
Case Report of Myelodysplastic Syndrome in a Sickle-Cell Disease Patient Treated with Hydroxyurea and Literature Review

 , ,
, ,
Abstract
:1. Introduction
2. Case Report
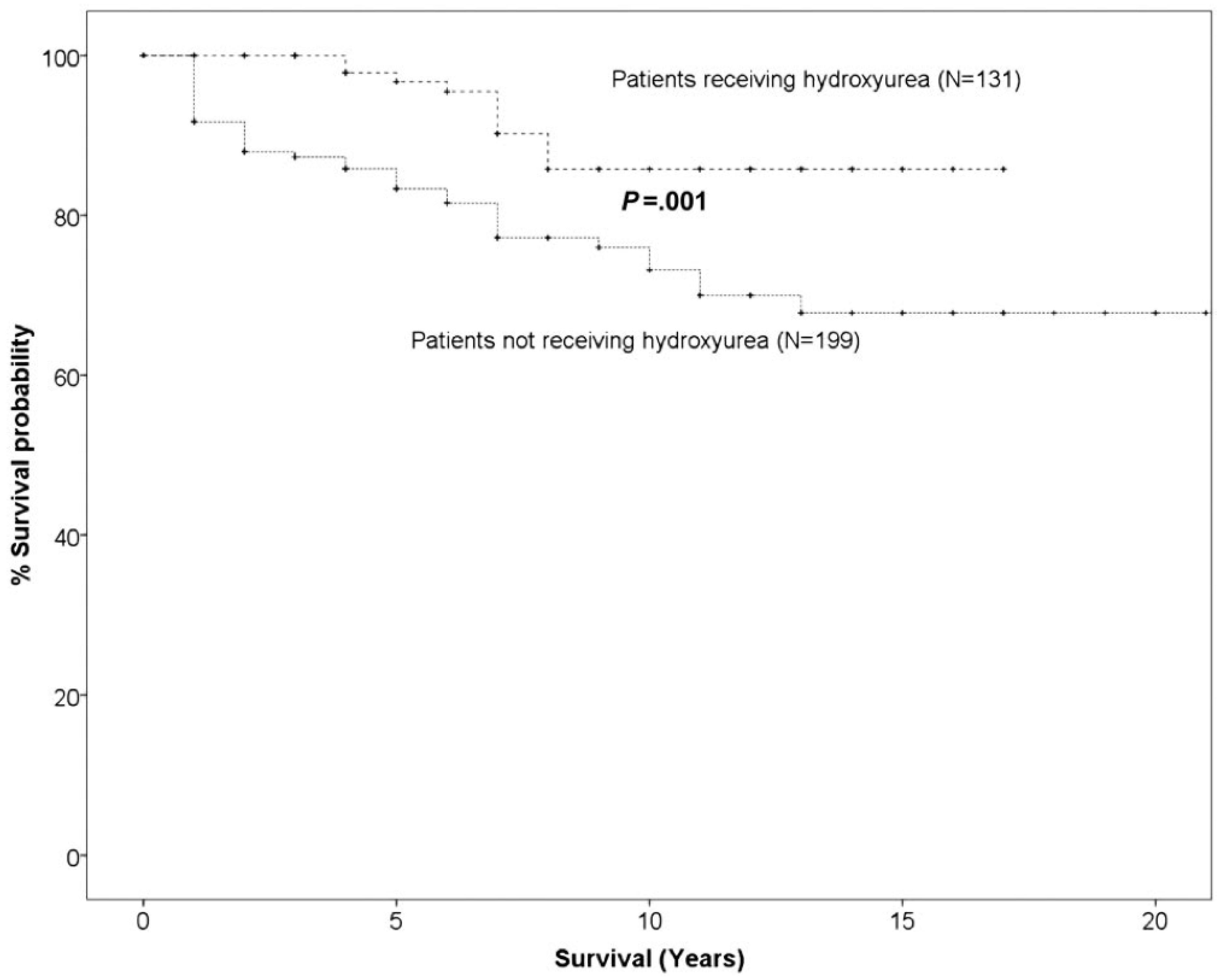
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brunson, A.; Keegan, T.H.M.; Bang, H.; Mahajan, A.; Paulukonis, S.; Wun, T. Increased risk of leukemia among sickle cell disease patients in California. Blood 2017, 130, 1597–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Montalembert, M.; Voskaridou, E.; Oevermann, L.; Cannas, G.; Habibi, A.; Loko, G.; Joseph, L.; Colombatti, R.; Bartolucci, P.; Brousse, V.; et al. Real-Life experience with hydroxyurea in patients with sickle cell disease: Results from the prospective ESCORT-HU cohort study. Am. J. Hematol. 2021, 96, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Singh, Z.N.; Huo, D.; Anastasi, J.; Smith, S.M.; Karrison, T.; Le Beau, M.M.; Larson, R.A.; Vardiman, J.W. Therapy-related myelodysplastic syndrome: Morphologic subclassification may not be clinically relevant. Am. J. Clin. Pathol. 2007, 127, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Soenen, V.; Preudhomme, C.; Roumier, C.; Daudignon, A.; Lai, J.L.; Fenaux, P. 17 p Deletion in acute myeloid leukemia and myelodysplastic syndrome. Analysis of breakpoints and deleted segments by fluorescence in situ. Blood 1998, 91, 1008–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ok, C.Y.; Patel, K.P.; Garcia-Manero, G.; Routbort, M.J.; Fu, B.; Tang, G.; Goswami, M.; Singh, R.; Kanagal-Shamanna, R.; Pierce, S.A.; et al. Mutational profiling of therapy-related myelodysplastic syndromes and acute myeloid leukemia by next generation sequencing, a comparison with de novo diseases. Leuk. Res. 2015, 39, 348–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNerney, M.E.; Godley, L.A.; Le Beau, M.M. Therapy-related myeloid neoplasms: When genetics and environment collide. Nat. Rev. Cancer 2017, 17, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.N.; Ramsingh, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch, J.S.; Lamprecht, T.L.; Shen, D.; Hundal, J.; Fulton, R.S.; et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 2015, 518, 552–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterkers, Y.; Preudhomme, C.; Lai, J.L.; Demory, J.L.; Caulier, M.T.; Wattel, E.; Bordessoule, D.; Bauters, F.; Fenaux, P. Acute myeloid leukemia and myelodysplastic syndromes following essential thrombocythemia treated with hydroxyurea: High proportion of cases with 17 p deletion. Blood 1998, 91, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Maia Filho, P.A.; Pereira, J.F.; Almeida Filho, T.P.; Cavalcanti, B.C.; Sousa, J.C.; Lemes, R.P.G. Is chronic use of hydroxyurea safe for patients with sickle cell anemia? An account of genotoxicity and mutagenicity. Environ. Mol. Mutagen 2019, 60, 302–304. [Google Scholar] [CrossRef] [PubMed]
