model of Alzheimer’s disease. Neuroscience 2009, 159, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Obregon, D.F.; Rezai-Zadeh, K.; Bai, Y.; Sun, N.; Hou, H.; Ehrhart, J.; Zeng, J.; Mori, T.; Arendash, G.W.; Shytle, D.; et al. Adam10 activation is required for green tea (-)-epigallocatechin-3-gallate-induced alpha-secretase cleavage of amyloid precursor protein. J. Biol. Chem. 2006, 281, 16419–16427. [Google Scholar] [CrossRef] [PubMed]
- Rezai-Zadeh, K.; Shytle, D.; Sun, N.; Mori, T.; Hou, H.; Jeanniton, D.; Ehrhart, J.; Townsend, K.; Zeng, J.; Morgan, D.; et al. Green tea epigallocatechin-3-gallate (egcg) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in Alzheimer transgenic mice. J. Neurosci. 2005, 25, 8807–8814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levites, Y.; Amit, T.; Mandel, S.; Youdim, M.B. Neuroprotection and neurorescue against abeta toxicity and pkc-dependent release of nonamyloidogenic soluble precursor protein by green tea polyphenol (-)-epigallocatechin-3-gallate. FASEB J. 2003, 17, 952–954. [Google Scholar] [CrossRef] [PubMed]
- Ettcheto, M.; Cano, A.; Manzine, P.R.; Busquets, O.; Verdaguer, E.; Castro-Torres, R.D.; Garcia, M.L.; Beas-Zarate, C.; Olloquequi, J.; Auladell, C.; et al. Epigallocatechin-3-gallate (egcg) improves cognitive deficits aggravated by an obesogenic diet through modulation of unfolded protein response in appswe/ps1de9 mice. Mol. Neurobiol. 2020, 57, 1814–1827. [Google Scholar] [CrossRef]
- Wang, Z.; Huang, X.; Zhao, P.; Zhao, L.; Wang, Z.Y. Catalpol inhibits amyloid-beta generation through promoting alpha-cleavage of app in swedish mutant app overexpressed n2a cells. Front. Aging Neurosci. 2018, 10, 66. [Google Scholar] [CrossRef] [PubMed]
- Janitschke, D.; Nelke, C.; Lauer, A.A.; Regner, L.; Winkler, J.; Thiel, A.; Grimm, H.S.; Hartmann, T.; Grimm, M.O.W. Effect of caffeine and other methylxanthines on abeta-homeostasis in sh-sy5y cells. Biomolecules 2019, 9, 689. [Google Scholar] [CrossRef] [Green Version]
- Hooijmans, C.R.; Van der Zee, C.E.; Dederen, P.J.; Brouwer, K.M.; Reijmer, Y.D.; van Groen, T.; Broersen, L.M.; Lutjohann, D.; Heerschap, A.; Kiliaan, A.J. Dha and cholesterol containing diets influence Alzheimer-like pathology, cognition and cerebral vasculature in appswe/ps1de9 mice. Neurobiol. Dis. 2009, 33, 482–498. [Google Scholar] [CrossRef]
- Hashimoto, M.; Hossain, S.; Agdul, H.; Shido, O. Docosahexaenoic acid-induced amelioration on impairment of memory learning in amyloid beta-infused rats relates to the decreases of amyloid beta and cholesterol levels in detergent-insoluble membrane fractions. Biochim. Biophys. Acta 2005, 1738, 91–98. [Google Scholar] [CrossRef]
- Stillwell, W.; Shaikh, S.R.; Zerouga, M.; Siddiqui, R.; Wassall, S.R. Docosahexaenoic acid affects cell signaling by altering lipid rafts. Reprod. Nutr. Dev. 2005, 45, 559–579. [Google Scholar] [CrossRef] [Green Version]
- Cole, G.M.; Frautschy, S.A. Docosahexaenoic acid protects from amyloid and dendritic pathology in an Alzheimer’s disease mouse model. Nutr. Health 2006, 18, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Green, K.N.; Martinez-Coria, H.; Khashwji, H.; Hall, E.B.; Yurko-Mauro, K.A.; Ellis, L.; LaFerla, F.M. Dietary docosahexaenoic acid and docosapentaenoic acid ameliorate amyloid-beta and tau pathology via a mechanism involving presenilin 1 levels. J. Neurosci. 2007, 27, 4385–4395. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.P.; Calon, F.; Morihara, T.; Yang, F.; Teter, B.; Ubeda, O.; Salem, N., Jr.; Frautschy, S.A.; Cole, G.M. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J. Neurosci. 2005, 25, 3032–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, S.E.; Berg, B.M.; Moore, K.A.; He, B.; Counts, S.E.; Fritz, J.J.; Hu, Y.S.; Lazarov, O.; Lah, J.J.; Mufson, E.J. Dha diet reduces ad pathology in young appswe/ps1 delta e9 transgenic mice: Possible gender effects. J. Neurosci. Res. 2010, 88, 1026–1040. [Google Scholar] [PubMed] [Green Version]
- Amtul, Z.; Uhrig, M.; Wang, L.; Rozmahel, R.F.; Beyreuther, K. Detrimental effects of arachidonic acid and its metabolites in cellular and mouse models of Alzheimer’s disease: Structural insight. Neurobiol. Aging 2012, 33, 831.e21–831.e31. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, L.; Conde-Knape, K.; Beher, D.; Shearman, M.S.; Shachter, N.S. Fatty acids increase presenilin-1 levels and [gamma]-secretase activity in pswt-1 cells. J. Lipid Res. 2004, 45, 2368–2376. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q. Natural forms of vitamin e: Metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic. Biol. Med. 2014, 72, 76–90. [Google Scholar] [CrossRef] [Green Version]
- Browne, D.; McGuinness, B.; Woodside, J.V.; McKay, G.J. Vitamin e and Alzheimer’s disease: What do we know so far? Clin. Interv. Aging 2019, 14, 1303–1317. [Google Scholar] [CrossRef] [Green Version]
- Sung, S.; Yao, Y.; Uryu, K.; Yang, H.; Lee, V.M.; Trojanowski, J.Q.; Pratico, D. Early vitamin e supplementation in young but not aged mice reduces abeta levels and amyloid deposition in a transgenic model of Alzheimer’s disease. FASEB J. 2004, 18, 323–325. [Google Scholar] [CrossRef]
- Wang, S.W.; Yang, S.G.; Liu, W.; Zhang, Y.X.; Xu, P.X.; Wang, T.; Ling, T.J.; Liu, R.T. Alpha-tocopherol quinine ameliorates spatial memory deficits by reducing beta-amyloid oligomers, neuroinflammation and oxidative stress in transgenic mice with Alzheimer’s disease. Behav. Brain Res. 2016, 296, 109–117. [Google Scholar] [CrossRef]
- Pilleron, S.; Desport, J.C.; Jesus, P.; Mbelesso, P.; Ndamba-Bandzouzi, B.; Dartigues, J.F.; Clement, J.P.; Preux, P.M.; Guerchet, M. Diet, alcohol consumption and cognitive disorders in central africa: A study from the epidemca program. J. Nutr. Health Aging 2015, 19, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Pedrini, S.; Chatterjee, P.; Hone, E.; Martins, R.N. High-density lipoprotein-related cholesterol metabolism in Alzheimer’s disease. J. Neurochem. 2021, 159, 343–377. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.; Lee, H.; Cho, S.; Seo, J. Apoe4-induced cholesterol dysregulation and its brain cell type-specific implications in the pathogenesis of Alzheimer’s disease. Mol. Cells 2019, 42, 739–746. [Google Scholar] [PubMed]
- Loera-Valencia, R.; Goikolea, J.; Parrado-Fernandez, C.; Merino-Serrais, P.; Maioli, S. Alterations in cholesterol metabolism as a risk factor for developing Alzheimer’s disease: Potential novel targets for treatment. J. Steroid. Biochem. Mol. Biol. 2019, 190, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kulas, J.A.; Wang, C.; Holtzman, D.M.; Ferris, H.A.; Hansen, S.B. Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. Proc. Natl. Acad. Sci. USA 2021, 118, e2102191118. [Google Scholar] [CrossRef]
- Fassbender, K.; Simons, M.; Bergmann, C.; Stroick, M.; Lutjohann, D.; Keller, P.; Runz, H.; Kuhl, S.; Bertsch, T.; von Bergmann, K.; et al. Simvastatin strongly reduces levels of Alzheimer’s disease beta -amyloid peptides abeta 42 and abeta 40 in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 5856–5861. [Google Scholar] [CrossRef] [Green Version]
- Grimm, M.O.; Grimm, H.S.; Tomic, I.; Beyreuther, K.; Hartmann, T.; Bergmann, C. Independent inhibition of Alzheimer disease beta- and gamma-secretase cleavage by lowered cholesterol levels. J. Biol. Chem. 2008, 283, 11302–11311. [Google Scholar] [CrossRef] [Green Version]
- Kao, Y.C.; Ho, P.C.; Tu, Y.K.; Jou, I.M.; Tsai, K.J. Lipids and Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 1505. [Google Scholar] [CrossRef]
- Briones, T.L.; Darwish, H. Vitamin d mitigates age-related cognitive decline through the modulation of pro-inflammatory state and decrease in amyloid burden. J. Neuroinflammation 2012, 9, 244. [Google Scholar] [CrossRef] [Green Version]
- Grimm, M.O.; Lehmann, J.; Mett, J.; Zimmer, V.C.; Grosgen, S.; Stahlmann, C.P.; Hundsdorfer, B.; Haupenthal, V.J.; Rothhaar, T.L.; Herr, C.; et al. Impact of vitamin d on amyloid precursor protein processing and amyloid-beta peptide degradation in Alzheimer’s disease. Neurodegener. Dis. 2014, 13, 75–81. [Google Scholar] [CrossRef]
- Wang, L.; Hara, K.; Van Baaren, J.M.; Price, J.C.; Beecham, G.W.; Gallins, P.J.; Whitehead, P.L.; Wang, G.; Lu, C.; Slifer, M.A.; et al. Vitamin d receptor and Alzheimer’s disease: A genetic and functional study. Neurobiol. Aging 2012, 33, 1844.e1841–1849. [Google Scholar] [CrossRef]
- Yu, J.; Gattoni-Celli, M.; Zhu, H.; Bhat, N.R.; Sambamurti, K.; Gattoni-Celli, S.; Kindy, M.S. Vitamin d3-enriched diet correlates with a decrease of amyloid plaques in the brain of abetapp transgenic mice. J. Alzheimer’s Dis. 2011, 25, 295–307. [Google Scholar] [CrossRef]
- Lai, R.H.; Hsu, C.C.; Yu, B.H.; Lo, Y.R.; Hsu, Y.Y.; Chen, M.H.; Juang, J.L. Vitamin d supplementation worsens Alzheimer’s progression: Animal model and human cohort studies. Aging Cell 2022, 21, e13670. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, J.M.; Pratico, D. Acceleration of brain amyloidosis in an Alzheimer’s disease mouse model by a folate, vitamin b6 and b12-deficient diet. Exp. Gerontol. 2010, 45, 195–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuso, A.; Nicolia, V.; Cavallaro, R.A.; Ricceri, L.; D’Anselmi, F.; Coluccia, P.; Calamandrei, G.; Scarpa, S. B-vitamin deprivation induces hyperhomocysteinemia and brain s-adenosylhomocysteine, depletes brain s-adenosylmethionine, and enhances ps1 and bace expression and amyloid-beta deposition in mice. Mol. Cell. Neurosci. 2008, 37, 731–746. [Google Scholar] [CrossRef]
- Fuso, A.; Nicolia, V.; Pasqualato, A.; Fiorenza, M.T.; Cavallaro, R.A.; Scarpa, S. Changes in presenilin 1 gene methylation pattern in diet-induced b vitamin deficiency. Neurobiol. Aging 2011, 32, 187–199. [Google Scholar] [CrossRef]
- Wei, W.; Liu, Y.H.; Zhang, C.E.; Wang, Q.; Wei, Z.; Mousseau, D.D.; Wang, J.Z.; Tian, Q.; Liu, G.P. Folate/vitamin-b12 prevents chronic hyperhomocysteinemia-induced tau hyperphosphorylation and memory deficits in aged rats. J. Alzheimer’s Dis. 2011, 27, 639–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Ni, S.; Li, Q.; Wang, J.Z.; Yang, Y. Folate/vitamin b alleviates hyperhomocysteinemia-induced Alzheimer-like pathologies in rat retina. Neurosci. Bull. 2019, 35, 325–335. [Google Scholar] [CrossRef]
- Adaikalakoteswari, A.; Finer, S.; Voyias, P.D.; McCarthy, C.M.; Vatish, M.; Moore, J.; Smart-Halajko, M.; Bawazeer, N.; Al-Daghri, N.M.; McTernan, P.G.; et al. Vitamin b12 insufficiency induces cholesterol biosynthesis by limiting s-adenosylmethionine and modulating the methylation of srebf1 and ldlr genes. Clin. Epigenetics 2015, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Perla-Kajan, J.; Wloczkowska, O.; Ziola-Frankowska, A.; Frankowski, M.; Smith, A.D.; de Jager, C.A.; Refsum, H.; Jakubowski, H. Paraoxonase 1, b vitamins supplementation, and mild cognitive impairment. J. Alzheimer’s Dis. 2021, 81, 1211–1229. [Google Scholar] [CrossRef]
