


Bromodomain and Extra-Terminal Proteins in Brain Physiology and Pathology: BET-ing on Epigenetic Regulation
 , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
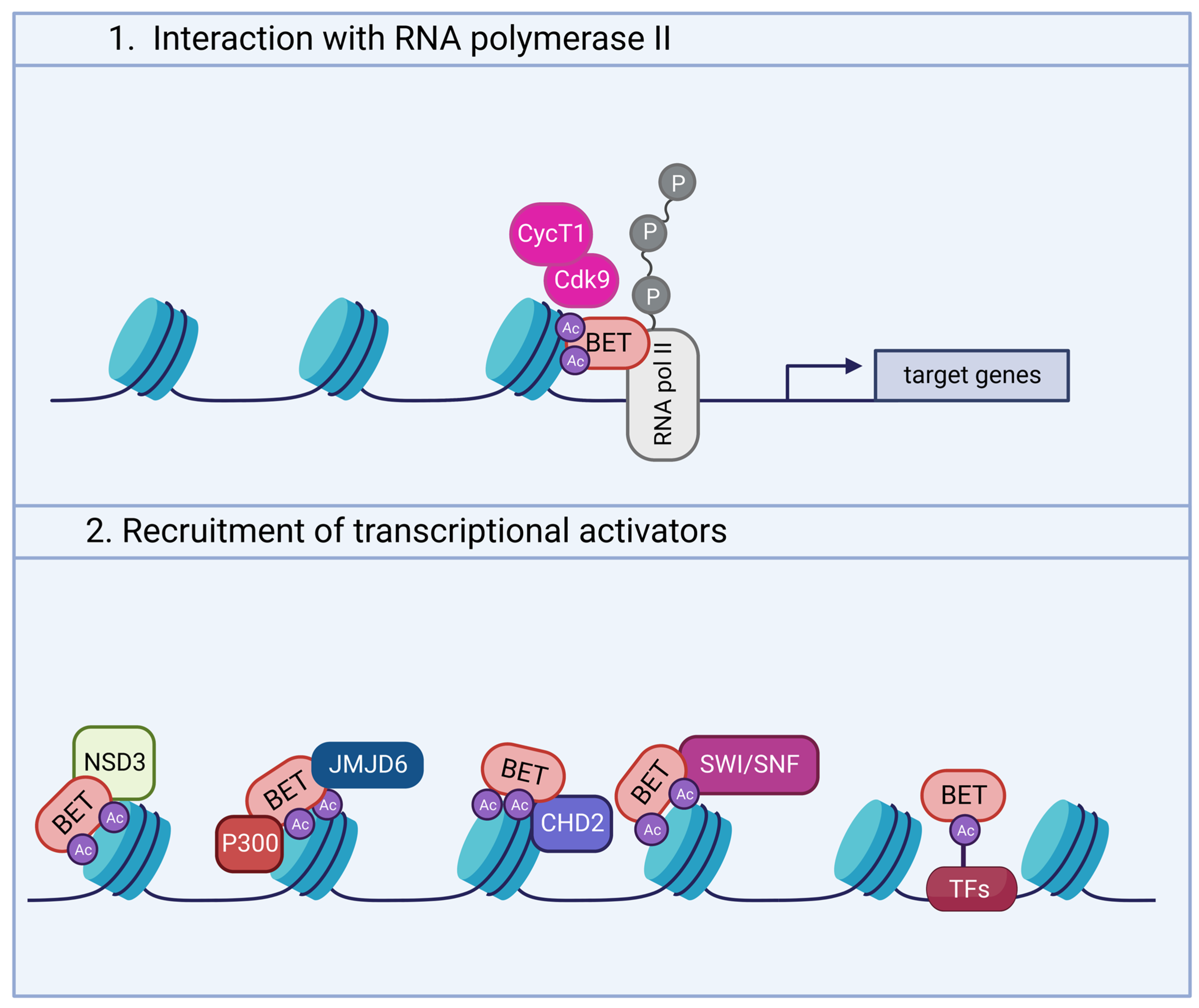
2. Structure and Functions of BET Proteins
Expression of BET Proteins in the Brain
3. BET Proteins in Brain Physiology
3.1. Neuronal Differentiation and Neurodevelopment
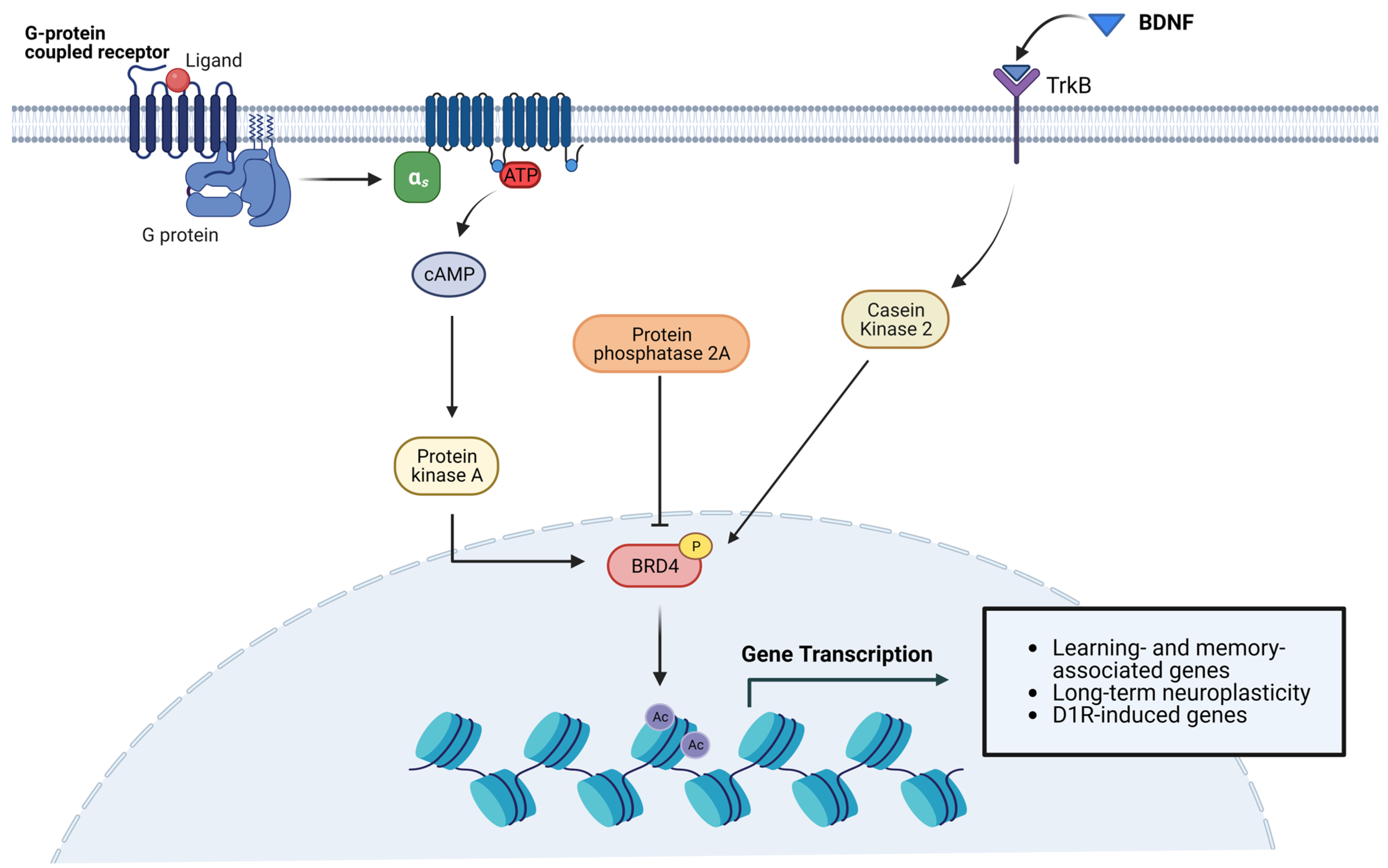
3.2. Cognitive Functions and Behavior
4. Involvement of BET Proteins in Neuropathological Conditions
4.1. Neurodevelopmental Disorders
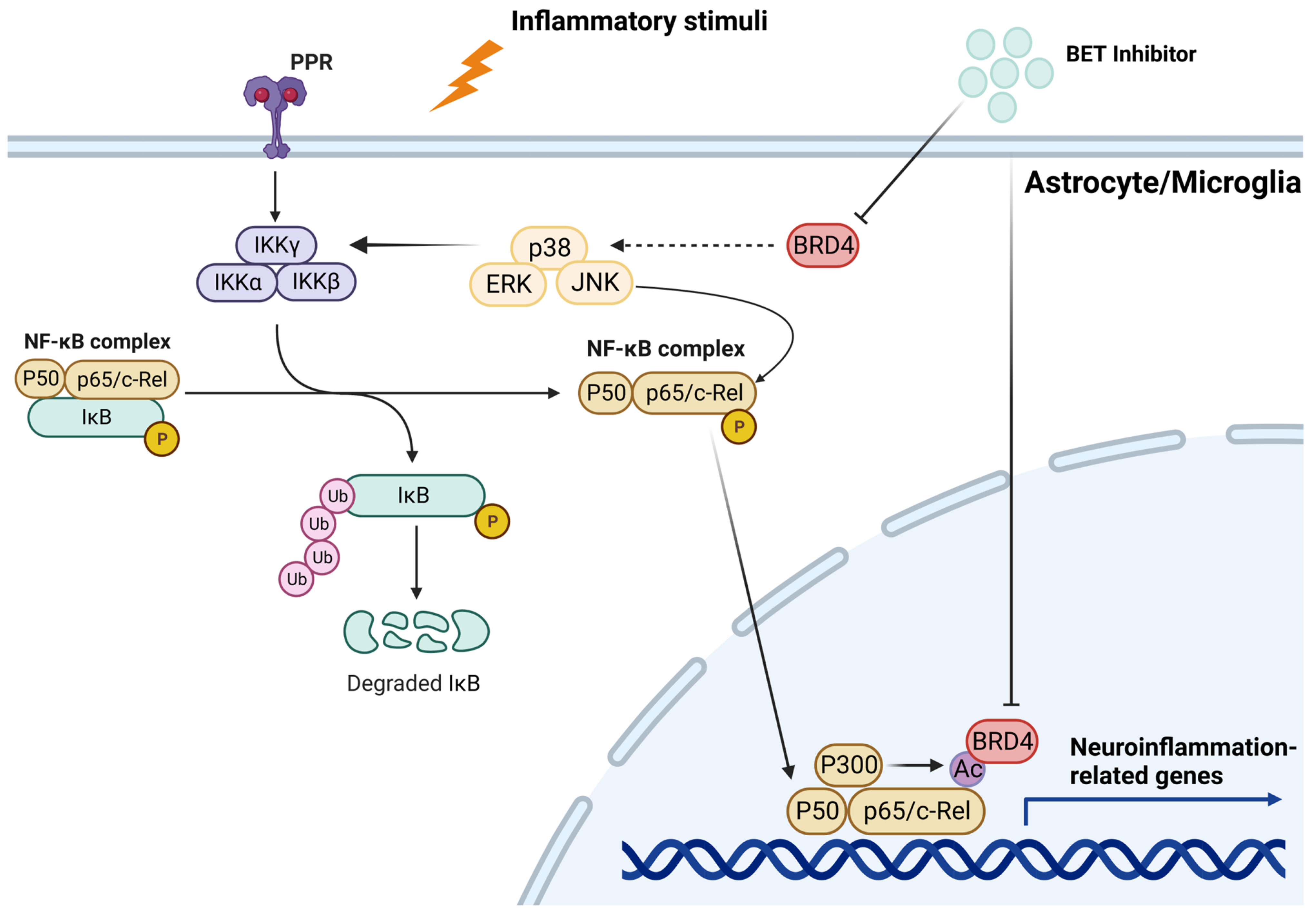
4.2. Neuroinflammation
4.3. Neurodegenerative Diseases

{kind=link}
{kind=link}
{kind=link}
| Disease | Experimental Model | BET Inhibition | Pathways/Processes | Functional Effects | References |
|---|---|---|---|---|---|
| AD | APP/PS1-21 mouse |
|
|
| [23] |
| 3xTg mouse |
|
|
| [17] | |
| Wistar rats |
|
|
| [143] | |
| Wistar rats |
|
|
| [56] | |
|
|
|
| [146] | |
| BV2 murine microglial cells |
|
|
| [145] | |
| Clinical study (NCT02586155) | Apabetalone (RVX-208) |
|
| [148] | |
| PD | 6-OHDA rat model |
|
|
| [149] |
| 6-OHDA rat model |
|
|
| [150] | |
| HD |
|
|
|
| [151] |
| ALS/ FTD | Cells derived from ALS patients |
|
|
| [152] |
|
|
|
| [153] |
4.4. Neuropsychiatric Disorders
| Disease | Experimental Model | BET Inhibition | Pathways/Processes | Functional Effects | References |
|---|---|---|---|---|---|
| SUD |
|
|
|
| [16] |
| Long Evans rats |
|
|
| [182] | |
| PTSD | C57BL/6 mice |
|
|
| [18] |
| C57BL/6 mice |
|
|
| [100] | |
| SZ |
|
|
|
| [192] |
| Wistar Han rats |
|
|
| [193] |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haynes, S.R.; Dollard, C.; Winston, F.; Beck, S.; Trowsdale, J.; Dawid, I.B. The Bromodomain: A Conserved Sequence Found in Human, Drosophila and Yeast Proteins. Nucleic Acids Res. 1992, 20, 2603. [Google Scholar] [CrossRef] [Green Version]
- Filippakopoulos, P.; Knapp, S. The Bromodomain Interaction Module. FEBS Lett. 2012, 586, 2692–2704. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.-P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvani, A.; Thiriet, C. Nucleosome Dancing at the Tempo of Histone Tail Acetylation. Genes 2015, 6, 607–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, L.; Zhou, M.-M. Bromodomain: An Acetyl-Lysine Binding Domain. FEBS Lett. 2002, 513, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.; Sowa, M.E.; Ottinger, M.; Smith, J.A.; Shi, Y.; Harper, J.W.; Howley, P.M. The Brd4 Extraterminal Domain Confers Transcription Activation Independent of PTEFb by Recruiting Multiple Proteins, Including NSD3. Mol. Cell. Biol. 2011, 31, 2641–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Ma, Q.; Wong, K.; Li, W.; Ohgi, K.; Zhang, J.; Aggarwal, A.K.; Rosenfeld, M.G. Brd4 and JMJD6-Associated Anti-Pause Enhancers in Regulation of Transcriptional Pause Release. Cell 2013, 155, 1581–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbieri, I.; Cannizzaro, E.; Dawson, M.A. Bromodomains as Therapeutic Targets in Cancer. Brief. Funct. Genom. 2013, 12, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Boehm, D.; Conrad, R.J.; Ott, M. Bromodomain Proteins in HIV Infection. Viruses 2013, 5, 1571–1586. [Google Scholar] [CrossRef]
- Prinjha, R.K.; Witherington, J.; Lee, K. Place Your BETs: The Therapeutic Potential of Bromodomains. Trends Pharmacol. Sci. 2012, 33, 146–153. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective Inhibition of BET Bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of Inflammation by a Synthetic Histone Mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segatto, M.; Fittipaldi, R.; Pin, F.; Sartori, R.; Dae Ko, K.; Zare, H.; Fenizia, C.; Zanchettin, G.; Pierobon, E.S.; Hatakeyama, S.; et al. Epigenetic Targeting of Bromodomain Protein BRD4 Counteracts Cancer Cachexia and Prolongs Survival. Nat. Commun. 2017, 8, 1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrieu, G.P.; Shafran, J.S.; Deeney, J.T.; Bharadwaj, K.R.; Rangarajan, A.; Denis, G. v BET Proteins in Abnormal Metabolism, Inflammation, and the Breast Cancer Microenvironment. J. Leukoc. Biol. 2018, 104, 265–274. [Google Scholar] [CrossRef]
- Korb, E.; Herre, M.; Zucker-Scharff, I.; Darnell, R.B.; Allis, C.D. BET Protein Brd4 Activates Transcription in Neurons and BET Inhibitor Jq1 Blocks Memory in Mice. Nat. Neurosci. 2015, 18, 1464–1473. [Google Scholar] [CrossRef]
- Sartor, G.C.; Powell, S.K.; Brothers, S.P.; Wahlestedt, C. Epigenetic Readers of Lysine Acetylation Regulate Cocaine-Induced Plasticity. J. Neurosci. 2015, 35, 15062–15072. [Google Scholar] [CrossRef] [Green Version]
- Magistri, M.; Velmeshev, D.; Makhmutova, M.; Patel, P.; Sartor, G.C.; Volmar, C.-H.; Wahlestedt, C.; Faghihi, M.A. The BET-Bromodomain Inhibitor JQ1 Reduces Inflammation and Tau Phosphorylation at Ser396 in the Brain of the 3xTg Model of Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 985–995. [Google Scholar] [CrossRef]
- Duan, Q.; Huang, F.L.; Li, S.J.; Chen, K.Z.; Gong, L.; Qi, J.; Yang, Z.H.; Yang, T.L.; Li, F.; Li, C.Q. BET Proteins Inhibitor JQ-1 Impaired the Extinction of Remote Auditory Fear Memory: An Effect Mediated by Insulin like Growth Factor 2. Neuropharmacology 2020, 177, 108255. [Google Scholar] [CrossRef]
- Sullivan, J.M.; Badimon, A.; Schaefer, U.; Ayata, P.; Gray, J.; Chung, C.; von Schimmelmann, M.; Zhang, F.; Garton, N.; Smithers, N.; et al. Autism-like Syndrome Is Induced by Pharmacological Suppression of BET Proteins in Young Mice. J. Exp. Med. 2015, 212, 1771–1781. [Google Scholar] [CrossRef] [Green Version]
- Umehara, T.; Nakamura, Y.; Wakamori, M.; Ozato, K.; Yokoyama, S.; Padmanabhan, B. Structural Implications for K5/K12-Di-Acetylated Histone H4 Recognition by the Second Bromodomain of BRD2. FEBS Lett. 2010, 584, 3901–3908. [Google Scholar] [CrossRef] [Green Version]
- Umehara, T.; Nakamura, Y.; Jang, M.K.; Nakano, K.; Tanaka, A.; Ozato, K.; Padmanabhan, B.; Yokoyama, S. Structural Basis for Acetylated Histone H4 Recognition by the Human BRD2 Bromodomain. J. Biol. Chem. 2010, 285, 7610–7618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeRoy, G.; Chepelev, I.; DiMaggio, P.A.; Blanco, M.A.; Zee, B.M.; Zhao, K.; Garcia, B.A. Proteogenomic Characterization and Mapping of Nucleosomes Decoded by Brd and HP1 Proteins. Genome Biol. 2012, 13, R68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benito, E.; Ramachandran, B.; Schroeder, H.; Schmidt, G.; Urbanke, H.; Burkhardt, S.; Capece, V.; Dean, C.; Fischer, A. The BET/BRD Inhibitor JQ1 Improves Brain Plasticity in WT and APP Mice. Transl. Psychiatry 2017, 7, e1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, R.; Meslamani, J.; Zhou, M.M. The Bromodomain: From Epigenome Reader to Druggable Target. Biochim. Biophys. Acta BBA—Gene Regul. Mech. 2014, 1839, 676–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.-M.; Zhou, M.-M. Structure and Ligand of a Histone Acetyltransferase Bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Salvia, M.; Esteller, M. Bromodomain Inhibitors and Cancer Therapy: From Structures to Applications. Epigenetics 2017, 12, 323–339. [Google Scholar] [CrossRef] [Green Version]