- Voskaridou, E.; Christoulas, D.; Bilalis, A.; Plata, E.; Varvagiannis, K.; Stamatopoulos, G.; Sinopoulou, K.; Balassopoulou, A.; Loukopoulos, D.; Terpos, E. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: Results of a 17-year, single-center trial (LaSHS). Blood 2010, 115, 2354–2363. [Google Scholar] [CrossRef] [PubMed]
- Baz, W.; Najfeld, V.; Yotsuya, M.; Talwar, J.; Terjanian, T.; Forte, F. Development of myelodysplastic syndrome and acute myeloid leukemia 15 years after hydroxyurea use in a patient with sickle cell anemia. Clin. Med. Insights Oncol. 2012, 6, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Zemenides, S.; Erblich, T.; Luqmani, A.; Bain, B.J. Peripheral blood features of acute myeloid leukemia with myelodysplasia-related changes developing in a patient with sickle cell anemia. Am. J. Hematol. 2014, 89, 1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aumont, C.; Driss, F.; Lazure, T.; Picard, V.; Creidy, R.; De Botton, S.; Saada, V.; Lambotte, O.; Bilhou-Nabera, C.; Tertian, G.; et al. Myelodysplastic syndrome with clonal cytogenetic abnormalities followed by fatal erythroid leukemia after 14 years of exposure to hydroxyurea for sickle cell anemia. Am. J. Hematol. 2015, 90, E131–E132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Maule, J.; Neff, J.L.; McCall, C.M.; Rapisardo, S.; Lagoo, A.S.; Yang, L.H.; Crawford, R.D.; Zhao, Y.; Wang, E. Myeloid neoplasms in the setting of sickle cell disease: An intrinsic association with the underlying condition rather than a coincidence; report of 4 cases and review of the literature. Mod. Pathol. 2019, 32, 1712–1726. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.M.; Bonner, M.; Pierciey, F.J.; Uchida, N.; Rottman, J.; Demopoulos, L.; Schmidt, M.; Kanter, J.; Walters, M.C.; Thompson, A.A.; et al. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv. 2020, 4, 2058–2063. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Case Nr, Ref, Country | Case 1, US [11] | Case 2, UK [12] | Case 3, FR [13] | Case 4, US [14] | Case 5, GR (Current Report) | Case 6, US [15] |
|---|---|---|---|---|---|---|
| Gender | Male | Male | Male | Male | Male | Male |
| Hb beta-chain genotype | Not specified | S/S | S/S | S/β0 | S/S | S/S |