- Zhang, C.; Luo, J.; Yuan, C.; Ding, D. Vitamin b12, b6, or folate and cognitive function in community-dwelling older adults: A systematic review and meta-analysis. J. Alzheimer’s Dis. 2020, 77, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Guo, Y.; Men, J.; Fu, H.; Xu, T. The preventive efficacy of vitamin b supplements on the cognitive decline of elderly adults: A systematic review and meta-analysis. BMC Geriatr. 2021, 21, 367. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Zhou, X.; Li, Q.; Zhao, J.; Song, A.; An, P.; Du, Y.; Xu, W.; Huang, G. Effects of folic acid and vitamin b12, alone and in combination on cognitive function and inflammatory factors in the elderly with mild cognitive impairment: A single-blind experimental design. Curr. Alzheimer Res. 2019, 16, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.Y.; Bae, K.; Seong, Y.H.; Song, K.S. Green tea catechins as a bace1 (beta-secretase) inhibitor. Bioorg. Med. Chem. Lett. 2003, 13, 3905–3908. [Google Scholar] [CrossRef] [PubMed]
- Marambaud, P.; Zhao, H.; Davies, P. Resveratrol promotes clearance of Alzheimer’s disease amyloid-beta peptides. J. Biol. Chem. 2005, 280, 37377–37382. [Google Scholar] [CrossRef] [Green Version]
- Choi, C.W.; Choi, Y.H.; Cha, M.R.; Kim, Y.S.; Yon, G.H.; Hong, K.S.; Park, W.K.; Kim, Y.H.; Ryu, S.Y. In vitro bace-1 inhibitory activity of resveratrol oligomers from the seed extract of paeonia lactiflora. Planta Med. 2011, 77, 374–376. [Google Scholar] [CrossRef]
- Koukoulitsa, C.; Villalonga-Barber, C.; Csonka, R.; Alexi, X.; Leonis, G.; Dellis, D.; Hamelink, E.; Belda, O.; Steele, B.R.; Micha-Screttas, M.; et al. Biological and computational evaluation of resveratrol inhibitors against Alzheimer’s disease. J. Enzym. Inhib. Med. Chem. 2016, 31, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Shi, G.W.; Liang, Z.M.; Sheng, S.Y.; Shi, Y.S.; Peng, L.; Wang, Y.P.; Wang, F.; Zhang, X.M. Resveratrol improves cognition and decreases amyloid plaque formation in tg6799 mice. Mol. Med. Rep. 2019, 19, 3783–3790. [Google Scholar] [CrossRef]
- Zhao, H.F.; Li, N.; Wang, Q.; Cheng, X.J.; Li, X.M.; Liu, T.T. Resveratrol decreases the insoluble abeta1-42 level in hippocampus and protects the integrity of the blood-brain barrier in ad rats. Neuroscience 2015, 310, 641–649. [Google Scholar] [CrossRef]
- Youn, K.; Park, J.H.; Lee, S.; Lee, S.; Lee, J.; Yun, E.Y.; Jeong, W.S.; Jun, M. Bace1 inhibition by genistein: Biological evaluation, kinetic analysis, and molecular docking simulation. J. Med. Food 2018, 21, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Cai, N.; Gu, L.; Yao, L.; Bi, D.; Fang, W.; Lin, Z.; Wu, Y.; Xu, H.; Li, H.; et al. Genipin attenuates tau phosphorylation and abeta levels in cellular models of Alzheimer’s disease. Mol. Neurobiol. 2021, 58, 4134–4144. [Google Scholar] [CrossRef]
- Talia, S.; Benarous, K.; Lamrani, M.; Yousfi, M. Lepidine b from lepidium sativum seeds as multi-functional anti- Alzheimer’s disease agent: In vitro and in silico studies. Curr. Comput. Aided Drug Des. 2021, 17, 360–377. [Google Scholar] [CrossRef]
- Kurochkin, I.V.; Guarnera, E.; Berezovsky, I.N. Insulin-degrading enzyme in the fight against Alzheimer’s disease. Trends Pharm. Sci. 2018, 39, 49–58. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Zhuravin, I.A.; Turner, A.J. Neprilysin expression and functions in development, ageing and disease. Mech. Ageing Dev. 2020, 192, 111363. [Google Scholar] [CrossRef]
- Grimm, M.O.; Mett, J.; Stahlmann, C.P.; Haupenthal, V.J.; Blumel, T.; Stotzel, H.; Grimm, H.S.; Hartmann, T. Eicosapentaenoic acid and docosahexaenoic acid increase the degradation of amyloid-beta by affecting insulin-degrading enzyme. Biochem. Cell Biol. 2016, 94, 534–542. [Google Scholar] [CrossRef]
- Mett, J.; Lauer, A.A.; Janitschke, D.; Griebsch, L.V.; Theiss, E.L.; Grimm, H.S.; Koivisto, H.; Tanila, H.; Hartmann, T.; Grimm, M.O.W. Medium-chain length fatty acids enhance abeta degradation by affecting insulin-degrading enzyme. Cells 2021, 10, 2941. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Stahlmann, C.P.; Mett, J.; Haupenthal, V.J.; Zimmer, V.C.; Lehmann, J.; Hundsdorfer, B.; Endres, K.; Grimm, H.S.; Hartmann, T. Vitamin e: Curse or benefit in Alzheimer’s disease? A systematic investigation of the impact of alpha-, gamma- and delta-tocopherol on ass generation and degradation in neuroblastoma cells. J. Nutr. Health Aging 2015, 19, 646–656. [Google Scholar] [CrossRef]
- Grimm, M.O.; Regner, L.; Mett, J.; Stahlmann, C.P.; Schorr, P.; Nelke, C.; Streidenberger, O.; Stoetzel, H.; Winkler, J.; Zaidan, S.R.; et al. Tocotrienol affects oxidative stress, cholesterol homeostasis and the amyloidogenic pathway in neuroblastoma cells: Consequences for Alzheimer’s disease. Int. J. Mol. Sci. 2016, 17, 1809. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhang, Y.; Deng, X.; Yin, F. Geniposide decreases the level of abeta1-42 in the hippocampus of streptozotocin-induced diabetic rats. Acta Biochim. Biophys. Sin. 2013, 45, 787–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.Z.; Wu, J.; Xiang, S.; Sheng, S.; Jiang, Y.; Yang, Z.; Hua, F. Catalpol preserves neural function and attenuates the pathology of Alzheimer’s disease in mice. Mol. Med. Rep. 2016, 13, 491–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Chen, K.; Lu, Y.; Fang, Z.; Yu, G. Catalpol provides a protective effect on fibrillary abeta1-42 -induced barrier disruption in an in vitro model of the blood-brain barrier. Phytother. Res. 2018, 32, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- El Shatshat, A.; Pham, A.T.; Rao, P.P.N. Interactions of polyunsaturated fatty acids with amyloid peptides abeta40 and abeta42. Arch. Biochem. Biophys. 2019, 663, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.S.; Garlind, A.; Berglind-Dehlin, F.; Karlsson, G.; Edwards, K.; Gellerfors, P.; Ekholm-Pettersson, F.; Palmblad, J.; Lannfelt, L. Docosahexaenoic acid stabilizes soluble amyloid-beta protofibrils and sustains amyloid-beta-induced neurotoxicity in vitro. FEBS J. 2007, 274, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Alam, P.; Siddiqi, M.K.; Chaturvedi, S.K.; Zaman, M.; Khan, R.H. Vitamin b12 offers neuronal cell protection by inhibiting abeta-42 amyloid fibrillation. Int. J. Biol. Macromol. 2017, 99, 477–482. [Google Scholar] [CrossRef]
- Andrade, S.; Loureiro, J.A.; Pereira, M.C. Vitamin b12 inhibits abeta fibrillation and disaggregates preformed fibrils in the presence of synthetic neuronal membranes. ACS Chem. Neurosci. 2021, 12, 2491–2502. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Yamada, M. Vitamin a and Alzheimer’s disease. Geriatr. Gerontol. Int. 2012, 12, 180–188. [Google Scholar] [CrossRef]
- Ono, K.; Yoshiike, Y.; Takashima, A.; Hasegawa, K.; Naiki, H.; Yamada, M. Vitamin a exhibits potent antiamyloidogenic and fibril-destabilizing effects in vitro. Exp. Neurol. 2004, 189, 380–392. [Google Scholar] [CrossRef]
- Takasaki, J.; Ono, K.; Yoshiike, Y.; Hirohata, M.; Ikeda, T.; Morinaga, A.; Takashima, A.; Yamada, M. Vitamin a has anti-oligomerization effects on amyloid-beta in vitro. J. Alzheimer’s Dis. 2011, 27, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Qiao, A.; Wang, Z.; Goodwin, J.S.; Lee, E.S.; Block, M.L.; Allsbrook, M.; McDonald, M.P.; Fan, G.H. Retinoic acid attenuates beta-amyloid deposition and rescues memory deficits in an Alzheimer’s disease transgenic mouse model. J. Neurosci. 2008, 28, 11622–11634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, P.; Chia, S.; Yan, X.; Perni, M.; Habchi, J.; Vendruscolo, M. Vitamin a and vitamin e metabolites comodulate amyloid-b aggregation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ringman, J.M.; Frautschy, S.A.; Cole, G.M.; Masterman, D.L.; Cummings, J.L. A potential role of the curry spice curcumin in Alzheimer’s disease. Curr. Alzheimer Res. 2005, 2, 131–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Curcumin has potent anti-amyloidogenic effects for Alzheimer’s beta-amyloid fibrils in vitro. J. Neurosci. Res. 2004, 75, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chainoglou, E.; Hadjipavlou-Litina, D. Curcumin in health and diseases: Alzheimer’s disease and curcumin analogues, derivatives, and hybrids. Int. J. Mol. Sci. 2020, 21, 1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Aucoin, D.; Ahmed, M.; Ziliox, M.; Van Nostrand, W.E.; Smith, S.O. Capping of abeta42 oligomers by small molecule inhibitors. Biochemistry 2014, 53, 7893–7903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mithu, V.S.; Sarkar, B.; Bhowmik, D.; Das, A.K.; Chandrakesan, M.; Maiti, S.; Madhu, P.K. Curcumin alters the salt bridge-containing turn region in amyloid beta(1-42) aggregates. J. Biol. Chem. 2014, 289, 11122–11131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doytchinova, I.; Atanasova, M.; Salamanova, E.; Ivanov, S.; Dimitrov, I. Curcumin inhibits the primary nucleation of amyloid-beta peptide: A molecular dynamics study. Biomolecules 2020, 10, 1323. [Google Scholar] [CrossRef]
- Garcia-Alloza, M.; Borrelli, L.A.; Rozkalne, A.; Hyman, B.T.; Bacskai, B.J. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J. Neurochem. 2007, 102, 1095–1104. [Google Scholar] [CrossRef]
- Ahmed, R.; VanSchouwen, B.; Jafari, N.; Ni, X.; Ortega, J.; Melacini, G. Molecular mechanism for the (-)-epigallocatechin gallate-induced toxic to nontoxic remodeling of abeta oligomers. J. Am. Chem. Soc. 2017, 139, 13720–13734. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. Egcg redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. [Google Scholar] [CrossRef]
- Zhan, C.; Chen, Y.; Tang, Y.; Wei, G. Green tea extracts egcg and egc display distinct mechanisms in disrupting abeta42 protofibril. ACS Chem. Neurosci. 2020, 11, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhan, C.; Dong, X.; Wei, G. Molecular mechanisms of resveratrol and egcg in the inhibition of abeta42 aggregation and disruption of abeta42 protofibril: Similarities and differences. Phys. Chem. Chem. Phys. 2021, 23, 18843–18854. [Google Scholar] [CrossRef]
- Zhang, S.; Zhu, Q.; Chen, J.Y.; OuYang, D.; Lu, J.H. The pharmacological activity of epigallocatechin-3-gallate (egcg) on Alzheimer’s disease animal model: A systematic review. Phytomedicine 2020, 79, 153316. [Google Scholar] [CrossRef] [PubMed]