- Zaware, N.; Zhou, M.-M. Chemical Modulators for Epigenome Reader Domains as Emerging Epigenetic Therapies for Cancer and Inflammation. Curr. Opin. Chem. Biol. 2017, 39, 116–125. [Google Scholar] [CrossRef]
- Jones, M.H.; Numata, M.; Shimane, M. Identification and Characterization of BRDT: A Testis-Specific Gene Related to the Bromodomain Genes RING3 and Drosophila Fsh. Genomics 1997, 45, 529–534. [Google Scholar] [CrossRef]
- Taniguchi, Y. The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. Int. J. Mol. Sci. 2016, 17, 1849. [Google Scholar] [CrossRef] [Green Version]
- Mujtaba, S.; Zeng, L.; Zhou, M.-M. Structure and Acetyl-Lysine Recognition of the Bromodomain. Oncogene 2007, 26, 5521–5527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, J.T.; Glass, K.C. Biological Function and Histone Recognition of Family IV Bromodomain-Containing Proteins. J. Cell. Physiol. 2018, 233, 1877–1886. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Chitsaz, F.; Abbasi, A.; Misteli, T.; Ozato, K. The Double Bromodomain Protein Brd4 Binds to Acetylated Chromatin during Interphase and Mitosis. Proc. Natl. Acad. Sci. USA 2003, 100, 8758–8763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morinière, J.; Rousseaux, S.; Steuerwald, U.; Soler-López, M.; Curtet, S.; Vitte, A.-L.; Govin, J.; Gaucher, J.; Sadoul, K.; Hart, D.J.; et al. Cooperative Binding of Two Acetylation Marks on a Histone Tail by a Single Bromodomain. Nature 2009, 461, 664–668. [Google Scholar] [CrossRef]
- Zhang, Q.; Zeng, L.; Shen, C.; Ju, Y.; Konuma, T.; Zhao, C.; Vakoc, C.R.; Zhou, M.-M. Structural Mechanism of Transcriptional Regulator NSD3 Recognition by the ET Domain of BRD4. Structure 2016, 24, 1201–1208. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.W.; Veschambre, P.; Erdjument-Bromage, H.; Tempst, P.; Conaway, J.W.; Conaway, R.C.; Kornberg, R.D. Mammalian Mediator of Transcriptional Regulation and Its Possible Role as an End-Point of Signal Transduction Pathways. Proc. Natl. Acad. Sci. USA 1998, 95, 8538–8543. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The Bromodomain Protein Brd4 Is a Positive Regulatory Component of P-TEFb and Stimulates RNA Polymerase II-Dependent Transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef]
- Barrero, M.J. Epigenetic Strategies to Boost Cancer Immunotherapies. Int. J. Mol. Sci. 2017, 18, 1108. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Li, T.; Price, D.H. RNA Polymerase II Elongation Control. Annu. Rev. Biochem. 2012, 81, 119–143. [Google Scholar] [CrossRef] [Green Version]
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved P-TEFb-Interacting Domain of BRD4 Inhibits HIV Transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 13690–13695. [Google Scholar] [CrossRef] [Green Version]
- Schröder, S.; Cho, S.; Zeng, L.; Zhang, Q.; Kaehlcke, K.; Mak, L.; Lau, J.; Bisgrove, D.; Schnölzer, M.; Verdin, E.; et al. Two-Pronged Binding with Bromodomain-Containing Protein 4 Liberates Positive Transcription Elongation Factor b from Inactive Ribonucleoprotein Complexes. J. Biol. Chem. 2012, 287, 1090–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Yik, J.H.N.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for Stimulation of Transcriptional Elongation by the Bromodomain Protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Kanno, Y.; LeRoy, G.; Campos, E.; Sun, H.-W.; Brooks, S.R.; Vahedi, G.; Heightman, T.D.; Garcia, B.A.; Reinberg, D.; et al. BRD4 Assists Elongation of Both Coding and Enhancer RNAs by Interacting with Acetylated Histones. Nat. Struct. Mol. Biol. 2014, 21, 1047–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adelman, K.; Lis, J.T. Promoter-Proximal Pausing of RNA Polymerase II: Emerging Roles in Metazoans. Nat. Rev. Genet. 2012, 13, 720–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.-Y.; Lee, A.-Y.; Lai, H.-T.; Zhang, H.; Chiang, C.-M. Phospho Switch Triggers Brd4 Chromatin Binding and Activator Recruitment for Gene-Specific Targeting. Mol. Cell 2013, 49, 843–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, G.E.; Mayer, A.; Buckley, D.L.; Erb, M.A.; Roderick, J.E.; Vittori, S.; Reyes, J.M.; di Iulio, J.; Souza, A.; Ott, C.J.; et al. BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol. Cell 2017, 67, 5–18.e19. [Google Scholar] [CrossRef] [Green Version]
- Bhagwat, A.S.; Roe, J.-S.; Mok, B.Y.L.; Hohmann, A.F.; Shi, J.; Vakoc, C.R. BET Bromodomain Inhibition Releases the Mediator Complex from Select Cis-Regulatory Elements. Cell Rep. 2016, 15, 519–530. [Google Scholar] [CrossRef] [Green Version]
- LeRoy, G.; Rickards, B.; Flint, S.J. The Double Bromodomain Proteins Brd2 and Brd3 Couple Histone Acetylation to Transcription. Mol. Cell 2008, 30, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Alpatov, R.; Lesch, B.J.; Nakamoto-Kinoshita, M.; Blanco, A.; Chen, S.; Stützer, A.; Armache, K.J.; Simon, M.D.; Xu, C.; Ali, M.; et al. A Chromatin-Dependent Role of the Fragile X Mental Retardation Protein FMRP in the DNA Damage Response. Cell 2014, 157, 869–881. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Whyte, W.A.; Zepeda-Mendoza, C.J.; Milazzo, J.P.; Shen, C.; Roe, J.S.; Minder, J.L.; Mercan, F.; Wang, E.; Eckersley-Maslin, M.A.; et al. Role of SWI/SNF in Acute Leukemia Maintenance and Enhancer-Mediated Myc Regulation. Genes Dev. 2013, 27, 2648–2662. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Kamikawa, Y.F.; Donohoe, M.E. Brd4′s Bromodomains Mediate Histone H3 Acetylation and Chromatin Remodeling in Pluripotent Cells through P300 and Brg1. Cell Rep. 2018, 25, 1756–1771. [Google Scholar] [CrossRef] [Green Version]
- Bai, P.; Wey, H.-Y.; Patnaik, D.; Lu, X.; Lan, Y.; Rokka, J.; Stephanie, F.; Haggarty, S.J.; Wang, C. Positron Emission Tomography Probes Targeting Bromodomain and Extra-Terminal (BET) Domains to Enable in Vivo Neuroepigenetic Imaging. Chem. Commun. 2019, 55, 12932–12935. [Google Scholar] [CrossRef]
- Gyuris, A.; Donovan, D.J.; Seymour, K.A.; Lovasco, L.A.; Smilowitz, N.R.; Halperin, A.L.P.; Klysik, J.E.; Freiman, R.N. The Chromatin-Targeting Protein Brd2 Is Required for Neural Tube Closure and Embryogenesis. Biochim. Biophys. Acta Gene Regul. Mech. 2009, 1789, 413–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padmanabhan, B.; Mathur, S.; Manjula, R.; Tripathi, S. Bromodomain and Extra-Terminal (BET) Family Proteins: New Therapeutic Targets in Major Diseases. J. Biosci. 2016, 41, 295–311. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.-S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.-H.; Joseph, N.; Gao, J.; Nieland, T.J.F.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 Negatively Regulates Memory Formation and Synaptic Plasticity. Nature 2009, 459, 55–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badrikoohi, M.; Esmaeli, A.; Babaei, P. Simultaneous Administration of Bromodomain and Histone Deacetylase I Inhibitors Alleviates Cognition Deficit in Alzheimer’s Model of Rats. Brain Res. Bull. 2022, 179, 49–56. [Google Scholar] [CrossRef]
- Baek, M.; Yoo, E.; Choi, H.I.; An, G.Y.; Chai, J.C.; Lee, Y.S.; Jung, K.H.; Chai, Y.G. The BET Inhibitor Attenuates the Inflammatory Response and Cell Migration in Human Microglial HMC3 Cell Line. Sci. Rep. 2021, 11, 8828. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yang, C.; Candelario-Jalil, E. Role of BET Proteins in Inflammation and CNS Diseases. Front. Mol. Biosci. 2021, 8, 748449. [Google Scholar] [CrossRef]
- Picaud, S.; Leonards, K.; Lambert, J.-P.; Dovey, O.; Wells, C.; Fedorov, O.; Monteiro, O.; Fujisawa, T.; Wang, C.-Y.; Lingard, H.; et al. Promiscuous Targeting of Bromodomains by Bromosporine Identifies BET Proteins as Master Regulators of Primary Transcription Response in Leukemia. Sci. Adv. 2023, 2, e1600760. [Google Scholar] [CrossRef] [Green Version]
- Schwalm, M.P.; Knapp, S. BET Bromodomain Inhibitors. Curr. Opin. Chem. Biol. 2022, 68, 102148. [Google Scholar] [CrossRef]
- Alqahtani, A.; Choucair, K.; Ashraf, M.; Hammouda, D.M.; Alloghbi, A.; Khan, T.; Senzer, N.; Nemunaitis, J. Bromodomain and Extra-Terminal Motif Inhibitors: A Review of Preclinical and Clinical Advances in Cancer Therapy. Future Sci. OA 2019, 5, FSO372. [Google Scholar] [CrossRef] [Green Version]
- Watson, R.J.; Bamborough, P.; Barnett, H.; Chung, C.; Davis, R.; Gordon, L.; Grandi, P.; Petretich, M.; Phillipou, A.; Prinjha, R.K.; et al. GSK789: A Selective Inhibitor of the First Bromodomains (BD1) of the Bromo and Extra Terminal Domain (BET) Proteins. J. Med. Chem. 2020, 63, 9045–9069. [Google Scholar] [CrossRef]
- Babigian, C.J.; Wiedner, H.J.; Wahlestedt, C.; Sartor, G.C. JQ1 Attenuates Psychostimulant- but Not Opioid-Induced Conditioned Place Preference. Behav. Brain Res. 2022, 418, 113644. [Google Scholar] [CrossRef]
- Saunders, A.; Macosko, E.Z.; Wysoker, A.; Goldman, M.; Krienen, F.M.; de Rivera, H.; Bien, E.; Baum, M.; Bortolin, L.; Wang, S.; et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 2018, 174, 1015–1030.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.B.; Sartor, G.C. BET Bromodomains as Novel Epigenetic Targets for Brain Health and Disease. Neuropharmacology 2020, 181, 108306. [Google Scholar] [CrossRef]
- Jones-Tabah, J.; Martin, R.D.; Chen, J.J.; Tanny, J.C.; Clarke, P.B.S.; Hébert, T.E. A Role for BET Proteins in Regulating Basal, Dopamine-Induced and CAMP/PKA-Dependent Transcription in Rat Striatal Neurons. Cell. Signal. 2022, 91, 110226. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Long, H.; Bu, Q.; Zhao, Y.; Wang, H.; Tian, J.; Cen, X. Role of BRD4 Phosphorylation in the Nucleus Accumbens in Relapse to Cocaine-Seeking Behavior in Mice. Addict. Biol. 2020, 25, e12808. [Google Scholar] [CrossRef]
- Wu, S.; Wang, L.; Zhang, L.; Xu, X.; Zhao, J. Molecular Dynamics Simulations Data of Six Compounds F3J-BRD4/CBP, EX1-BRD4/CBP, and E2T-BRD4/CBP. Data Brief 2021, 36, 107009. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.S.; Hong, S.H.; Sim, S.; Cho, K.S.; Kim, J.-W.; Yang, S.M.; Jeon, S.J.; You, J.S.; Shin, C.Y. The Epigenetic Reader BRD2 as a Specific Modulator of PAI-1 Expression in Lipopolysaccharide-Stimulated Mouse Primary Astrocytes. Neurochem. Res. 2015, 40, 2211–2219. [Google Scholar] [CrossRef]
- Chiang, C.M. Nonequivalent Response to Bromodomain-Targeting BET Inhibitors in Oligodendrocyte Cell Fate Decision. Chem. Biol. 2014, 21, 804–806. [Google Scholar] [CrossRef] [Green Version]
- Cowan, W.M.; Cowan, W.M.; Jessell, T.M.; Zipursky, S.L. Molecular and Cellular Approaches to Neural Development; Oxford University Press: New York, NY, USA, 1998; ISBN 9780199865833. [Google Scholar]
- Reese, D.; Drapeau, P. Neurite Growth Patterns Leading to Functional Synapses in an Identified Embryonic Neuron. J. Neurosci. 1998, 18, 5652–5662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirabayashi, Y.; Gotoh, Y. Epigenetic Control of Neural Precursor Cell Fate during Development. Nat. Rev. Neurosci. 2010, 11, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Juliandi, B.; Abematsu, M.; Nakashima, K. Epigenetic Regulation in Neural Stem Cell Differentiation. Dev. Growth Differ. 2010, 52, 493–504. [Google Scholar] [CrossRef]
- Li, J.; Ma, J.; Meng, G.; Lin, H.; Wu, S.; Wang, J.; Luo, J.; Xu, X.; Tough, D.; Lindon, M.; et al. BET Bromodomain Inhibition Promotes Neurogenesis While Inhibiting Gliogenesis in Neural Progenitor Cells. Stem Cell Res. 2016, 17, 212–221. [Google Scholar] [CrossRef] [Green Version]
- Westphal, M.; Sant, P.; Hauser, A.T.; Jung, M.; Driever, W. Chemical Genetics Screen Identifies Epigenetic Mechanisms Involved in Dopaminergic and Noradrenergic Neurogenesis in Zebrafish. Front. Genet. 2020, 11, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsume, M.; Kimura-Yoshida, C.; Mochida, K.; Shibukawa, Y.; Amazaki, S.; Wada, Y.; Hiramatsu, R.; Shimokawa, K.; Matsuo, I. Brd2 Is Required for Cell Cycle Exit and Neuronal Differentiation through the E2F1 Pathway in Mouse Neuroepithelial Cells. Biochem. Biophys. Res. Commun. 2012, 425, 762–768. [Google Scholar] [CrossRef]
- Crowley, T.E.; Brunori, M.; Rhee, K.; Wang, X.; Wolgemuth, D.J. Change in Nuclear-Cytoplasmic Localization of a Double-Bromodomain Protein during Proliferation and Differentiation of Mouse Spinal Cord and Dorsal Root Ganglia. Dev. Brain Res. 2004, 149, 93–101. [Google Scholar] [CrossRef]