- Hirohata, M.; Ono, K.; Takasaki, J.; Takahashi, R.; Ikeda, T.; Morinaga, A.; Yamada, M. Anti-amyloidogenic effects of soybean isoflavones in vitro: Fluorescence spectroscopy demonstrating direct binding to abeta monomers, oligomers and fibrils. Biochim. Biophys. Acta 2012, 1822, 1316–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Smith, J.V.; Paramasivam, V.; Burdick, A.; Curry, K.J.; Buford, J.P.; Khan, I.; Netzer, W.J.; Xu, H.; Butko, P. Inhibition of amyloid-beta aggregation and caspase-3 activation by the ginkgo biloba extract egb761. Proc. Natl. Acad. Sci. USA 2002, 99, 12197–12202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, R.; Li, L.; Yu, H.; Liu, M.; Zhao, W. Melanopsin retinal ganglion cell loss and circadian dysfunction in Alzheimer’s disease (review). Mol. Med. Rep. 2016, 13, 3397–3400. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.E.; Tian, Q.; Wei, W.; Peng, J.H.; Liu, G.P.; Zhou, X.W.; Wang, Q.; Wang, D.W.; Wang, J.Z. Homocysteine induces tau phosphorylation by inactivating protein phosphatase 2a in rat hippocampus. Neurobiol. Aging 2008, 29, 1654–1665. [Google Scholar] [CrossRef]
- Rafiee, S.; Asadollahi, K.; Riazi, G.; Ahmadian, S.; Saboury, A.A. Vitamin b12 inhibits tau fibrillization via binding to cysteine residues of tau. ACS Chem. Neurosci. 2017, 8, 2676–2682. [Google Scholar] [CrossRef]
- Zhu, W.; Zhao, L.; Li, T.; Xu, H.; Ding, Y.; Cui, G. Docosahexaenoic acid ameliorates traumatic brain injury involving jnk-mediated tau phosphorylation signaling. Neurosci. Res. 2020, 157, 44–50. [Google Scholar] [CrossRef]
- Ma, Q.L.; Yang, F.; Rosario, E.R.; Ubeda, O.J.; Beech, W.; Gant, D.J.; Chen, P.P.; Hudspeth, B.; Chen, C.; Zhao, Y.; et al. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-jun n-terminal kinase signaling: Suppression by omega-3 fatty acids and curcumin. J. Neurosci. 2009, 29, 9078–9089. [Google Scholar] [CrossRef] [Green Version]
- Qu, M.H.; Yang, X.; Wang, Y.; Tang, Q.; Han, H.; Wang, J.; Wang, G.D.; Xue, C.; Gao, Z. Docosahexaenoic acid-phosphatidylcholine improves cognitive deficits in an abeta23-35-induced Alzheimer’s disease rat model. Curr. Top. Med. Chem. 2016, 16, 558–564. [Google Scholar] [CrossRef]
- Bie, N.; Li, J.; Li, C.; Lian, R.; Qin, L.; Wang, C. Protective effect and mechanism of docosahexaenoic acid on the cognitive function in female app/ps1 mice. Food Funct. 2021, 12, 11435–11448. [Google Scholar] [CrossRef]
- Xu, J.; Ni, B.; Ma, C.; Rong, S.; Gao, H.; Zhang, L.; Xiang, X.; Huang, Q.; Deng, Q.; Huang, F. Docosahexaenoic acid enhances hippocampal insulin sensitivity to promote cognitive function of aged rats on a high-fat diet. J. Adv. Res. 2022. [Google Scholar] [CrossRef]
- Zussy, C.; John, R.; Urgin, T.; Otaegui, L.; Vigor, C.; Acar, N.; Canet, G.; Vitalis, M.; Morin, F.; Planel, E.; et al. Intranasal administration of nanovectorized docosahexaenoic acid (dha) improves cognitive function in two complementary mouse models of Alzheimer’s disease. Antioxidants 2022, 11, 838. [Google Scholar] [CrossRef]
- Wu, T.Y.; Zhao, L.X.; Zhang, Y.H.; Fan, Y.G. Activation of vitamin d receptor inhibits tau phosphorylation is associated with reduction of iron accumulation in app/ps1 transgenic mice. Neurochem. Int. 2022, 153, 105260. [Google Scholar] [CrossRef]
- Saad El-Din, S.; Rashed, L.; Medhat, E.; Emad Aboulhoda, B.; Desoky Badawy, A.; Mohammed ShamsEldeen, A.; Abdelgwad, M. Active form of vitamin d analogue mitigates neurodegenerative changes in Alzheimer’s disease in rats by targeting keap1/nrf2 and mapk-38p/erk signaling pathways. Steroids 2020, 156, 108586. [Google Scholar] [CrossRef] [PubMed]
- Briones, T.L.; Darwish, H. Decrease in age-related tau hyperphosphorylation and cognitive improvement following vitamin d supplementation are associated with modulation of brain energy metabolism and redox state. Neuroscience 2014, 262, 143–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.I.; Chang, Y.C.; Kao, N.J.; Lee, W.J.; Cross, T.W.; Lin, S.H. 1,25(oh)2d3 alleviates abeta(25-35)-induced tau hyperphosphorylation, excessive reactive oxygen species, and apoptosis through interplay with glial cell line-derived neurotrophic factor signaling in sh-sy5y cells. Int. J. Mol. Sci. 2020, 21, 4215. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.Z.; Valeur, J.; Saltyte Benth, J.; Knapskog, A.B.; Selbaek, G.; Bogdanovic, N.; Pettersen, R. Associations between intrathecal levels of vitamin d, cytokines, and core biomarkers of Alzheimer’s disease: A cross-sectional study. J. Alzheimer’s Dis. 2022, 89, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Vina, J. Abeta and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: Protective role of vitamin e. Redox. Biol. 2014, 2, 873–877. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.; Rogers, E.; Shea, T.B. Dietary deficiency in folate and vitamin e under conditions of oxidative stress increases phospho-tau levels: Potentiation by apoe4 and alleviation by s-adenosylmethionine. J. Alzheimer’s Dis. 2009, 17, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Zaulkffali, A.S.; Md Razip, N.N.; Syed Alwi, S.S.; Abd Jalil, A.; Abd Mutalib, M.S.; Gopalsamy, B.; Chang, S.K.; Zainal, Z.; Ibrahim, N.N.; Zakaria, Z.A.; et al. Vitamins d and e stimulate the pi3k-akt signalling pathway in insulin-resistant sk-n-sh neuronal cells. Nutrients 2019, 11, 2525. [Google Scholar] [CrossRef]
- Kunzler, A.; Kolling, E.A.; da Silva, J.D., Jr.; Gasparotto, J.; de Bittencourt Pasquali, M.A.; Moreira, J.C.F.; Gelain, D.P. Retinol (vitamin a) increases alpha-synuclein, beta-amyloid peptide, tau phosphorylation and rage content in human sh-sy5y neuronal cell line. Neurochem. Res. 2017, 42, 2788–2797. [Google Scholar] [CrossRef] [PubMed]
- Rezai-Zadeh, K.; Arendash, G.W.; Hou, H.; Fernandez, F.; Jensen, M.; Runfeldt, M.; Shytle, R.D.; Tan, J. Green tea epigallocatechin-3-gallate (egcg) reduces beta-amyloid mediated cognitive impairment and modulates tau pathology in Alzheimer transgenic mice. Brain Res. 2008, 1214, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Henry, E.C.; Gasiewicz, T.A. (-)-epigallocatechin-3-gallate is a novel hsp90 inhibitor. Biochemistry 2009, 48, 336–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Tan, L.; Yu, J.T.; Tan, L. Tau in Alzheimer’s disease: Mechanisms and therapeutic strategies. Curr. Alzheimer Res. 2018, 15, 283–300. [Google Scholar] [CrossRef]
- Zhang, Y.; Yin, F.; Liu, J.; Liu, Z.; Guo, L.; Xia, Z.; Zidichouski, J. Geniposide attenuates insulin-deficiency-induced acceleration of beta-amyloidosis in an app/ps1 transgenic model of Alzheimer’s disease. Neurochem. Int. 2015, 89, 7–16. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Z.; Zhang, Y.; Yin, F. Leptin signaling plays a critical role in the geniposide-induced decrease of tau phosphorylation. Acta Biochim. Biophys. Sin. 2015, 47, 1018–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Zhang, Y.; Liu, J.; Yin, F. Geniposide attenuates the level of abeta1-42 via enhancing leptin signaling in cellular and app/ps1 transgenic mice. Arch. Pharm. Res. 2017, 40, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Findings in redox biology: From h2o2 to oxidative stress. J. Biol. Chem. 2020, 295, 13458–13473. [Google Scholar] [CrossRef]
- Yang, J.; Yang, J.; Liang, S.H.; Xu, Y.; Moore, A.; Ran, C. Imaging hydrogen peroxide in Alzheimer’s disease via cascade signal amplification. Sci. Rep. 2016, 6, 35613. [Google Scholar] [CrossRef] [Green Version]
- Milton, N.G. Role of hydrogen peroxide in the aetiology of Alzheimer’s disease: Implications for treatment. Drugs Aging 2004, 21, 81–100. [Google Scholar] [CrossRef]
- Kaminsky, Y.G.; Kosenko, E.A. Effects of amyloid-beta peptides on hydrogen peroxide-metabolizing enzymes in rat brain in vivo. Free Radic. Res. 2008, 42, 564–573. [Google Scholar] [CrossRef]
- Tonnies, E.; Trushina, E. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [Green Version]
- Sinyor, B.; Mineo, J.; Ochner, C. Alzheimer’s disease, inflammation, and the role of antioxidants. J. Alzheimer’s Dis. Rep. 2020, 4, 175–183. [Google Scholar] [CrossRef]
- Nasrabady, S.E.; Rizvi, B.; Goldman, J.E.; Brickman, A.M. White matter changes in Alzheimer’s disease: A focus on myelin and oligodendrocytes. Acta Neuropathol. Commun. 2018, 6, 22. [Google Scholar] [CrossRef] [Green Version]
- Aborode, A.T.; Pustake, M.; Awuah, W.A.; Alwerdani, M.; Shah, P.; Yarlagadda, R.; Ahmad, S.; Silva Correia, I.F.; Chandra, A.; Nansubuga, E.P.; et al. Targeting oxidative stress mechanisms to treat Alzheimer’s and parkinson’s disease: A critical review. Oxid. Med. Cell. Longev. 2022, 2022, 7934442. [Google Scholar] [CrossRef]
- Lloret, A.; Esteve, D.; Monllor, P.; Cervera-Ferri, A.; Lloret, A. The effectiveness of vitamin e treatment in Alzheimer’s disease. Int. J. Mol. Sci. 2019, 20, 879. [Google Scholar] [CrossRef] [Green Version]
- Joshi, Y.B.; Pratico, D. Vitamin e in aging, dementia, and Alzheimer’s disease. Biofactors 2012, 38, 90–97. [Google Scholar] [CrossRef]
- Wolf, G. The discovery of the antioxidant function of vitamin e: The contribution of henry a. Mattill. J. Nutr. 2005, 135, 363–366. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Quinn, P.J. Vitamin e and its function in membranes. Prog. Lipid Res. 1999, 38, 309–336. [Google Scholar] [CrossRef]
- Wimalawansa, S.J. Vitamin d deficiency: Effects on oxidative stress, epigenetics, gene regulation, and aging. Biology 2019, 8, 30. [Google Scholar] [CrossRef]
- Ali, A.; Shah, S.A.; Zaman, N.; Uddin, M.N.; Khan, W.; Ali, A.; Riaz, M.; Kamil, A. Vitamin d exerts neuroprotection via sirt1/nrf-2/ nf-kb signaling pathways against d-galactose-induced memory impairment in adult mice. Neurochem. Int. 2021, 142, 104893. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.G.; Pang, Z.Q.; Wu, T.Y.; Zhang, Y.H.; Xuan, W.Q.; Wang, Z.; Yu, X.; Li, Y.C.; Guo, C.; Wang, Z.Y. Vitamin d deficiency exacerbates Alzheimer-like pathologies by reducing antioxidant capacity. Free Radic. Biol. Med. 2020, 161, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Theiss, E.L.; Griebsch, L.V.; Lauer, A.A.; Janitschke, D.; Erhardt, V.K.J.; Haas, E.C.; Kuppler, K.N.; Radermacher, J.; Walzer, O.; Portius, D.; et al. Vitamin b12 attenuates changes in phospholipid levels related to oxidative stress in sh-sy5y cells. Cells 2022, 11, 2574. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Mi, X.; Ni, C.; Liu, T.; Yang, N.; Hong, J.; Li, Y.; Li, Z.; Han, D.; Guo, X. Protective effect of vitamin c on DNA damage in surgery-induced cognitive dysfunction in app/ps1 mice. Neurosci. Lett. 2022, 784, 136740. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.R. The pecking order of free radicals and antioxidants: Lipid peroxidation, alpha-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.Q.; Shen, T.T.; Wang, F.; Wu, P.F.; Chen, J.G. Preventive and therapeutic potential of vitamin c in mental disorders. Curr. Med. Sci. 2018, 38, 1–10. [Google Scholar] [CrossRef]