- Garcia-Gutierrez, P.; Mundi, M.; Garcia-Dominguez, M. Association of Bromodomain BET Proteins with Chromatin Requires Dimerization through the Conserved Motif B. J. Cell Sci. 2012, 125, 3671–3680. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Gutierrez, P.; Juarez-Vicente, F.; Wolgemuth, D.J.; Garcia-Dominguez, M. Pleiotrophin Antagonizes Brd2 during Neuronal Differentiation. J. Cell Sci. 2014, 127, 2554–2564. [Google Scholar] [CrossRef] [Green Version]
- Linares-Saldana, R.; Kim, W.; Bolar, N.A.; Zhang, H.; Koch-Bojalad, B.A.; Yoon, S.; Shah, P.P.; Karnay, A.; Park, D.S.; Luppino, J.M.; et al. BRD4 Orchestrates Genome Folding to Promote Neural Crest Differentiation. Nat. Genet. 2021, 53, 1480–1492. [Google Scholar] [CrossRef]
- Houzelstein, D.; Bullock, S.L.; Lynch, D.E.; Grigorieva, E.F.; Wilson, V.A.; Beddington, R.S.P. Growth and Early Postimplantation Defects in Mice Deficient for the Bromodomain-Containing Protein Brd4. Mol. Cell. Biol. 2002, 22, 3794–3802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa, J.S.; Luo, L. Timing Neurogenesis and Differentiation: Insights from Quantitative Clonal Analyses of Cerebellar Granule Cells. J. Neurosci. 2008, 28, 2301–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penas, C.; Maloof, M.E.; Stathias, V.; Long, J.; Tan, S.K.; Mier, J.; Fang, Y.; Valdes, C.; Rodriguez-Blanco, J.; Chiang, C.M.; et al. Time Series Modeling of Cell Cycle Exit Identifies Brd4 Dependent Regulation of Cerebellar Neurogenesis. Nat. Commun. 2019, 10, 3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Zundert, B.; Montecino, M. Epigenetic Changes and Chromatin Reorganization in Brain Function: Lessons from Fear Memory Ensemble and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 12081. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.A.; Li, Y.; Bilal, A.H.M.; Qin, T.; Yuan, Z.; Zhao, W. A Comprehensive Review of BET Protein Biochemistry, Physiology, and Pathological Roles. Front. Pharmacol. 2022, 13, 818891. [Google Scholar] [CrossRef] [PubMed]
- Sartor, G.C.; Malvezzi, A.M.; Kumar, A.; Andrade, N.S.; Wiedner, H.J.; Vilca, S.J.; Janczura, K.J.; Bagheri, A.; Al-Ali, H.; Powell, S.K.; et al. Enhancement of BDNF Expression and Memory by HDAC Inhibition Requires BET Bromodomain Reader Proteins. J. Neurosci. 2019, 39, 612–626. [Google Scholar] [CrossRef] [Green Version]
- Volmar, C.H.; Wahlestedt, C. Histone Deacetylases (HDACs) and Brain Function. Neuroepigenetics 2015, 1, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Pulya, S.; Mahale, A.; Bobde, Y.; Routholla, G.; Patel, T.; Swati; Biswas, S.; Sharma, V.; Kulkarni, O.P.; Ghosh, B. PT3: A Novel Benzamide Class Histone Deacetylase 3 Inhibitor Improves Learning and Memory in Novel Object Recognition Mouse Model. ACS Chem. Neurosci. 2021, 12, 883–892. [Google Scholar] [CrossRef]
- Kwapis, J.L.; Alaghband, Y.; López, A.J.; White, A.O.; Campbell, R.R.; Dang, R.T.; Rhee, D.; Tran, A.V.; Carl, A.E.; Matheos, D.P.; et al. Context and Auditory Fear Are Differentially Regulated by HDAC3 Activity in the Lateral and Basal Subnuclei of the Amygdala. Neuropsychopharmacology 2017, 42, 1284–1294. [Google Scholar] [CrossRef] [Green Version]
- Bieszczad, K.M.; Bechay, K.; Rusche, J.R.; Jacques, V.; Kudugunti, S.; Miao, W.; Weinberger, N.M.; McGaugh, J.L.; Wood, M.A. Histone Deacetylase Inhibition via RGFP966 Releases the Brakes on Sensory Cortical Plasticity and the Specificity of Memory Formation. J. Neurosci. 2015, 35, 13124–13132. [Google Scholar] [CrossRef] [Green Version]
- Malvaez, M.; Greenfield, V.Y.; Matheos, D.P.; Angelillis, N.A.; Murphy, M.D.; Kennedy, P.J.; Wood, M.A.; Wassum, K.M. Habits Are Negatively Regulated by Histone Deacetylase 3 in the Dorsal Striatum. Biol. Psychiatry 2018, 84, 383–392. [Google Scholar] [CrossRef]
- Kim, S.-K.; Liu, X.; Park, J.; Um, D.; Kilaru, G.; Chiang, C.-M.; Kang, M.; Huber, K.M.; Kang, K.; Kim, T.-K. Functional Coordination of BET Family Proteins Underlies Altered Transcription Associated with Memory Impairment in Fragile X Syndrome. Sci. Adv. 2021, 7, eabf7346. [Google Scholar] [CrossRef]
- Briscione, M.A.; Jovanovic, T.; Norrholm, S.D. Conditioned Fear Associated Phenotypes as Robust, Translational Indices of Trauma-, Stressor-, and Anxiety-Related Behaviors. Front. Psychiatry 2014, 5, 88. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.R.; McGuire, J.; Lazarus, R.; Palmer, A.A. Pavlovian Fear Memory Circuits and Phenotype Models of PTSD. Neuropharmacology 2012, 62, 638–646. [Google Scholar] [CrossRef]
- Mahan, A.L.; Ressler, K.J. Fear Conditioning, Synaptic Plasticity and the Amygdala: Implications for Posttraumatic Stress Disorder. Trends Neurosci. 2012, 35, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Hemstedt, T.J.; Lattal, K.M.; Wood, M.A. Reconsolidation and Extinction: Using Epigenetic Signatures to Challenge Conventional Wisdom. Neurobiol. Learn. Mem. 2017, 142, 55–65. [Google Scholar] [CrossRef]
- Whittle, N.; Singewald, N. HDAC Inhibitors as Cognitive Enhancers in Fear, Anxiety and Trauma Therapy: Where Do We Stand? Biochem. Soc. Trans. 2014, 42, 569–581. [Google Scholar] [CrossRef]
- Bousiges, O.; Neidl, R.; Majchrzak, M.; Muller, M.-A.; Barbelivien, A.; Pereira de Vasconcelos, A.; Schneider, A.; Loeffler, J.-P.; Cassel, J.-C.; Boutillier, A.-L. Detection of Histone Acetylation Levels in the Dorsal Hippocampus Reveals Early Tagging on Specific Residues of H2B and H4 Histones in Response to Learning. PLoS ONE 2013, 8, e57816. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.L.; Li, F.; Zhang, W.J.; Li, S.J.; Yang, Z.H.; Yang, T.L.; Qi, J.; Duan, Q.; Li, C.Q. Brd4 Participates in Epigenetic Regulation of the Extinction of Remote Auditory Fear Memory. Neurobiol. Learn. Mem. 2021, 179, 107383. [Google Scholar] [CrossRef]
- Stathis, A.; Bertoni, F. BET Proteins as Targets for Anticancer Treatment. Cancer Discov. 2018, 8, 24–36. [Google Scholar] [CrossRef] [Green Version]
- Korb, E.; Herre, M.; Zucker-Scharff, I.; Gresack, J.; Allis, C.D.; Darnell, R.B. Excess Translation of Epigenetic Regulators Contributes to Fragile X Syndrome and Is Alleviated by Brd4 Inhibition. Cell 2017, 170, 1209–1223.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Tanaka, Y.; Patterson, B.; Hwang, S.M.; Hysolli, E.; Cakir, B.; Kim, K.Y.; Wang, W.; Kang, Y.J.; Clement, E.M.; et al. Dysregulation of BRD4 Function Underlies the Functional Abnormalities of MeCP2 Mutant Neurons. Mol. Cell 2020, 79, 84–98.e9. [Google Scholar] [CrossRef]
- Bassell, G.J.; Warren, S.T. Fragile X Syndrome: Loss of Local MRNA Regulation Alters Synaptic Development and Function. Neuron 2008, 60, 201–214. [Google Scholar] [CrossRef] [Green Version]