- Dixit, S.; Bernardo, A.; Walker, J.M.; Kennard, J.A.; Kim, G.Y.; Kessler, E.S.; Harrison, F.E. Vitamin c deficiency in the brain impairs cognition, increases amyloid accumulation and deposition, and oxidative stress in app/psen1 and normally aging mice. ACS Chem. Neurosci. 2015, 6, 570–581. [Google Scholar] [CrossRef] [Green Version]
- Kook, S.Y.; Lee, K.M.; Kim, Y.; Cha, M.Y.; Kang, S.; Baik, S.H.; Lee, H.; Park, R.; Mook-Jung, I. High-dose of vitamin c supplementation reduces amyloid plaque burden and ameliorates pathological changes in the brain of 5xfad mice. Cell Death Dis. 2014, 5, e1083. [Google Scholar] [CrossRef] [Green Version]
- Li, F.J.; Shen, L.; Ji, H.F. Dietary intakes of vitamin e, vitamin c, and beta-carotene and risk of Alzheimer’s disease: A meta-analysis. J. Alzheimer’s Dis. 2012, 31, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, J.; Hino, K. Plasmalogen in the brain: Effects on cognitive functions and behaviors attributable to its properties. Brain Res. Bull. 2022, 188, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Stadelmann-Ingrand, S.; Favreliere, S.; Fauconneau, B.; Mauco, G.; Tallineau, C. Plasmalogen degradation by oxidative stress: Production and disappearance of specific fatty aldehydes and fatty alpha-hydroxyaldehydes. Free Radic. Biol. Med. 2001, 31, 1263–1271. [Google Scholar] [CrossRef]
- Oboh, G.; Ademiluyi, A.O.; Akinyemi, A.J. Inhibition of acetylcholinesterase activities and some pro-oxidant induced lipid peroxidation in rat brain by two varieties of ginger (zingiber officinale). Exp. Toxicol. Pathol. 2012, 64, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Park, G.H.; Kim, C.Y.; Jang, J.H. [6]-gingerol attenuates beta-amyloid-induced oxidative cell death via fortifying cellular antioxidant defense system. Food Chem. Toxicol. 2011, 49, 1261–1269. [Google Scholar] [CrossRef]
- Shim, S.; Kwon, J. Effects of [6]-shogaol on cholinergic signaling in ht22 cells following neuronal damage induced by hydrogen peroxide. Food Chem. Toxicol. 2012, 50, 1454–1459. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xie, N.; Li, L.; Zou, Y.; Zhang, X.; Dong, M. Puerarin alleviates cognitive impairment and oxidative stress in app/ps1 transgenic mice. Int. J. Neuropsychopharmacol. 2014, 17, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Hole, K.L.; Williams, R.J. Flavonoids as an intervention for Alzheimer’s disease: Progress and hurdles towards defining a mechanism of action. Brain Plast. 2021, 6, 167–192. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.L.; Yen, G.C. Modulation of akt, jnk, and p38 activation is involved in citrus flavonoid-mediated cytoprotection of pc12 cells challenged by hydrogen peroxide. J. Agric. Food Chem. 2009, 57, 2576–2582. [Google Scholar] [CrossRef]
- Pallauf, K.; Duckstein, N.; Hasler, M.; Klotz, L.O.; Rimbach, G. Flavonoids as putative inducers of the transcription factors nrf2, foxo, and ppargamma. Oxid. Med. Cell. Longev. 2017, 2017, 4397340. [Google Scholar] [CrossRef] [Green Version]
- Grimm, M.O.W.; Lauer, A.A.; Grosgen, S.; Thiel, A.; Lehmann, J.; Winkler, J.; Janitschke, D.; Herr, C.; Beisswenger, C.; Bals, R.; et al. Profiling of Alzheimer’s disease related genes in mild to moderate vitamin d hypovitaminosis. J. Nutr. Biochem. 2019, 67, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Kajiwara, Y.; McKenzie, A.; Dorr, N.; Gama Sosa, M.A.; Elder, G.; Schmeidler, J.; Dickstein, D.L.; Bozdagi, O.; Zhang, B.; Buxbaum, J.D. The human-specific casp4 gene product contributes to Alzheimer-related synaptic and behavioural deficits. Hum. Mol. Genet. 2016, 25, 4315–4327. [Google Scholar] [CrossRef]
- Endres, K.; Fahrenholz, F.; Lotz, J.; Hiemke, C.; Teipel, S.; Lieb, K.; Tuscher, O.; Fellgiebel, A. Increased csf apps-alpha levels in patients with Alzheimer disease treated with acitretin. Neurology 2014, 83, 1930–1935. [Google Scholar] [CrossRef]
- Dos Santos Guilherme, M.; Stoye, N.M.; Rose-John, S.; Garbers, C.; Fellgiebel, A.; Endres, K. The synthetic retinoid acitretin increases il-6 in the central nervous system of Alzheimer disease model mice and human patients. Front. Aging Neurosci. 2019, 11, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Liu, S.; Ji, L.; Wu, T.; Ji, Y.; Zhou, Y.; Zheng, M.; Zhang, M.; Xu, W.; Huang, G. Folic acid supplementation mitigates Alzheimer’s disease by reducing inflammation: A randomized controlled trial. Mediat. Inflamm. 2016, 2016, 5912146. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.X.; Zhu, Y.F.; Wang, C.C.; Yang, J.Y.; Xue, C.H.; Huang, Q.R.; Wang, Y.M.; Zhang, T.T. Epa-enriched plasmalogen attenuates the cytotoxic effects of lps-stimulated microglia on the sh-sy5y neuronal cell line. Brain Res. Bull. 2022, 186, 143–152. [Google Scholar] [CrossRef]
- Parolini, C. Marine n-3 polyunsaturated fatty acids: Efficacy on inflammatory-based disorders. Life Sci. 2020, 263, 118591. [Google Scholar] [CrossRef]
- Borsini, A.; Nicolaou, A.; Camacho-Munoz, D.; Kendall, A.C.; Di Benedetto, M.G.; Giacobbe, J.; Su, K.P.; Pariante, C.M. Omega-3 polyunsaturated fatty acids protect against inflammation through production of lox and cyp450 lipid mediators: Relevance for major depression and for human hippocampal neurogenesis. Mol. Psychiatry 2021, 26, 6773–6788. [Google Scholar] [CrossRef]
- Zgorzynska, E.; Dziedzic, B.; Markiewicz, M.; Walczewska, A. Omega-3 pufas suppress il-1beta-induced hyperactivity of immunoproteasomes in astrocytes. Int. J. Mol. Sci. 2021, 22, 5410. [Google Scholar] [CrossRef]
- Dong, Y.; Xu, M.; Kalueff, A.V.; Song, C. Dietary eicosapentaenoic acid normalizes hippocampal omega-3 and 6 polyunsaturated fatty acid profile, attenuates glial activation and regulates bdnf function in a rodent model of neuroinflammation induced by central interleukin-1beta administration. Eur. J. Nutr. 2018, 57, 1781–1791. [Google Scholar] [CrossRef]
- Ha, S.K.; Moon, E.; Ju, M.S.; Kim, D.H.; Ryu, J.H.; Oh, M.S.; Kim, S.Y. 6-shogaol, a ginger product, modulates neuroinflammation: A new approach to neuroprotection. Neuropharmacology 2012, 63, 211–223. [Google Scholar] [CrossRef]
- Moon, M.; Kim, H.G.; Choi, J.G.; Oh, H.; Lee, P.K.; Ha, S.K.; Kim, S.Y.; Park, Y.; Huh, Y.; Oh, M.S. 6-shogaol, an active constituent of ginger, attenuates neuroinflammation and cognitive deficits in animal models of dementia. Biochem. Biophys. Res. Commun. 2014, 449, 8–13. [Google Scholar] [CrossRef]
- Grzanna, R.; Phan, P.; Polotsky, A.; Lindmark, L.; Frondoza, C.G. Ginger extract inhibits beta-amyloid peptide-induced cytokine and chemokine expression in cultured thp-1 monocytes. J. Altern. Complement. Med. 2004, 10, 1009–1013. [Google Scholar] [CrossRef] [PubMed]
- Spagnuolo, C.; Moccia, S.; Russo, G.L. Anti-inflammatory effects of flavonoids in neurodegenerative disorders. Eur. J. Med. Chem. 2018, 153, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.R.; Feng, X.Q.; Li, N.; Qu, J.X.; Feng, L.; Chen, L.; Chen, W.F. G protein-coupled estrogen receptor is involved in the anti-inflammatory effects of genistein in microglia. Phytomedicine 2018, 43, 11–20. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Yan, S.; Zheng, J.; Gao, Y.; Zhang, S.; Liu, Z.; Liu, X.; Xiao, C. Eriodictyol attenuates lps-induced neuroinflammation, amyloidogenesis, and cognitive impairments via the inhibition of nf-kappab in male c57bl/6j mice and bv2 microglial cells. J. Agric. Food Chem. 2018, 66, 10205–10214. [Google Scholar] [CrossRef]
- Uddin, M.S.; Kabir, M.T.; Al Mamun, A.; Behl, T.; Mansouri, R.A.; Aloqbi, A.A.; Perveen, A.; Hafeez, A.; Ashraf, G.M. Exploring potential of alkaloidal phytochemicals targeting neuroinflammatory signaling of Alzheimer’s disease. Curr. Pharm. Des. 2021, 27, 357–366. [Google Scholar] [CrossRef]
- Thu Thuy Nguyen, V.; Endres, K. Targeting gut microbiota to alleviate neuroinflammation in Alzheimer’s disease. Adv. Drug Deliv. Rev. 2022, 188, 114418. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Huang, X.; Zhen, J.; Van Halm-Lutterodt, N.; Wang, J.; Zhou, C.; Yuan, L. Dietary vitamin e status dictates oxidative stress outcomes by modulating effects of fish oil supplementation in Alzheimer disease model appswe/ps1de9 mice. Mol. Neurobiol. 2018, 55, 9204–9219. [Google Scholar] [CrossRef]
- Badia-Soteras, A.; de Vries, J.; Dykstra, W.; Broersen, L.M.; Verkuyl, J.M.; Smit, A.B.; Verheijen, M.H.G. High-throughput analysis of astrocyte cultures shows prevention of reactive astrogliosis by the multi-nutrient combination fortasyn connect. Cells 2022, 11, 1428. [Google Scholar] [CrossRef] [PubMed]
- Bottero, V.; Potashkin, J.A. A comparison of gene expression changes in the blood of individuals consuming diets supplemented with olives, nuts or long-chain omega-3 fatty acids. Nutrients 2020, 12, 3765. [Google Scholar] [CrossRef] [PubMed]
- Fiala, M.; Kooij, G.; Wagner, K.; Hammock, B.; Pellegrini, M. Modulation of innate immunity of patients with Alzheimer’s disease by omega-3 fatty acids. FASEB J. 2017, 31, 3229–3239. [Google Scholar] [CrossRef] [PubMed]
- van Wijk, N.; Broersen, L.M.; de Wilde, M.C.; Hageman, R.J.; Groenendijk, M.; Sijben, J.W.; Kamphuis, P.J. Targeting synaptic dysfunction in Alzheimer’s disease by administering a specific nutrient combination. J. Alzheimer’s Dis. 2014, 38, 459–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janickova, H.; Rudajev, V.; Dolejsi, E.; Koivisto, H.; Jakubik, J.; Tanila, H.; El-Fakahany, E.E.; Dolezal, V. Lipid-based diets improve muscarinic neurotransmission in the hippocampus of transgenic appswe/ps1de9 mice. Curr. Alzheimer Res. 2015, 12, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, J. The lipididiet trial: What does it add to the current evidence for fortasyn connect in early Alzheimer’s disease? Clin. Interv. Aging 2019, 14, 1481–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soininen, H.; Solomon, A.; Visser, P.J.; Hendrix, S.B.; Blennow, K.; Kivipelto, M.; Hartmann, T.; LipiDiDiet clinical study, g. 36-month lipididiet multinutrient clinical trial in prodromal Alzheimer’s disease. Alzheimer’s Dement. 2021, 17, 29–40. [Google Scholar] [CrossRef]
- Ali, A.A.; Abd El-Fattah, A.I.; Abu-Elfotuh, K.; Elariny, H.A. Natural antioxidants enhance the power of physical and mental activities versus risk factors inducing progression of Alzheimer’s disease in rats. Int. Immunopharmacol. 2021, 96, 107729. [Google Scholar] [CrossRef]
- World Health Organization. The European Health Report 2005: Public Health Action for Healthier Children and Populations; World Health Organization, Regional Office for Europe: Geneva, Switzerland, 2005.
- Finger, J.D.; Mensink, G.B.M.; Lange, C.; Manz, K. Gesundheitsfördernde körperliche aktivität in der freizeit bei erwachsenen in deutschland. J. Health Monit. 2017, 2, 37–44. [Google Scholar]
- Lee, I.M.; Shiroma, E.J.; Lobelo, F.; Puska, P.; Blair, S.N.; Katzmarzyk, P.T.; Lancet Physical Activity Series Working, G. Effect of physical inactivity on major non-communicable diseases worldwide: An analysis of burden of disease and life expectancy. Lancet 2012, 380, 219–229. [Google Scholar] [CrossRef] [Green Version]
- Luck, T.; Riedel-Heller, S.G. Prevention of Alzheimer’s dementia in germany: A projection of the possible potential of reducing selected risk factors. Nervenarzt 2016, 87, 1194–1200. [Google Scholar] [CrossRef]
- Aarsland, D.; Sardahaee, F.S.; Anderssen, S.; Ballard, C.; Alzheimer’s Society Systematic Review, g. Is physical activity a potential preventive factor for vascular dementia? A systematic review. Aging Ment. Health 2010, 14, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Saltin, B. Exercise as medicine—Evidence for prescribing exercise as therapy in 26 different chronic diseases. Scand. J. Med. Sci. Sports 2015, 25 (Suppl. S3), 1–72. [Google Scholar] [PubMed] [Green Version]
- Nyberg, J.; Aberg, M.A.; Schioler, L.; Nilsson, M.; Wallin, A.; Toren, K.; Kuhn, H.G. Cardiovascular and cognitive fitness at age 18 and risk of early-onset dementia. Brain 2014, 137, 1514–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, D.; Forbes, S.C.; Blake, C.M.; Thiessen, E.J.; Forbes, S. Exercise programs for people with dementia. Cochrane Database Syst. Rev. 2015, 4, CD006489. [Google Scholar] [CrossRef] [Green Version]
- Kemoun, G.; Thibaud, M.; Roumagne, N.; Carette, P.; Albinet, C.; Toussaint, L.; Paccalin, M.; Dugue, B. Effects of a physical training programme on cognitive function and walking efficiency in elderly persons with dementia. Dement. Geriatr. Cogn. Disord. 2010, 29, 109–114. [Google Scholar] [CrossRef]
- Rolland, Y.; Pillard, F.; Klapouszczak, A.; Reynish, E.; Thomas, D.; Andrieu, S.; Riviere, D.; Vellas, B. Exercise program for nursing home residents with Alzheimer’s disease: A 1-year randomized, controlled trial. J. Am. Geriatr. Soc. 2007, 55, 158–165. [Google Scholar] [CrossRef]
- Steinberg, M.; Leoutsakos, J.M.; Podewils, L.J.; Lyketsos, C.G. Evaluation of a home-based exercise program in the treatment of Alzheimer’s disease: The maximizing independence in dementia (mind) study. Int. J. Geriatr. Psychiatry 2009, 24, 680–685. [Google Scholar] [CrossRef] [Green Version]
- Baker, L.D.; Frank, L.L.; Foster-Schubert, K.; Green, P.S.; Wilkinson, C.W.; McTiernan, A.; Plymate, S.R.; Fishel, M.A.; Watson, G.S.; Cholerton, B.A.; et al. Effects of aerobic exercise on mild cognitive impairment: A controlled trial. Arch. Neurol. 2010, 67, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Lautenschlager, N.T.; Cox, K.L.; Flicker, L.; Foster, J.K.; van Bockxmeer, F.M.; Xiao, J.; Greenop, K.R.; Almeida, O.P. Effect of physical activity on cognitive function in older adults at risk for Alzheimer disease: A randomized trial. JAMA 2008, 300, 1027–1037. [Google Scholar] [CrossRef] [Green Version]
- Erickson, K.I.; Voss, M.W.; Prakash, R.S.; Basak, C.; Szabo, A.; Chaddock, L.; Kim, J.S.; Heo, S.; Alves, H.; White, S.M.; et al. Exercise training increases size of hippocampus and improves memory. Proc. Natl. Acad. Sci. USA 2011, 108, 3017–3022. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Swartwood, R.M. Feasibility and perception of the impact from aerobic exercise in older adults with Alzheimer’s disease. Am. J. Alzheimer’s Dis. Other Demen. 2012, 27, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Freberg, E.; Taglialatela, G. Exercise as a potential therapeutic strategy to target the clinical link between depression and Alzheimer’s disease: A narrative review. J. Alzheimer’s Dis. 2022, 89, 759–767. [Google Scholar] [CrossRef]
- Sun, L.; Liu, T.; Liu, J.; Gao, C.; Zhang, X. Physical exercise and mitochondrial function: New therapeutic interventions for psychiatric and neurodegenerative disorders. Front. Neurol. 2022, 13, 929781. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, E.J.R.; Ibrahim, H.J.; Chitolina Schetinger, M.R.; de Andrade, C.M.; Cardoso, A.M. Modulation of inflammatory mediators and microglial activation through physical exercise in Alzheimer’s and parkinson’s diseases. Neurochem. Res. 2022, 47, 3221–3240. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Risk reduction of cognitive decline and dementia. In WHO Guidelines; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Chen, B.; Fu, Y.; Song, G.; Zhong, W.; Guo, J. Research trends and hotspots of exercise for Alzheimer’s disease: A bibliometric analysis. Front. Aging Neurosci. 2022, 14, 984705. [Google Scholar] [CrossRef]
- Barha, C.K.; Davis, J.C.; Falck, R.S.; Nagamatsu, L.S.; Liu-Ambrose, T. Sex differences in exercise efficacy to improve cognition: A systematic review and meta-analysis of randomized controlled trials in older humans. Front. Neuroendocr. 2017, 46, 71–85. [Google Scholar] [CrossRef]
- de Souto Barreto, P.; Demougeot, L.; Vellas, B.; Rolland, Y. Exercise training for preventing dementia, mild cognitive impairment, and clinically meaningful cognitive decline: A systematic review and meta-analysis. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 1504–1511. [Google Scholar] [CrossRef]
- Northey, J.M.; Cherbuin, N.; Pumpa, K.L.; Smee, D.J.; Rattray, B. Exercise interventions for cognitive function in adults older than 50: A systematic review with meta-analysis. Br. J. Sports Med. 2018, 52, 154–160. [Google Scholar] [CrossRef] [Green Version]
- Song, D.; Yu, D.S.F.; Li, P.W.C.; Lei, Y. The effectiveness of physical exercise on cognitive and psychological outcomes in individuals with mild cognitive impairment: A systematic review and meta-analysis. Int. J. Nurs. Stud. 2018, 79, 155–164. [Google Scholar] [CrossRef]
- World Health Organization. Guidelines on Physical Activity and Sedentary Behaviour; World Health Organization: Geneva, Switzerland, 2020.
- Adzhar, M.A.; Manlapaz, D.; Singh, D.K.A.; Mesbah, N. Exercise to improve postural stability in older adults with Alzheimer’s disease: A systematic review of randomized control trials. Int. J. Environ. Res. Public Health 2022, 19, 10350. [Google Scholar] [CrossRef]
- Bahar-Fuchs, A.; Clare, L.; Woods, B. Cognitive training and cognitive rehabilitation for mild to moderate Alzheimer’s disease and vascular dementia. Cochrane Database Syst. Rev. 2013, 6, CD003260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, É.; Spector, A. Cognitive stimulation therapy. In Practical Guide Fondation Médéric Alzheimer; Fondation Médéric Alzheimer: Paris, France, 2021; pp. 40–47. [Google Scholar]
- Capotosto, E.; Belacchi, C.; Gardini, S.; Faggian, S.; Piras, F.; Mantoan, V.; Salvalaio, E.; Pradelli, S.; Borella, E. Cognitive stimulation therapy in the italian context: Its efficacy in cognitive and non-cognitive measures in older adults with dementia. Int. J. Geriatr. Psychiatry 2017, 32, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Carbone, E.; Piras, F.; Pastore, M.; Borella, E. The role of individual characteristics in predicting short- and long-term cognitive and psychological benefits of cognitive stimulation therapy for mild-to-moderate dementia. Front. Aging Neurosci. 2021, 13, 811127. [Google Scholar] [CrossRef] [PubMed]
- Lobbia, A.; Carbone, E.; Faggian, S.; Gardini, S.; Piras, F.; Spector, A.; Borella, E. The efficacy of cognitive stimulation therapy (cst) for people with mild-to-moderate dementia. Eur. Psychol. 2019, 24, 257–277. [Google Scholar] [CrossRef] [Green Version]
- Clare, L. Rehabilitation for people living with dementia: A practical framework of positive support. PLoS Med. 2017, 14, e1002245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudlicka, A. Cognitive rehabilitationn. In Practical Guide Fondation Médéric Alzheimer; Fondation Médéric Alzheimer: Paris, France, 2021; pp. 32–39. [Google Scholar]
- Gates, N.J.; Vernooij, R.W.; Di Nisio, M.; Karim, S.; March, E.; Martínez, G.; Rutjes, A.W. Computerised cognitive training for preventing dementia in people with mild cognitive impairment. Cochrane Database Syst. Rev. 2019, 3, CD012279. [Google Scholar] [CrossRef]
- Hafdi, M.; Hoevenaar-Blom, M.P.; Richard, E. Multi-domain interventions for the prevention of dementia and cognitive decline. Cochrane Database Syst. Rev. 2021, 11, CD013572. [Google Scholar] [CrossRef]
- Liu, T.; Spector, A.; Mograbi, D.C.; Cheung, G.; Wong, G.H.Y. Changes in default mode network connectivity in resting-state fmri in people with mild dementia receiving cognitive stimulation therapy. Brain Sci. 2021, 11, 1137. [Google Scholar] [CrossRef]
- Stern, Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 2012, 11, 1006–1012. [Google Scholar] [CrossRef] [Green Version]
- Swan, K.; Hopper, M.; Wenke, R.; Jackson, C.; Till, T.; Conway, E. Speech-language pathologist interventions for communication in moderate-severe dementia: A systematic review. Am. J. Speech-Lang. Pathol. 2018, 27, 836–852. [Google Scholar] [CrossRef]
- Gates, N.J.; Rutjes, A.W.; Di Nisio, M.; Karim, S.; Chong, L.-Y.; March, E.; Martínez, G.; Vernooij, R.W. Computerised cognitive training for 12 or more weeks for maintaining cognitive function in cognitively healthy people in late life. Cochrane Database Syst. Rev. 2020, 2, CD012277. [Google Scholar] [CrossRef] [PubMed]
- Clare, L.; Kudlicka, A.; Oyebode, J.R.; Jones, R.W.; Bayer, A.; Leroi, I.; Kopelman, M.; James, I.A.; Culverwell, A.; Pool, J.; et al. Individual goal-oriented cognitive rehabilitation to improve everyday functioning for people with early-stage dementia: A multicentre randomised controlled trial (the great trial). Int. J. Geriatr. Psychiatry 2019, 34, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amieva, H.; Robert, P.H.; Grandoulier, A.-S.; Meillon, C.; Rotrou, J.d.; Andrieu, S.; Berr, C.; Desgranges, B.; Dubois, B.; Girtanner, C.; et al. Group and individual cognitive therapies in Alzheimer’s disease: The etna3 randomized trial. Int. Psychogeriatr. 2016, 28, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Scott, I.; Cooper, C.; Leverton, M.; Burton, A.; Beresford-Dent, J.; Rockwood, K.; Butler, L.; Rapaport, P. Effects of nonpharmacological interventions on functioning of people living with dementia at home: A systematic review of randomised controlled trials. Int. J. Geriatr. Psychiatry 2019, 34, 1386–1402. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.; Hunt, A.; Quinn, C.; Martyr, A.; Pentecost, C.; Clare, L. Methods and approaches for enhancing communication with people with moderate-to-severe dementia that can facilitate their inclusion in research and service evaluation: Findings from the ideal programme. Dementia 2022, 21, 1135–1153. [Google Scholar] [CrossRef]
- Nguyen, H.; Terry, D.; Phan, H.; Vickers, J.; McInerney, F. Communication training and its effects on carer and care-receiver outcomes in dementia settings: A systematic review. J. Clin. Nurs. 2019, 28, 1050–1069. [Google Scholar] [CrossRef]
- Morello, A.N.d.C.; Lima, T.M.; Brandão, L. Language and communication non-pharmacological interventions in patients with Alzheimer’s disease: A systematic review. Communication intervention in Alzheimer. Dement. Neuropsychol. 2017, 11, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Carbone, E.; Gardini, S.; Pastore, M.; Piras, F.; Vincenzi, M.; Borella, E. Cognitive stimulation therapy for older adults with mild-to-moderate dementia in italy: Effects on cognitive functioning, and on emotional and neuropsychiatric symptoms. J. Gerontol. Ser. B Psychol. Sci. Soc. Sci. 2021, 76, 1700–1710. [Google Scholar] [CrossRef]