- Bear, M.F.; Huber, K.M.; Warren, S.T. The MGluR Theory of Fragile X Mental Retardation. Trends Neurosci. 2004, 27, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Niere, F.; Wilkerson, J.R.; Huber, K.M. Evidence for a Fragile X Mental Retardation Protein-Mediated Translational Switch in Metabotropic Glutamate Receptor-Triggered Arc Translation and Long-Term Depression. J. Neurosci. 2012, 32, 5924–5936. [Google Scholar] [CrossRef] [Green Version]
- Spencer, C.M.; Alekseyenko, O.; Serysheva, E.; Yuva-Paylor, L.A.; Paylor, R. Altered Anxiety-Related and Social Behaviors in the Fmr1 Knockout Mouse Model of Fragile X Syndrome. Genes Brain Behav. 2005, 4, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Tonkin, E.T.; Wang, T.-J.; Lisgo, S.; Bamshad, M.J.; Strachan, T. NIPBL, Encoding a Homolog of Fungal Scc2-Type Sister Chromatid Cohesion Proteins and Fly Nipped-B, Is Mutated in Cornelia de Lange Syndrome. Nat. Genet. 2004, 36, 636–641. [Google Scholar] [CrossRef] [Green Version]
- Olley, G.; Ansari, M.; Bengani, H.; Grimes, G.R.; Rhodes, J.; von Kriegsheim, A.; Blatnik, A.; Stewart, F.J.; Wakeling, E.; Carroll, N.; et al. BRD4 Interacts with NIPBL and BRD4 Is Mutated in a Cornelia de Lange-like Syndrome. Nat. Genet. 2018, 50, 329–332. [Google Scholar] [CrossRef]
- Alesi, V.; Dentici, M.L.; Loddo, S.; Genovese, S.; Orlando, V.; Calacci, C.; Pompili, D.; Dallapiccola, B.; Digilio, M.C.; Novelli, A. Confirmation of BRD4 Haploinsufficiency Role in Cornelia de Lange–like Phenotype and Delineation of a 19p13.12p13.11 Gene Contiguous Syndrome. Ann. Hum. Genet. 2019, 83, 100–109. [Google Scholar] [CrossRef]
- Rentas, S.; Rathi, K.S.; Kaur, M.; Raman, P.; Krantz, I.D.; Sarmady, M.; Tayoun, A.A. Diagnosing Cornelia de Lange Syndrome and Related Neurodevelopmental Disorders Using RNA Sequencing. Genet. Med. 2020, 22, 927–936. [Google Scholar] [CrossRef]
- Luna-Peláez, N.; March-Díaz, R.; Ceballos-Chávez, M.; Guerrero-Martínez, J.A.; Grazioli, P.; García-Gutiérrez, P.; Vaccari, T.; Massa, V.; Reyes, J.C.; García-Domínguez, M. The Cornelia de Lange Syndrome-Associated Factor NIPBL Interacts with BRD4 ET Domain for Transcription Control of a Common Set of Genes. Cell Death Dis. 2019, 10, 548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawauchi, S.; Calof, A.L.; Santos, R.; Lopez-Burks, M.E.; Young, C.M.; Hoang, M.P.; Chua, A.; Lao, T.; Lechner, M.S.; Daniel, J.A.; et al. Multiple Organ System Defects and Transcriptional Dysregulation in the Nipbl+/− Mouse, a Model of Cornelia de Lange Syndrome. PLoS Genet. 2009, 5, e1000650. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M. v Astrocyte Barriers to Neurotoxic Inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B. v Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Bandopadhyay, R.; Singh, P.K.; Mishra, P.S.; Sharma, N.; Khurana, N. Neuroinflammation in Neurological Disorders: Pharmacotherapeutic Targets from Bench to Bedside. Metab. Brain Dis. 2021, 36, 1591–1626. [Google Scholar] [CrossRef] [PubMed]
- Psenicka, M.W.; Smith, B.C.; Tinkey, R.A.; Williams, J.L. Connecting Neuroinflammation and Neurodegeneration in Multiple Sclerosis: Are Oligodendrocyte Precursor Cells a Nexus of Disease? Front. Cell. Neurosci. 2021, 15, 654284. [Google Scholar] [CrossRef] [PubMed]
- Belkina, A.C.; Nikolajczyk, B.S.; Denis, G. v BET Protein Function Is Required for Inflammation: Brd2 Genetic Disruption and BET Inhibitor JQ1 Impair Mouse Macrophage Inflammatory Responses. J. Immunol. 2013, 190, 3670–3678. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Vakoc, C.R. The Mechanisms behind the Therapeutic Activity of BET Bromodomain Inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Vakoc, C.R. Brd4 Is on the Move during Inflammation. Trends Cell Biol. 2014, 24, 615–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, Y.; Wu, X.; Chen, J.; Hu, X.; Zeng, F.; Cheng, J.; Jin, H.; Lin, X.; Chen, L.-F. Brd4 Modulates the Innate Immune Response through Mnk2-EIF4E Pathway-Dependent Translational Control of IκBα. Proc. Natl. Acad. Sci. USA 2017, 114, E3993–E4001. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Yang, X.-D.; Zhou, M.-M.; Ozato, K.; Chen, L.-F. Brd4 Coactivates Transcriptional Activation of NF-KappaB via Specific Binding to Acetylated RelA. Mol. Cell Biol. 2009, 29, 1375–1387. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Huang, W.; Liang, M.; Shi, Y.; Zhang, C.; Li, Q.; Liu, M.; Shou, Y.; Yin, H.; Zhu, X.; et al. (+)-JQ1 Attenuated LPS-Induced Microglial Inflammation via MAPK/NFκB Signaling. Cell Biosci. 2018, 8, 60. [Google Scholar] [CrossRef]
- Hajmirza, A.; Emadali, A.; Gauthier, A.; Casasnovas, O.; Gressin, R.; Callanan, M.B. BET Family Protein BRD4: An Emerging Actor in NFκB Signaling in Inflammation and Cancer. Biomedicines 2018, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Hertz, L.; Chen, Y. Editorial: All 3 Types of Glial Cells Are Important for Memory Formation. Front. Integr. Neurosci. 2016, 10, 31. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, G.; Colín-González, A.L.; Rangel-López, E.; Chavarría, A.; Santamaría, A. Redox Signaling, Neuroinflammation, and Neurodegeneration. Antioxid. Redox Signal. 2017, 28, 1626–1651. [Google Scholar] [CrossRef]
- DeMars, K.M.; Yang, C.; Castro-Rivera, C.I.; Candelario-Jalil, E. Selective Degradation of BET Proteins with DBET1, a Proteolysis-Targeting Chimera, Potently Reduces pro-Inflammatory Responses in Lipopolysaccharide-Activated Microglia. Biochem. Biophys. Res. Commun. 2018, 497, 410–415. [Google Scholar] [CrossRef]
- Dou, Y.; Wu, H.; Li, H.; Qin, S.; Wang, Y.; Li, J.; Lou, H.; Chen, Z.; Li, X.; Luo, Q.; et al. Microglial Migration Mediated by ATP-Induced ATP Release from Lysosomes. Cell Res. 2012, 22, 1022–1033. [Google Scholar] [CrossRef] [Green Version]
- Smolders, S.M.T.; Kessels, S.; Vangansewinkel, T.; Rigo, J.M.; Legendre, P.; Brône, B. Microglia: Brain Cells on the Move. Prog. Neurobiol. 2019, 178, 101612. [Google Scholar] [CrossRef]
- van de Craen, B.; Declerck, P.J.; Gils, A. The Biochemistry, Physiology and Pathological Roles of PAI-1 and the Requirements for PAI-1 Inhibition in Vivo. Thromb. Res. 2012, 130, 576–585. [Google Scholar] [CrossRef]
- Liu, M.; Lou, H.; Huang, M.; Ma, G.; Li, X. BET Protein BRD4 as a New Therapeutic Target in Cerebral Ischemic Stroke. Int. J. Clin. Exp. Pathol. 2017, 10, 258–265. [Google Scholar]
- Zhou, Y.; Gu, Y.; Liu, J. BRD4 Suppression Alleviates Cerebral Ischemia-Induced Brain Injury by Blocking Glial Activation via the Inhibition of Inflammatory Response and Pyroptosis. Biochem. Biophys. Res. Commun. 2019, 519, 481–488. [Google Scholar] [CrossRef]
- Liu, L.; Yang, C.; Lavayen, B.P.; Tishko, R.J.; Larochelle, J.; Candelario-Jalil, E. Targeted BRD4 Protein Degradation by DBET1 Ameliorates Acute Ischemic Brain Injury and Improves Functional Outcomes Associated with Reduced Neuroinflammation and Oxidative Stress and Preservation of Blood–Brain Barrier Integrity. J. Neuroinflammation 2022, 19, 168. [Google Scholar] [CrossRef]
- DeMars, K.M.; Yang, C.; Candelario-Jalil, E. Neuroprotective Effects of Targeting BET Proteins for Degradation with DBET1 in Aged Mice Subjected to Ischemic Stroke. Neurochem. Int. 2019, 127, 94–102. [Google Scholar] [CrossRef]
- Chatterjee, N.; Bohmann, D. BET-Ting on Nrf2: How Nrf2 Signaling Can Influence the Therapeutic Activities of BET Protein Inhibitors. Bioessays 2018, 40, e1800007. [Google Scholar] [CrossRef]
- Segatto, M.; Szokoll, R.; Fittipaldi, R.; Bottino, C.; Nevi, L.; Mamchaoui, K.; Filippakopoulos, P.; Caretti, G. BETs Inhibition Attenuates Oxidative Stress and Preserves Muscle Integrity in Duchenne Muscular Dystrophy. Nat. Commun. 2020, 11, 6108. [Google Scholar] [CrossRef]
- Li, X.; Zhu, H.; Wen, J.; Huang, J.; Chen, Y.; Tian, M.; Ren, J.; Zhou, L.; Yang, Q. Inhibition of BRD4 Decreases Fibrous Scarring after Ischemic Stroke in Rats by Inhibiting the Phosphorylation of Smad2/3. Brain Res. 2022, 1797, 148126. [Google Scholar] [CrossRef]
- Zhong, X.; Chen, Z.; Wang, Y.; Mao, M.; Deng, Y.; Shi, M.; Xu, Y.; Chen, L.; Cao, W. JQ1 Attenuates Neuroinflammation by Inhibiting the Inflammasome-Dependent Canonical Pyroptosis Pathway in SAE. Brain Res. Bull. 2022, 189, 174–183. [Google Scholar] [CrossRef]
- Kurtishi, A.; Rosen, B.; Patil, K.S.; Alves, G.W.; Møller, S.G. Cellular Proteostasis in Neurodegeneration. Mol. Neurobiol. 2019, 56, 3676–3689. [Google Scholar] [CrossRef]
- Katsnelson, A.; de Strooper, B.; Zoghbi, H.Y. Neurodegeneration: From Cellular Concepts to Clinical Applications. Sci. Transl. Med. 2016, 8, 364ps18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, Z.; Jia, J.; Du, T.; Zhang, N.; Tang, Y.; Fang, Y.; Fang, D. Overview of histone modification. In Histone Mutations and Cancer; Fang, D., Han, J., Eds.; Springer: Singapore, 2021; Volume 1283, pp. 1–16. ISBN 978-981-15-8104-5. [Google Scholar]
- Berson, A.; Nativio, R.; Berger, S.L.; Bonini, N.M. Epigenetic Regulation in Neurodegenerative Diseases. Trends Neurosci. 2018, 41, 587–598. [Google Scholar] [CrossRef]
- Nikkar, R.; Esmaeili-bandboni, A.; Badrikoohi, M.; Babaei, P. Effects of Inhibiting Astrocytes and BET/BRD4 Chromatin Reader on Spatial Memory and Synaptic Proteins in Rats with Alzheimer’s Disease. Metab. Brain Dis. 2022, 37, 1119–1131. [Google Scholar] [CrossRef]
- Taylor, X.; Cisternas, P.; Jury, N.; Martinez, P.; Huang, X.; You, Y.; Redding-Ochoa, J.; Vidal, R.; Zhang, J.; Troncoso, J.; et al. Activated Endothelial Cells Induce a Distinct Type of Astrocytic Reactivity. Commun. Biol. 2022, 5, 282. [Google Scholar] [CrossRef]
- Matuszewska, M.; Cieślik, M.; Wilkaniec, A.; Strawski, M.; Czapski, G.A. The Role of Bromodomain and Extraterminal (BET) Proteins in Controlling the Phagocytic Activity of Microglia In Vitro: Relevance to Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Bai, P.; Lei, D.; Liang, Y.; Zhen, S.; Bakiasi, G.; Pang, H.; Choi, S.H.; Wang, C.; Tanzi, R.E.; et al. Degradation and Inhibition of Epigenetic Regulatory Protein BRD4 Exacerbate Alzheimer’s Disease-Related Neuropathology in Cell Models. J. Biol. Chem. 2022, 298, 101794. [Google Scholar] [CrossRef]
- Ray, K.K.; Nicholls, S.J.; Ginsberg, H.D.; Johansson, J.O.; Kalantar-Zadeh, K.; Kulikowski, E.; Toth, P.P.; Wong, N.; Cummings, J.L.; Sweeney, M.; et al. Effect of Selective BET Protein Inhibitor Apabetalone on Cardiovascular Outcomes in Patients with Acute Coronary Syndrome and Diabetes: Rationale, Design, and Baseline Characteristics of the BETonMACE Trial. Am. Heart J. 2019, 217, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Schwartz, G.G.; Nicholls, S.J.; Khan, A.; Halliday, C.; Toth, P.P.; Sweeney, M.; Johansson, J.O.; Wong, N.C.W.; Kulikowski, E.; et al. Cognitive Effects of the BET Protein Inhibitor Apabetalone: A Prespecified Montreal Cognitive Assessment Analysis Nested in the BETonMACE Randomized Controlled Trial. J. Alzheimers Dis. 2021, 83, 1703–1715. [Google Scholar] [CrossRef]
- Figge, D.A.; Standaert, D.G. Dysregulation of BET Proteins in Levodopa-Induced Dyskinesia. Neurobiol. Dis. 2017, 102, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Han, L.; Rong, L.; Yang, S.; Song, L.; Wu, N.; Liu, Z.; Gan, J. Inhibition of BET Protein Function Suppressed the Overactivation of the Canonical NF-ΚB Signaling Pathway in 6-OHDA-Lesioned Rat Model of Levodopa-Induced Dyskinesia. Front. Neurosci. 2022, 16, 896322. [Google Scholar] [CrossRef]
- Kedaigle, A.J.; Reidling, J.C.; Lim, R.G.; Adam, M.; Wu, J.; Wassie, B.; Stocksdale, J.T.; Casale, M.S.; Fraenkel, E.; Thompson, L.M. Treatment with JQ1, a BET Bromodomain Inhibitor, Is Selectively Detrimental to R6/2 Huntington’s Disease Mice. Hum. Mol. Genet. 2020, 29, 202–215. [Google Scholar] [CrossRef]
- Zeier, Z.; Esanov, R.; Belle, K.C.; Volmar, C.H.; Johnstone, A.L.; Halley, P.; DeRosa, B.A.; Khoury, N.; van Blitterswijk, M.; Rademakers, R.; et al. Bromodomain Inhibitors Regulate the C9ORF72 Locus in ALS. Exp. Neurol. 2015, 271, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Quezada, E.; Cappelli, C.; Diaz, I.; Jury, N.; Wightman, N.; Brown, R.H.; Montecino, M.; van Zundert, B. BET Bromodomain Inhibitors PFI-1 and JQ1 Are Identified in an Epigenetic Compound Screen to Enhance C9ORF72 Gene Expression and Shown to Ameliorate C9ORF72-Associated Pathological and Behavioral Abnormalities in a C9ALS/FTD Model. Clin. Epigenetics 2021, 13, 56. [Google Scholar] [CrossRef]
- Neergaard, J.S.; Dragsbæk, K.; Christiansen, C.; Nielsen, H.B.; Brix, S.; Karsdal, M.A.; Henriksen, K. Metabolic Syndrome, Insulin Resistance, and Cognitive Dysfunction: Does Your Metabolic Profile Affect Your Brain? Diabetes 2017, 66, 1957–1963. [Google Scholar] [CrossRef] [Green Version]
- Liang, E.; Ma, M.; Wang, L.; Liu, X.; Xu, J.; Zhang, M.; Yang, R.; Zhao, Y. The BET/BRD Inhibitor JQ1 Attenuates Diabetes-Induced Cognitive Impairment in Rats by Targeting Nox4-Nrf2 Redox Imbalance. Biochem. Biophys. Res. Commun. 2018, 495, 204–211. [Google Scholar] [CrossRef]
- Zheng, C.-Q.; Fan, H.-X.; Li, X.-X.; Li, J.-J.; Sheng, S.; Zhang, F. Resveratrol Alleviates Levodopa-Induced Dyskinesia in Rats. Front. Immunol. 2021, 12, 683577. [Google Scholar] [CrossRef] [PubMed]