- Juárez-Cedillo, T.; Gutiérrez-Gutiérrez, L.; Sánchez-Hurtado, L.A.; Martínez-Rodríguez, N.; Juarez-Cedillo, E. Randomized controlled trial of multi-component cognitive stimulation therapy (sadem) in community-dwelling demented adults. J. Alzheimer’s Dis. JAD 2020, 78, 1033–1045. [Google Scholar] [CrossRef]
- Spector, A.; Thorgrimsen, L.; Woods, B.; Royan, L.; Davies, S.; Butterworth, M.; Orrell, M. Efficacy of an evidence-based cognitive stimulation therapy programme for people with dementia: Randomised controlled trial. Br. J. Psychiatry J. Ment. Sci. 2003, 183, 248–254. [Google Scholar] [CrossRef]
- Henderson, S.K.; Peterson, K.A.; Patterson, K.; Lambon Ralph, M.A.; Rowe, J.B. Verbal fluency tests assess global cognitive status but have limited diagnostic differentiation: Evidence from a large-scale examination of six neurodegenerative diseases. medRxiv, 2022; preprint. [Google Scholar]
- Spector, A.; Orrell, M.; Woods, B. Cognitive stimulation therapy (cst): Effects on different areas of cognitive function for people with dementia. Int. J. Geriatr. Psychiatry 2010, 25, 1253–1258. [Google Scholar] [CrossRef]
- Saragih, I.D.; Tonapa, S.I.; Yao, C.-T.; Saragih, I.S.; Lee, B.-O. Effects of reminiscence therapy in people with dementia: A systematic review and meta-analysis. J. Psychiatr. Ment. Health Nurs. 2022, 29, 883–903. [Google Scholar] [CrossRef]
- Woods, B.; O’Philbin, L.; Farrell, E.M.; Spector, A.E.; Orrell, M. Reminiscence therapy for dementia. Cochrane Database Syst. Rev. 2018, 3, CD001120. [Google Scholar] [CrossRef] [PubMed]
- Pietro, M.J.S.; Boczko, F. The breakfast club: Results of a study examining the effectiveness of a multi-modality group communication treatment. Am. J. Alzheimer’s Dis. 1998, 13, 146–158. [Google Scholar] [CrossRef]
- Rogalski, E.J.; Khayum, B. A life participation approach to primary progressive aphasia intervention. Semin. Speech Lang. 2018, 39, 284–296. [Google Scholar] [PubMed]
- Chalfont, G.; Milligan, C.; Simpson, J. A mixed methods systematic review of multimodal non-pharmacological interventions to improve cognition for people with dementia. Dementia 2020, 19, 1086–1130. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, K.J.; Copland, D.A.; van Hees, S.; Byrne, G.J.; Angwin, A.J. Semantic feature training for the treatment of anomia in Alzheimer disease: A preliminary investigation. Cogn. Behav. Neurol. Off. J. Soc. Behav. Cogn. Neurol. 2016, 29, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Flurie, M.; Ungrady, M.; Reilly, J. Evaluating a maintenance-based treatment approach to preventing lexical dropout in progressive anomia. J. Speech Lang. Hear. Res. JSLHR 2020, 63, 4082–4095. [Google Scholar] [CrossRef]
- Jelcic, N.; Cagnin, A.; Meneghello, F.; Turolla, A.; Ermani, M.; Dam, M. Effects of lexical-semantic treatment on memory in early Alzheimer disease: An observer-blinded randomized controlled trial. Neurorehabilit. Neural Repair 2012, 26, 949–956. [Google Scholar] [CrossRef]
- Beales, A.; Whitworth, A.; Cartwright, J. A review of lexical retrieval intervention in primary progressive aphasia and Alzheimer’s disease: Mechanisms of change, generalisation, and cognition. Aphasiology 2018, 32, 1360–1387. [Google Scholar] [CrossRef]
- Bourgeois, M.S. Memory and Communication Aids for People with Dementia; Health Professions Press: Baltimore, MD, USA, 2014; p. 150. [Google Scholar]
- Bourgeois, M.S. Caregiving for persons with dementia: Evidence-based resources for slps. Top. Lang. Disord. 2019, 39, 89–103. [Google Scholar] [CrossRef]
- Bourgeois, M.S.; Camp, C.J.; Antenucci, V.; Fox, K. Voicemychoice™: Facilitating understanding of preferences of residents with dementia. Adv. Aging Res. 2016, 05, 131–141. [Google Scholar]
- Clare, L.; Wilson, B.A.; Carter, G.; Breen, K.; Gosses, A.; Hodges, J.R. Intervening with everyday memory problems in dementia of Alzheimer type: An errorless learning approach. J. Clin. Exp. Neuropsychol. 2000, 22, 132–146. [Google Scholar] [CrossRef]
- Haslam, C.; Hodder, K.I.; Yates, P.J. Errorless learning and spaced retrieval: How do these methods fare in healthy and clinical populations? J. Clin. Exp. Neuropsychol. 2011, 33, 432–447. [Google Scholar] [CrossRef] [PubMed]
- Clare, L.; Jones, R.S.P. Errorless learning in the rehabilitation of memory impairment: A critical review. Neuropsychol. Rev. 2008, 18, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Voigt-Radloff, S.; Werd, M.M.E.d.; Leonhart, R.; Boelen, D.H.E.; Olde Rikkert, M.G.M.; Fliessbach, K.; Klöppel, S.; Heimbach, B.; Fellgiebel, A.; Dodel, R.; et al. Structured relearning of activities of daily living in dementia: The randomized controlled redali-dem trial on errorless learning. Alzheimer’s Res. Ther. 2017, 9, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beales, A.; Whitworth, A.; Cartwright, J.; Panegyres, P.K.; Kane, R.T. Making the right connections: Maximizing lexical generalization in lexical impairments in primary progressive aphasia and Alzheimer’s disease. Am. J. Speech-Lang. Pathol. 2021, 30, 697–712. [Google Scholar] [CrossRef]
- Jokel, R.; Meltzer, J.; D.R., J.; D.M., L.; J.C., J.; A.N., E.; D.T., C. Group intervention for individuals with primary progressive aphasia and their spouses: Who comes first? J. Commun. Disord. 2017, 66, 51–64. [Google Scholar] [CrossRef]
- Rogalski, E.; Roberts, A.; Salley, E.; Saxon, M.; Fought, A.; Esparza, M.; Blaze, E.; Coventry, C.; Mesulam, M.-M.; Weintraub, S.; et al. Communication partner engagement: A relevant factor for functional outcomes in speech-language therapy for aphasic dementia. J. Gerontol. Ser. B Psychol. Sci. Soc. Sci. 2022, 77, 1017–1025. [Google Scholar] [CrossRef]
- Eggenberger, E.; Heimerl, K.; Bennett, M.I. Communication skills training in dementia care: A systematic review of effectiveness, training content, and didactic methods in different care settings. Int. Psychogeriatr. 2013, 25, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, M.S.; Hickey, E.M. Dementia: From Diagnosis to Management—A Functional Approach; Taylor & Francis: New York, NY, USA, 2009. [Google Scholar]
- Volkmer, A. Assessment and Therapy for Language and Cognitive Communication Difficulties in Dementia and Other Progressive Diseases; J. R. Press: Guildford, UK, 2013. [Google Scholar]
- Knels, C.; Grün, F.; Schuster, P. Sprache und Ernährung bei Demenz; Georg Thieme Verlag: Stuttgart, Germany, 2018. [Google Scholar]
- Wu, L.; Zhang, S.-Q.; Zhao, L.; Ren, Z.-H.; Hu, C.-Y. Global, regional, and national burden of periodontitis from 1990 to 2019: Results from the global burden of disease study 2019. J. Periodontol. 2022, 93, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Papapanou, P.N.; Sanz, M.; Buduneli, N.; Dietrich, T.; Feres, M.; Fine, D.H.; Flemmig, T.F.; Garcia, R.; Giannobile, W.V.; Graziani, F.; et al. Periodontitis: Consensus report of workgroup 2 of the 2017 world workshop on the classification of periodontal and peri-implant diseases and conditions. J. Periodontol. 2018, 89 (Suppl. S1), S173–S182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, R.C.; Offenbacher, S.; Schroeder, H.E.; Seymour, G.J.; Kornman, K.S. Advances in the pathogenesis of periodontitis: Summary of developments, clinical implications and future directions. Periodontology 2000 1997, 14, 216–248. [Google Scholar] [CrossRef] [PubMed]
- Slots, J. Periodontitis: Facts, fallacies and the future. Periodontology 2000 2017, 75, 7–23. [Google Scholar] [CrossRef]
- Socransky, S.S.; Haffajee, A.D. Periodontal microbial ecology. Periodontology 2000 2005, 38, 135–187. [Google Scholar] [CrossRef]
- Camelo-Castillo, A.J.; Mira, A.; Pico, A.; Nibali, L.; Henderson, B.; Donos, N.; Tomás, I. Subgingival microbiota in health compared to periodontitis and the influence of smoking. Front. Microbiol. 2015, 6, 119. [Google Scholar] [CrossRef] [Green Version]
- Haffajee, A.D.; Socransky, S.S. Relationship of cigarette smoking to the subgingival microbiota. J. Clin. Periodontol. 2001, 28, 377–388. [Google Scholar] [CrossRef]
- Luthra, S.; Orlandi, M.; Hussain, S.B.; Leira, Y.; Botelho, J.; Machado, V.; Mendes, J.J.; Marletta, D.; Harden, S.; D’Aiuto, F. Treatment of periodontitis and c-reactive protein: A systematic review and meta-analysis of randomized clinical trials. J. Clin. Periodontol. 2022. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Horigome, Y.; Endo, K.; Komagata, M.; Komai, S.; Komaki, K.; Miyata, H.; Sugano, K.; Ito, S.; Itabashi, S.; et al. Caregiver-reported dementia as a predictor of oral health among patients receiving home-visit dental treatment: A retrospective cohort study. Clin. Exp. Dent. Res. 2021, 7, 49–55. [Google Scholar] [CrossRef]
- Lopez-Jornet, P.; Zamora Lavella, C.; Pons-Fuster Lopez, E.; Tvarijonaviciute, A. Oral health status in older people with dementia: A case-control study. J. Clin. Med. 2021, 10, 477. [Google Scholar] [CrossRef] [PubMed]
- Leira, Y.; Domínguez, C.; Seoane, J.; Seoane-Romero, J.; Pías-Peleteiro, J.M.; Takkouche, B.; Blanco, J.; Aldrey, J.M. Is periodontal disease associated with Alzheimer’s disease? A systematic review with meta-analysis. Neuroepidemiology 2017, 48, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Leira, Y.; Seoane, J.; Blanco, M.; Rodríguez-Yáñez, M.; Takkouche, B.; Blanco, J.; Castillo, J. Association between periodontitis and ischemic stroke: A systematic review and meta-analysis. Eur. J. Epidemiol. 2017, 32, 43–53. [Google Scholar] [CrossRef]
- Kapellas, K. The association between periodontal disease and dementia: A systematic review and meta-analysis. Dent. Oral Biol. Craniofacial Res. 2019. [Google Scholar] [CrossRef]
- Nadim, R.; Tang, J.; Dilmohamed, A.; Yuan, S.; Wu, C.; Bakre, A.T.; Partridge, M.; Ni, J.; Copeland, J.R.; Anstey, K.J.; et al. Influence of periodontal disease on risk of dementia: A systematic literature review and a meta-analysis. Eur. J. Epidemiol. 2020, 35, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J. The inflammatory response in Alzheimer’s disease. J. Periodontol. 2008, 79, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Riviere, G.R.; Riviere, K.H.; Smith, K.S. Molecular and immunological evidence of oral treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol. Immunol. 2002, 17, 113–118. [Google Scholar] [CrossRef]
- Kamer, A.R.; Craig, R.G.; Pirraglia, E.; Dasanayake, A.P.; Norman, R.G.; Boylan, R.J.; Nehorayoff, A.; Glodzik, L.; Brys, M.; de Leon, M.J. Tnf-alpha and antibodies to periodontal bacteria discriminate between Alzheimer’s disease patients and normal subjects. J. Neuroimmunol. 2009, 216, 92–97. [Google Scholar] [CrossRef] [Green Version]
- Sparks Stein, P.; Steffen, M.J.; Smith, C.; Jicha, G.; Ebersole, J.L.; Abner, E.; Dawson, D. Serum antibodies to periodontal pathogens are a risk factor for Alzheimer’s disease. Alzheimer’s Dement. 2012, 8, 196–203. [Google Scholar] [CrossRef] [Green Version]
- Noble, J.M.; Scarmeas, N.; Celenti, R.S.; Elkind, M.S.; Wright, C.B.; Schupf, N.; Papapanou, P.N. Serum igg antibody levels to periodontal microbiota are associated with incident Alzheimer disease. PLoS ONE 2014, 9, e114959. [Google Scholar] [CrossRef] [Green Version]
- Singhrao, S.K.; Olsen, I. Assessing the role of porphyromonas gingivalis in periodontitis to determine a causative relationship with Alzheimer’s disease. J. Oral Microbiol. 2019, 11, 1563405. [Google Scholar] [CrossRef] [Green Version]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef] [Green Version]
- Cortexyme Inc. Gain Trial: Phase 2/3 Study of Cor388 in Subjects with Alzheimer’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT03823404 (accessed on 11 September 2022).