- Svenningsson, P.; Nishi, A.; Fisone, G.; Girault, J.-A.; Nairn, A.C.; Greengard, P. DARPP-32: An Integrator of Neurotransmission. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 269–296. [Google Scholar] [CrossRef]
- Santini, E.; Valjent, E.; Usiello, A.; Carta, M.; Borgkvist, A.; Girault, J.-A.; Hervé, D.; Greengard, P.; Fisone, G. Critical Involvement of CAMP/DARPP-32 and Extracellular Signal-Regulated Protein Kinase Signaling in L-DOPA-Induced Dyskinesia. J. Neurosci. 2007, 27, 6995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, C.A.; Tabrizi, S.J. Huntington’s Disease: From Molecular Pathogenesis to Clinical Treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Valor, L.M. Transcription, Epigenetics and Ameliorative Strategies in Huntington’s Disease: A Genome-Wide Perspective. Mol. Neurobiol. 2015, 51, 406–423. [Google Scholar] [CrossRef] [Green Version]
- Valor, L.M. Understanding Histone Deacetylation in Huntington’s Disease. Oncotarget 2017, 8, 5660–5661. [Google Scholar] [CrossRef] [PubMed]
- Hockly, E.; Richon, V.M.; Woodman, B.; Smith, D.L.; Zhou, X.; Rosa, E.; Sathasivam, K.; Ghazi-Noori, S.; Mahal, A.; Lowden, P.A.S.; et al. Suberoylanilide Hydroxamic Acid, a Histone Deacetylase Inhibitor, Ameliorates Motor Deficits in a Mouse Model of Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2041–2046. [Google Scholar] [CrossRef] [Green Version]
- van Langenhove, T.; van der Zee, J.; van Broeckhoven, C. The Molecular Basis of the Frontotemporal Lobar Degeneration–Amyotrophic Lateral Sclerosis Spectrum. Ann. Med. 2012, 44, 817–828. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.-C.; Polymenidou, M.; Cleveland, D.W. Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From Genes to Mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Ash, P.E.A.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.-L.; DeJesus-Hernandez, M.; van Blitterswijk, M.M.; Jansen-West, K.; Paul III, J.W.; Rademakers, R.; et al. Unconventional Translation of C9ORF72 GGGGCC Expansion Generates Insoluble Polypeptides Specific to C9FTD/ALS/ALS. Neuron 2013, 77, 639–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeJesus-Hernandez, M.; Finch, N.A.; Wang, X.; Gendron, T.F.; Bieniek, K.F.; Heckman, M.G.; Vasilevich, A.; Murray, M.E.; Rousseau, L.; Weesner, R.; et al. In-Depth Clinico-Pathological Examination of RNA Foci in a Large Cohort of C9ORF72 Expansion Carriers. Acta Neuropathol. 2017, 134, 255–269. [Google Scholar] [CrossRef] [Green Version]
- Haeusler, A.R.; Donnelly, C.J.; Rothstein, J.D. The Expanding Biology of the C9orf72 Nucleotide Repeat Expansion in Neurodegenerative Disease. Nat. Rev. Neurosci. 2016, 17, 383–395. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Esanov, R.; Cabrera, G.T.; Andrade, N.S.; Gendron, T.F.; Brown, R.H.; Benatar, M.; Wahlestedt, C.; Mueller, C.; Zeier, Z. A C9ORF72 BAC Mouse Model Recapitulates Key Epigenetic Perturbations of ALS/FTD. Mol. Neurodegener. 2017, 12, 46. [Google Scholar] [CrossRef] [Green Version]
- Belzil, V.V.; Bauer, P.O.; Prudencio, M.; Gendron, T.F.; Stetler, C.T.; Yan, I.K.; Pregent, L.; Daughrity, L.; Baker, M.C.; Rademakers, R.; et al. Reduced C9orf72 Gene Expression in C9FTD/ALS Is Caused by Histone Trimethylation, an Epigenetic Event Detectable in Blood. Acta Neuropathol. 2013, 126, 895–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belzil, V.V.; Bauer, P.O.; Gendron, T.F.; Murray, M.E.; Dickson, D.; Petrucelli, L. Characterization of DNA Hypermethylation in the Cerebellum of C9FTD/ALS Patients. Brain Res. 2014, 1584, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Boivin, M.; Pfister, V.; Gaucherot, A.; Ruffenach, F.; Negroni, L.; Sellier, C.; Charlet-Berguerand, N. Reduced Autophagy upon C9ORF72 Loss Synergizes with Dipeptide Repeat Protein Toxicity in G4C2 Repeat Expansion Disorders. EMBO J. 2020, 39, e100574. [Google Scholar] [CrossRef] [PubMed]
- Tremolizzo, L.; Carboni, G.; Ruzicka, W.B.; Mitchell, C.P.; Sugaya, I.; Tueting, P.; Sharma, R.; Grayson, D.R.; Costa, E.; Guidotti, A. An Epigenetic Mouse Model for Molecular and Behavioral Neuropathologies Related to Schizophrenia Vulnerability. Proc. Natl. Acad. Sci. USA 2002, 99, 17095–17100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Choi, K.H.; Renthal, W.; Tsankova, N.M.; Theobald, D.E.H.; Truong, H.T.; Russo, S.J.; LaPlant, Q.; Sasaki, T.S.; Whistler, K.N.; et al. Chromatin Remodeling Is a Key Mechanism Underlying Cocaine-Induced Plasticity in Striatum. Neuron 2005, 48, 303–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renthal, W.; Maze, I.; Krishnan, V.; Covington, H.E.; Xiao, G.; Kumar, A.; Russo, S.J.; Graham, A.; Tsankova, N.; Kippin, T.E.; et al. Histone Deacetylase 5 Epigenetically Controls Behavioral Adaptations to Chronic Emotional Stimuli. Neuron 2007, 56, 517–529. [Google Scholar] [CrossRef] [Green Version]
- Malvaez, M.; Sanchis-Segura, C.; Vo, D.; Lattal, K.M.; Wood, M.A. Modulation of Chromatin Modification Facilitates Extinction of Cocaine-Induced Conditioned Place Preference. Biol. Psychiatry 2010, 67, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurita, M.; Holloway, T.; García-Bea, A.; Kozlenkov, A.; Friedman, A.K.; Moreno, J.L.; Heshmati, M.; Golden, S.A.; Kennedy, P.J.; Takahashi, N.; et al. HDAC2 Regulates Atypical Antipsychotic Responses through the Modulation of MGlu2 Promoter Activity. Nat. Neurosci. 2012, 15, 1245–1254. [Google Scholar] [CrossRef] [Green Version]
- Stafford, J.M.; Raybuck, J.D.; Ryabinin, A.E.; Lattal, K.M. Increasing Histone Acetylation in the Hippocampus-Infralimbic Network Enhances Fear Extinction. Biol. Psychiatry 2012, 72, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Rogge, G.A.; Wood, M.A. The Role of Histone Acetylation in Cocaine-Induced Neural Plasticity and Behavior. Neuropsychopharmacology 2013, 38, 94–110. [Google Scholar] [CrossRef] [Green Version]
- Egervari, G.; Landry, J.; Callens, J.; Fullard, J.F.; Roussos, P.; Keller, E.; Hurd, Y.L. Striatal H3K27 Acetylation Linked to Glutamatergic Gene Dysregulation in Human Heroin Abusers Holds Promise as Therapeutic Target. Biol. Psychiatry 2017, 81, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Renthal, W.; Kumar, A.; Xiao, G.; Wilkinson, M.; Covington, H.E., 3rd; Maze, I.; Sikder, D.; Robison, A.J.; LaPlant, Q.; Dietz, D.M.; et al. Genome-Wide Analysis of Chromatin Regulation by Cocaine Reveals a Role for Sirtuins. Neuron 2009, 62, 335–348. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Wang, L.; Jiang, B.; Hui, B.; Lv, Z.; Ma, L. The Effects of Sodium Butyrate, an Inhibitor of Histone Deacetylase, on the Cocaine- and Sucrose-Maintained Self-Administration in Rats. Neurosci. Lett. 2008, 441, 72–76. [Google Scholar] [CrossRef]