- Jungbauer, G.; Stähli, A.; Zhu, X.; Auber Alberi, L.; Sculean, A.; Eick, S. Periodontal microorganisms and Alzheimer disease—A causative relationship? Periodontology 2000 2022, 89, 59–82. [Google Scholar] [CrossRef]
- Rolim, T.d.S.; Fabri, G.M.C.; Nitrini, R.; Anghinah, R.; Teixeira, M.J.; Siqueira, J.T.T.d.; Cesari, J.A.F.; Siqueira, S.R.D.T.d. Evaluation of patients with Alzheimer’s disease before and after dental treatment. Arq. Neuro-Psiquiatr. 2014, 72, 919–924. [Google Scholar] [CrossRef] [Green Version]
- Paganini-Hill, A.; White, S.C.; Atchison, K.A. Dentition, dental health habits, and dementia: The leisure world cohort study. J. Am. Geriatr. Soc. 2012, 60, 1556–1563. [Google Scholar] [CrossRef]
- Graetz, C.; Fawzy El-Sayed, K.; Sälzer, S.; Dörfer, C.E. Häusliches mechanisches biofilmmanagement in der prävention und therapie der gingivitis: Awmf-registernummer 083-022. 2018. Available online: https://register.awmf.org/de/leitlinien/detail/083-022 (accessed on 1 November 2022).
- Auschill, T.; Sälzer, S.; Arweiler, N. Häusliches chemisches biofilmmanagement in der prävention und therapie der gingivitis: Awmf-registernummer: 083-016. 2018. Available online: https://register.awmf.org/de/leitlinien/detail/083-016 (accessed on 1 November 2022).
- Zenthöfer, A.; Baumgart, D.; Cabrera, T.; Rammelsberg, P.; Schröder, J.; Corcodel, N.; Hassel, A.J. Poor dental hygiene and periodontal health in nursing home residents with dementia: An observational study. Odontology 2017, 105, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Barbe, A.G.; Kottmann, H.E.; Derman, S.H.M.; Noack, M.J. Efficacy of regular professional brushing by a dental nurse for 3 months in nursing home residents-a randomized, controlled clinical trial. Int. J. Dent. Hyg. 2019, 17, 327–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, M.; Herrera, D.; Kebschull, M.; Chapple, I.; Jepsen, S.; Beglundh, T.; Sculean, A.; Tonetti, M.S. Treatment of stage i–iii periodontitis-the efp s3 level clinical practice guideline. J. Clin. Periodontol. 2020, 47 (Suppl. 22), 4–60. [Google Scholar] [CrossRef]
- Desta, N.T. Pathophysiological association between periodontal disease and Alzheimer’s disease: Importance of periodontal health in the elderly. J. Oral Biosci. 2021, 63, 351–359. [Google Scholar] [CrossRef]
- Li, J.; Xu, H.; Pan, W.; Wu, B. Association between tooth loss and cognitive decline: A 13-year longitudinal study of chinese older adults. PLoS ONE 2017, 12, e0171404. [Google Scholar] [CrossRef] [PubMed]
- Makiura, T.; Ikeda, Y.; Hirai, T.; Terasawa, H.; Hamaue, N.; Minami, M. Influence of diet and occlusal support on learning memory in rats behavioral and biochemical studies. Res. Commun. Mol. Pathol. Pharmacol. 2000, 107, 269–277. [Google Scholar] [PubMed]
- Wang, X.; Hu, J.; Jiang, Q. Tooth loss-associated mechanisms that negatively affect cognitive function: A systematic review of animal experiments based on occlusal support loss and cognitive impairment. Front. Neurosci. 2022, 16, 811335. [Google Scholar] [CrossRef] [PubMed]
- Dioguardi, M.; Di Gioia, G.; Caloro, G.A.; Capocasale, G.; Zhurakivska, K.; Troiano, G.; Lo Russo, L.; Lo Muzio, L. The association between tooth loss and Alzheimer’s disease: A systematic review with meta-analysis of case control studies. Dent. J. 2019, 7, 49. [Google Scholar] [CrossRef] [Green Version]
- Furuta, M.; Komiya-Nonaka, M.; Akifusa, S.; Shimazaki, Y.; Adachi, M.; Kinoshita, T.; Kikutani, T.; Yamashita, Y. Interrelationship of oral health status, swallowing function, nutritional status, and cognitive ability with activities of daily living in japanese elderly people receiving home care services due to physical disabilities. Community Dent. Oral Epidemiol. 2013, 41, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Faggion, C.M. Critical appraisal of evidence supporting the placement of dental implants in patients with neurodegenerative diseases. Gerodontology 2016, 33, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Park, H.N.; Yoon, J.Y. Relationship between oral health and social activity among community-dwelling older adults in korea: Focusing on the mediating effect of depressive symptoms. Geriatr. Gerontol. Int. 2022, 22, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.E.; Mohan, J.; Kalaignan, P.; Kandasamy, S.; Raju, R.; Champakesan, B. Influence of dental prostheses on cognitive functioning in elderly population: A systematic review. J. Pharm. Bioallied Sci. 2021, 13, S788–S794. [Google Scholar] [CrossRef] [PubMed]
- Ming, Y.; Hsu, S.-W.; Yen, Y.-Y.; Lan, S.-J. Association of oral health-related quality of life and Alzheimer disease: A systematic review. J. Prosthet. Dent. 2020, 124, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Locker, D.; Slade, G. Association between clinical and subjective indicators of oral health status in an older adult population. Gerodontology 1994, 11, 108–114. [Google Scholar] [CrossRef]
- Axelsson, P.; Nyström, B.; Lindhe, J. The long-term effect of a plaque control program on tooth mortality, caries and periodontal disease in adults. Results after 30 years of maintenance. J. Clin. Periodontol. 2004, 31, 749–757. [Google Scholar] [CrossRef]
- Shigihara, Y.; Hoshi, H.; Shinada, K.; Okada, T.; Kamada, H. Non-pharmacological treatment changes brain activity in patients with dementia. Sci. Rep. 2020, 10, 6744. [Google Scholar] [CrossRef] [Green Version]
- Zucchella, C.; Sinforiani, E.; Tamburin, S.; Federico, A.; Mantovani, E.; Bernini, S.; Casale, R.; Bartolo, M. The multidisciplinary approach to Alzheimer’s disease and dementia. A narrative review of non-pharmacological treatment. Front. Neurol. 2018, 9, 1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Rue, A.; Felten, K.; Turkstra, L. Intervention of multi-modal activities for older adults with dementia translation to rural communities. Am. J. Alzheimer’s Dis. Other Dement. 2015, 30, 468–477. [Google Scholar] [CrossRef]
- Ngandu, T.; Lehtisalo, J.; Solomon, A.; Levälahti, E.; Ahtiluoto, S.; Antikainen, R.; Bäckman, L.; Hänninen, T.; Jula, A.; Laatikainen, T.; et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (finger): A randomised controlled trial. Lancet 2015, 385, 2255–2263. [Google Scholar] [CrossRef]
- Salzman, T.; Sarquis-Adamson, Y.; Son, S.; Montero-Odasso, M.; Fraser, S. Associations of multidomain interventions with improvements in cognition in mild cognitive impairment: A systematic review and meta-analysis. JAMA Netw. Open 2022, 5, e226744. [Google Scholar] [CrossRef] [PubMed]
- Sikkes, S.A.M.; Tang, Y.; Jutten, R.J.; Wesselman, L.M.P.; Turkstra, L.S.; Brodaty, H.; Clare, L.; Cassidy-Eagle, E.; Cox, K.L.; Chételat, G.; et al. Toward a theory-based specification of non-pharmacological treatments in aging and dementia: Focused reviews and methodological recommendations. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2021, 17, 255–270. [Google Scholar] [CrossRef]
- Han, K.; Tang, Z.; Bai, Z.; Su, W.; Zhang, H. Effects of combined cognitive and physical intervention on enhancing cognition in older adults with and without mild cognitive impairment: A systematic review and meta-analysis. Front. Aging Neurosci. 2022, 14, 878025. [Google Scholar] [CrossRef] [PubMed]
- Sharew, N.T. The effect of multimodal non-pharmacological interventions on cognitive function improvement for people with dementia: A systematic review. Front. Public Health 2022, 10, 894930. [Google Scholar] [CrossRef] [PubMed]
- Prick, A.-E.; Lange, J.d.; Scherder, E.; Twisk, J.; Pot, A.M. The effects of a multicomponent dyadic intervention on the mood, behavior, and physical health of people with dementia: A randomized controlled trial. Clin. Interv. Aging 2016, 11, 383–395. [Google Scholar] [CrossRef] [Green Version]
- Tappen, R.M.; Williams, C.L.; Barry, C.; Disesa, D. Conversation intervention with Alzheimer’s patients: Increasing the relevance of communication. Clin. Gerontol. 2002, 24, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Walter, E.; Pinquart, M. How effective are dementia caregiver interventions? An updated comprehensive meta-analysis. Gerontologist 2020, 60, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Luttenberger, K.; Hofner, B.; Graessel, E. Are the effects of a non-drug multimodal activation therapy of dementia sustainable? Follow-up study 10 months after completion of a randomised controlled trial. BMC Neurol. 2012, 12, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kivipelto, M.; Mangialasche, F.; Ngandu, T. Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Imaoka, M.; Nakao, H.; Nakamura, M.; Tazaki, F.; Maebuchi, M.; Ibuki, M.; Takeda, M. Effect of multicomponent exercise and nutrition support on the cognitive function of older adults: A randomized controlled trial. Clin. Interv. Aging 2019, 14, 2145–2153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moll van Charante, E.P.; Richard, E.; Eurelings, L.S.; van Dalen, J.W.; Ligthart, S.A.; van Bussel, E.F.; Hoevenaar-Blom, M.P.; Vermeulen, M.; van Gool, W.A. Effectiveness of a 6-year multidomain vascular care intervention to prevent dementia (prediva): A cluster-randomised controlled trial. Lancet 2016, 388, 797–805. [Google Scholar] [CrossRef]
- Andrieu, S.; Guyonnet, S.; Coley, N.; Cantet, C.; Bonnefoy, M.; Bordes, S.; Bories, L.; Cufi, M.N.; Dantoine, T.; Dartigues, J.F.; et al. Effect of long-term omega 3 polyunsaturated fatty acid supplementation with or without multidomain intervention on cognitive function in elderly adults with memory complaints (mapt): A randomised, placebo-controlled trial. Lancet Neurol. 2017, 16, 377–389. [Google Scholar] [CrossRef]
- Rosenberg, A.; Ngandu, T.; Rusanen, M.; Antikainen, R.; Backman, L.; Havulinna, S.; Hanninen, T.; Laatikainen, T.; Lehtisalo, J.; Levalahti, E.; et al. Multidomain lifestyle intervention benefits a large elderly population at risk for cognitive decline and dementia regardless of baseline characteristics: The finger trial. Alzheimer’s Dement. 2018, 14, 263–270. [Google Scholar] [CrossRef]
- Rosenberg, A.; Mangialasche, F.; Ngandu, T.; Solomon, A.; Kivipelto, M. Multidomain interventions to prevent cognitive impairment, Alzheimer’s disease, and dementia: From finger to world-wide fingers. J. Prev. Alzheimer’s Dis. 2020, 7, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Mangialasche, F.; Snyder, H.M.; Allegri, R.; Andrieu, S.; Arai, H.; Baker, L.; Belleville, S.; Brodaty, H.; Brucki, S.M.; et al. World-wide fingers network: A global approach to risk reduction and prevention of dementia. Alzheimer’s Dement. 2020, 16, 1078–1094. [Google Scholar] [CrossRef] [PubMed]
- Sindi, S.; Thunborg, C.; Rosenberg, A.; Andersen, P.; Andrieu, S.; Broersen, L.M.; Coley, N.; Couderc, C.; Duval, C.Z.; Faxen-Irving, G.; et al. Multimodal preventive trial for Alzheimer’s disease: Mind-admini pilot trial study design and progress. J. Prev. Alzheimer’s Dis. 2022, 9, 30–39. [Google Scholar] [CrossRef]
- Sugimoto, T.; Sakurai, T.; Akatsu, H.; Doi, T.; Fujiwara, Y.; Hirakawa, A.; Kinoshita, F.; Kuzuya, M.; Lee, S.; Matsuo, K.; et al. The japan-multimodal intervention trial for prevention of dementia (j-mint): The study protocol for an 18-month, multicenter, randomized, controlled trial. J. Prev. Alzheimer’s Dis. 2021, 8, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Jockusch, J.; Wiedemeier, D.; Nitschke, I. The orbid (oral health, bite force and dementia) pilot study: A study protocol for new approaches to masticatory muscle training and efficient recruitment for longitudinal studies in people with dementia. Int. J. Environ. Res. Public Health 2022, 19, 3700. [Google Scholar] [CrossRef] [PubMed]
- Bayles, K.A.; Tomoeda, C.K.; Dharmaperwira-Prins, R. Abcd: Arizona Battery for Communication Disorders of Dementia; Canyonlands Publishing: Bellemont, AZ, USA, 1993. [Google Scholar]
- Arkin, S.; Mahendra, N. Discourse analysis of Alzheimer’s patients before and after intervention: Methodology and outcomes. Aphasiology 2001, 15, 533–569. [Google Scholar] [CrossRef]
- Yu, J.T.; Xu, W.; Tan, C.C.; Andrieu, S.; Suckling, J.; Evangelou, E.; Pan, A.; Zhang, C.; Jia, J.; Feng, L.; et al. Evidence-based prevention of Alzheimer’s disease: Systematic review and meta-analysis of 243 observational prospective studies and 153 randomised controlled trials. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Stephen, R.; Ngandu, T.; Liu, Y.; Peltonen, M.; Antikainen, R.; Kemppainen, N.; Laatikainen, T.; Lötjönen, J.; Rinne, J.; Strandberg, T.; et al. Change in caide dementia risk score and neuroimaging biomarkers during a 2-year multidomain lifestyle randomized controlled trial: Results of a post-hoc subgroup analysis. J. Gerontol. Ser. A 2021, 76, 1407–1414. [Google Scholar] [CrossRef]
- Cheng, A.; Hou, Y.; Mattson, M.P. Mitochondria and neuroplasticity. ASN Neuro 2010, 2, e00045. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.; Grosgen, S.; Mett, J.; Zimmer, V.C.; Haupenthal, V.J.; Hundsdorfer, B.; Stahlmann, C.P.; Slobodskoy, Y.; Muller, U.C.; Hartmann, T.; et al. Upregulation of pgc-1alpha expression by Alzheimer’s disease-associated pathway: Presenilin 1/amyloid precursor protein (app)/intracellular domain of app. Aging Cell 2014, 13, 263–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chianese, R.; Coccurello, R.; Viggiano, A.; Scafuro, M.; Fiore, M.; Coppola, G.; Operto, F.F.; Fasano, S.; Laye, S.; Pierantoni, R.; et al. Impact of dietary fats on brain functions. Curr. Neuropharmacol. 2018, 16, 1059–1085. [Google Scholar] [CrossRef]
- Wurtman, R.J.; Ulus, I.H.; Cansev, M.; Watkins, C.J.; Wang, L.; Marzloff, G. Synaptic proteins and phospholipids are increased in gerbil brain by administering uridine plus docosahexaenoic acid orally. Brain Res. 2006, 1088, 83–92. [Google Scholar] [CrossRef]
- Wurtman, R.J.; Cansev, M.; Sakamoto, T.; Ulus, I.H. Use of phosphatide precursors to promote synaptogenesis. Annu. Rev. Nutr. 2009, 29, 59–87. [Google Scholar] [CrossRef]
- Stephen, R.; Liu, Y.; Ngandu, T.; Antikainen, R.; Hulkkonen, J.; Koikkalainen, J.; Kemppainen, N.; Lötjönen, J.; Levälahti, E.; Parkkola, R.; et al. Brain volumes and cortical thickness on mri in the finnish geriatric intervention study to prevent cognitive impairment and disability (finger). Alzheimer’s Res. Ther. 2019, 11, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicente de Sousa, O.; Soares Guerra, R.; Sousa, A.S.; Pais Henriques, B.; Pereira Monteiro, A.; Amaral, T.F. Impact of nutritional supplementation and a psychomotor program on patients with Alzheimer’s disease. Am. J. Alzheimer’s Dis. Other Dement. 2017, 32, 329–341. [Google Scholar]
- He, J.; Wang, J.; Zhong, H.; Guan, C. The effectiveness of multi-component interventions on the positive and negative aspects of well-being among informal caregivers of people with dementia: A systematic review and meta-analysis. Int. J. Environ. Res. Public Health 2022, 19, 6973. [Google Scholar]
- Harding, A.; Robinson, S.; Crean, S.; Singhrao, S.K. Can better management of periodontal disease delay the onset and progression of Alzheimer’s disease? J. Alzheimer’s Dis. 2017, 58, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Athanasaki, A.; Melanis, K.; Tsantzali, I.; Stefanou, M.I.; Ntymenou, S.; Paraskevas, S.G.; Kalamatianos, T.; Boutati, E.; Lambadiari, V.; Voumvourakis, K.I.; et al. Type 2 diabetes mellitus as a risk factor for Alzheimer’s disease: Review and meta-analysis. Biomedicines 2022, 10, 778. [Google Scholar] [CrossRef] [PubMed]
- Janoutova, J.; Machaczka, O.; Zatloukalova, A.; Janout, V. Is Alzheimer’s disease a type 3 diabetes? A review. Cent. Eur. J. Public Health 2022, 30, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Stohr, J.; Barbaresko, J.; Neuenschwander, M.; Schlesinger, S. Bidirectional association between periodontal disease and diabetes mellitus: A systematic review and meta-analysis of cohort studies. Sci. Rep. 2021, 11, 13686. [Google Scholar] [CrossRef]
- Smith, P.J.; Blumenthal, J.A.; Babyak, M.A.; Craighead, L.; Welsh-Bohmer, K.A.; Browndyke, J.N.; Strauman, T.A.; Sherwood, A. Effects of the dietary approaches to stop hypertension diet, exercise, and caloric restriction on neurocognition in overweight adults with high blood pressure. Hypertension 2010, 55, 1331–1338. [Google Scholar]
- Nitschke, I.; Hahnel, S. Zahnmedizinische versorgung älterer menschen: Chancen und herausforderungen. Bundesgesundheitsblatt Gesundh. Gesundh. 2021, 64, 802–811. [Google Scholar] [CrossRef]







| WHO Recommendations | |
|---|---|
| Strong recommendation | - Physical activity (aerobic, resistance training or multicomponent physical activity) - Tobacco cessation |
| Conditional recommendation | - Mediterranean-like diet - Interventions to treat alcohol use disorders - Cognitive training - Weight management - Management of hypertension - Management of diabetes - Management of dyslipidaemia |
| 6-Step Program | ||
|---|---|---|
| Step | Involved Disciplines | Content Clarification and Coordination |
| 1 | Oral health/dentist | - Potential risk factors that interfere with food intake? - Dental status sufficient that food intake is not perceived as unpleasant? - Xerostomia, dysphagia present? - Problems present in the mechanical comminution of food? - Current antibiotic treatment due to periodontitis etc.? |
| 2 | Speech and language therapy | - Clarification of existing dysphagia/dysphagia intervention - Language/reading comprehension present? - What to consider when communicating? - Participation in social/communication groups? |
| 3 | Sport science and physiotherapy | - Age corrected grip strength test to estimate malnutrition available? - WHO recommended sports load possible? (How many times/duration per week) - Sport practice preferably under sunlight? - Participation in regular sport activity groups? - Short individual sport program for home available? |
| 4 | Medical performance | - Signs of malnutrition present based on blood test (albumin, vitamin D, selene, vitamin B12)? - Any antibiotic treatments? (e.g., antibiotics and calcium; vitamin K and anticoagulant drugs) - Any medication that interferes with drug-degrading enzymes? (such as Cyp) - Any redocumentations that interfere with vitamin uptake or uptake of essential fatty acid? (proton-pump inhibitors) |
| 5 | Relatives/professional caregivers | - Any food intolerances or personal preferences such as vegetarian diets? - Regular food intake helps to structure the daily routine - Feeding or food intake should be done in an upright position and not lying down to avoid aspiration pneumonias - Utilize food intake, cooking classes, cooking in general to prevent social isolation - Use (of liked) foods to create sensory stimuli - Participation in social groups? E.g., regular meetings for a joint breakfast? |
| 6 | Nutritional advice | - Adjust caloric intake to achieve an age-appropriate normal weight - Avoid unintentional weight loss - When dental health affects food intake, additive use of liquid food might be necessary - Explain the Mediterranean diet: rich in antioxidants, vitamins, PUFA, polyphenols, phytochemicals, and vegetables in general - Explain potential beneficial use of medium chain fatty acids (coconut oil) - Supplementation of vitamin B12, vitamin D - Consider LipiDiDiet-based supplementation - Ensure sufficient intake of omega 3 fatty acids such as DHA (e.g., two times/week fatty sea fish) - Avoid trans fatty acid and highly processed food in general - In case of antibiotic treatment: make sure that the microbiome is built up afterwards through nutrition (increase fiber intake, probiotics if necessary) - Consider further phytochemicals as supplementation such as EGCG (or green tea extract), Ginseng, Gingko, resveratrol, etc. - Moderate coffee consumption |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ablinger, I.; Dressel, K.; Rott, T.; Lauer, A.A.; Tiemann, M.; Batista, J.P.; Taddey, T.; Grimm, H.S.; Grimm, M.O.W. Interdisciplinary Approaches to Deal with Alzheimer’s Disease—From Bench to Bedside: What Feasible Options Do Already Exist Today? Biomedicines 2022, 10, 2922. https://doi.org/10.3390/biomedicines10112922
Ablinger I, Dressel K, Rott T, Lauer AA, Tiemann M, Batista JP, Taddey T, Grimm HS, Grimm MOW. Interdisciplinary Approaches to Deal with Alzheimer’s Disease—From Bench to Bedside: What Feasible Options Do Already Exist Today? Biomedicines. 2022; 10(11):2922. https://doi.org/10.3390/biomedicines10112922
Chicago/Turabian StyleAblinger, Irene, Katharina Dressel, Thea Rott, Anna Andrea Lauer, Michael Tiemann, João Pedro Batista, Tim Taddey, Heike Sabine Grimm, and Marcus Otto Walter Grimm. 2022. "Interdisciplinary Approaches to Deal with Alzheimer’s Disease—From Bench to Bedside: What Feasible Options Do Already Exist Today?" Biomedicines 10, no. 11: 2922. https://doi.org/10.3390/biomedicines10112922
APA StyleAblinger, I., Dressel, K., Rott, T., Lauer, A. A., Tiemann, M., Batista, J. P., Taddey, T., Grimm, H. S., & Grimm, M. O. W. (2022). Interdisciplinary Approaches to Deal with Alzheimer’s Disease—From Bench to Bedside: What Feasible Options Do Already Exist Today? Biomedicines, 10(11), 2922. https://doi.org/10.3390/biomedicines10112922