- Im, H.-I.; Hollander, J.A.; Bali, P.; Kenny, P.J. MeCP2 Controls BDNF Expression and Cocaine Intake through Homeostatic Interactions with MicroRNA-212. Nat. Neurosci. 2010, 13, 1120–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malvaez, M.; Mhillaj, E.; Matheos, D.P.; Palmery, M.; Wood, M.A. CBP in the Nucleus Accumbens Regulates Cocaine-Induced Histone Acetylation and Is Critical for Cocaine-Associated Behaviors. J. Neurosci. 2011, 31, 16941–16948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malvaez, M.; McQuown, S.C.; Rogge, G.A.; Astarabadi, M.; Jacques, V.; Carreiro, S.; Rusche, J.R.; Wood, M.A. HDAC3-Selective Inhibitor Enhances Extinction of Cocaine-Seeking Behavior in a Persistent Manner. Proc. Natl. Acad. Sci. USA 2013, 110, 2647–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Lv, Z.; Hu, Z.; Sheng, J.; Hui, B.; Sun, J.; Ma, L. Chronic Cocaine-Induced H3 Acetylation and Transcriptional Activation of CaMKIIalpha in the Nucleus Accumbens Is Critical for Motivation for Drug Reinforcement. Neuropsychopharmacology 2010, 35, 913–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, P.J.; Feng, J.; Robison, A.J.; Maze, I.; Badimon, A.; Mouzon, E.; Chaudhury, D.; Damez-Werno, D.M.; Haggarty, S.J.; Han, M.-H.; et al. Class I HDAC Inhibition Blocks Cocaine-Induced Plasticity by Targeted Changes in Histone Methylation. Nat. Neurosci. 2013, 16, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Rogge, G.A.; Singh, H.; Dang, R.; Wood, M.A. HDAC3 Is a Negative Regulator of Cocaine-Context-Associated Memory Formation. J. Neurosci. 2013, 33, 6623–6632. [Google Scholar] [CrossRef] [Green Version]
- van den Oever, M.C.; Goriounova, N.A.; Wan Li, K.; van der Schors, R.C.; Binnekade, R.; Schoffelmeer, A.N.M.; Mansvelder, H.D.; Smit, A.B.; Spijker, S.; de Vries, T.J. Prefrontal Cortex AMPA Receptor Plasticity Is Crucial for Cue-Induced Relapse to Heroin-Seeking. Nat. Neurosci. 2008, 11, 1053–1058. [Google Scholar] [CrossRef]
- Farrelly, L.A.; Zheng, S.; Schrode, N.; Topol, A.; Bhanu, N.V.; Bastle, R.M.; Ramakrishnan, A.; Chan, J.C.; Cetin, B.; Flaherty, E.; et al. Chromatin Profiling in Human Neurons Reveals Aberrant Roles for Histone Acetylation and BET Family Proteins in Schizophrenia. Nat. Commun. 2022, 13, 2195. [Google Scholar] [CrossRef]
- Bilecki, W.; Wawrzczak-Bargieła, A.; Majcher-Maślanka, I.; Chmelova, M.; Maćkowiak, M. Inhibition of BET Proteins during Adolescence Affects Prefrontal Cortical Development: Relevance to Schizophrenia. Int. J. Mol. Sci. 2021, 22, 8710. [Google Scholar] [CrossRef] [PubMed]
- Kenny, P.J.; Chartoff, E.; Roberto, M.; Carlezon, W.A., Jr.; Markou, A. NMDA Receptors Regulate Nicotine-Enhanced Brain Reward Function and Intravenous Nicotine Self-Administration: Role of the Ventral Tegmental Area and Central Nucleus of the Amygdala. Neuropsychopharmacology 2009, 34, 266–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klebaur, J.E.; Ostrander, M.M.; Norton, C.S.; Watson, S.J.; Akil, H.; Robinson, T.E. The Ability of Amphetamine to Evoke Arc (Arg 3.1) MRNA Expression in the Caudate, Nucleus Accumbens and Neocortex Is Modulated by Environmental Context. Brain Res. 2002, 930, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; O’Brien, D.; Kovacs, K.M.; Wang, R.; Zavadil, J.; Vadasz, C. Nicotine-Induced Sensitization in Mice: Changes in Locomotor Activity and Mesencephalic Gene Expression. Neurochem. Res. 2005, 30, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.B.; Babigian, C.J.; Sartor, G.C. Domain-Selective BET Inhibition Attenuates Transcriptional and Behavioral Responses to Cocaine. Neuropharmacology 2022, 210, 109040. [Google Scholar] [CrossRef] [PubMed]
- Barbier, E.; Johnstone, A.L.; Khomtchouk, B.B.; Tapocik, J.D.; Pitcairn, C.; Rehman, F.; Augier, E.; Borich, A.; Schank, J.R.; Rienas, C.A.; et al. Dependence-Induced Increase of Alcohol Self-Administration and Compulsive Drinking Mediated by the Histone Methyltransferase PRDM2. Mol. Psychiatry 2017, 22, 1746–1758. [Google Scholar] [CrossRef]
- Zuber, V.; Bettella, F.; Witoelar, A.; Eeles, R.; Easton, D.; Kote-Jarai, Z.; al Olama, A.A.; Benlloch, S.; Muir, K.; Giles, G.G.; et al. Bromodomain Protein 4 Discriminates Tissue-Specific Super-Enhancers Containing Disease-Specific Susceptibility Loci in Prostate and Breast Cancer. BMC Genom. 2017, 18, 270. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, R.; Jiang, Z.; Li, H.; Zhu, Z.; Khalid, A.; Liu, D.; Pan, F. Inhibiting Brd4 Alleviated PTSD-like Behaviors and Fear Memory through Regulating Immediate Early Genes Expression and Neuroinflammation in Rats. J. Neurochem. 2021, 158, 912–927. [Google Scholar] [CrossRef]
- Tsujikawa, L.M.; Fu, L.; Das, S.; Halliday, C.; Rakai, B.D.; Stotz, S.C.; Sarsons, C.D.; Gilham, D.; Daze, E.; Wasiak, S.; et al. Apabetalone (RVX-208) Reduces Vascular Inflammation in Vitro and in CVD Patients by a BET-Dependent Epigenetic Mechanism. Clin. Epigenetics 2019, 11, 102. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martella, N.; Pensabene, D.; Varone, M.; Colardo, M.; Petraroia, M.; Sergio, W.; La Rosa, P.; Moreno, S.; Segatto, M. Bromodomain and Extra-Terminal Proteins in Brain Physiology and Pathology: BET-ing on Epigenetic Regulation. Biomedicines 2023, 11, 750. https://doi.org/10.3390/biomedicines11030750
Martella N, Pensabene D, Varone M, Colardo M, Petraroia M, Sergio W, La Rosa P, Moreno S, Segatto M. Bromodomain and Extra-Terminal Proteins in Brain Physiology and Pathology: BET-ing on Epigenetic Regulation. Biomedicines. 2023; 11(3):750. https://doi.org/10.3390/biomedicines11030750
Chicago/Turabian StyleMartella, Noemi, Daniele Pensabene, Michela Varone, Mayra Colardo, Michele Petraroia, William Sergio, Piergiorgio La Rosa, Sandra Moreno, and Marco Segatto. 2023. "Bromodomain and Extra-Terminal Proteins in Brain Physiology and Pathology: BET-ing on Epigenetic Regulation" Biomedicines 11, no. 3: 750. https://doi.org/10.3390/biomedicines11030750
APA StyleMartella, N., Pensabene, D., Varone, M., Colardo, M., Petraroia, M., Sergio, W., La Rosa, P., Moreno, S., & Segatto, M. (2023). Bromodomain and Extra-Terminal Proteins in Brain Physiology and Pathology: BET-ing on Epigenetic Regulation. Biomedicines, 11(3), 750. https://doi.org/10.3390/biomedicines11030750