| Treatment (daily dose) | Transfusions, HU (1.5 to 2.0 g) | none | Transfusions, HU (1 to 1.5 g) | Transfusion, HU (0.4 to 1.5 g) then matched-sibling donor HSCT with unspecified non-myelo-ablative therapy and radiation, then supportive therapy | Regular transfusions HU | HU then autologous HSCT with lentiviral vector encoding anti-sickling beta-globin, with busulfan myeloablative conditioning |
| Cumulative HU exposure | 15 years | N/A | 14 years (i.e., excluding 3 years of discontinuation) | 2 years | 20 years | 8 + 2 years |
| HU starting age | 26 years | N/A | 29 years | 25 years (?) | 20 years | 34 years |
| Age at MDS diagnosis | 41 years | 55 years | 47 years | 34 years | 40 years | 45 years |
| Time since HU start (time since HSCT) | 15 years | N/A | 18 years | 9 years (7 years) | 20 years | 11 years (3 years) |
| 2016 WHO classification | MDS-EB2/AML (authors RAEB-2/AML) | MDS-EB1 (authors: AML with myelodysplasia-related changes) | MDS-MLD/AML (authors: MDS/erythroid leukemia | MDS-MLD | MDS-EB1 | MDS-EB-2 |
| Presentation | Refractory pancytopenia | anaemia and erythroblastosis | Severe macrocytic nonregenerative anemia | Progressive anaemia, thrombocyto-penia | Progressive anaemia, thrombocyto-penia | Anemia, neutropenia |
| Peripheral blood smear | Not detailed | Agranular blast cells with high nucleocytoplasmic ratio, hypochromic erythrocytes, some with Pappenheimer bodies, ring sideroblasts | Holly-Jowel bodies, poikilocytosis, no blast | Red blood cell distortion, nucleated red blood cells, no blast | Holly-Jowel highly hypochromic erythrocytes, neutropenia, NRBC, no blasts | 3–9% blast-like cells |
| Bone marrow examination | 15% myeloblasts (15% non-erythroid non- lymphoid cells) | hypercellularity (66%), 65% erythroblasts, 8 % myeloblasts (55% non-erythroid non-lymphoid cells), dysgranulopoiesis | no excess blasts, dysmyelopoiesis in 3 cell lineages | hypercellularity (95%), 2% blasts, erythroid and megakaryocytic dysplasia, erythroid hyperplasia and left-shifted myelopoiesis | Lipocytes almost absent. All three lineages represented with hyperplasia and dyserythropo-iesis of the red blood cell lineage (granulocytes/RBC ¼), maturation of the granulocytes with left shift and dysmegakaryopoiesis, with elevated number of megakaryocytes. 6% blasts of the granulocyte lineage (CD34 stain). Intermediate bone marrow infiltration, 12–15%, by T lymphocytes CD3, CD2, CD8, CD5, CD56 (partially)- positive with CD7 loss and CD57, TdT, TIA-1 negative. A few small B lymphocytes | 10% malignant myeloblasts |
| Chromosomal analysis | 42XY with complex cytogenetics including t(5:18), del(7)(q21) and monosomy 17 | Complex abnormality with monosomy 5 and 7, del(17 p) | Monosomy 20, abnormalities 5 q, 17 p, 17 q | 45,XY,-2, der(7)(2pter- > 2p11.2::7p11.1- >7q22::?2q11.2- > 2qter),inv(9)(p11q13)c [18]/45, idem, ?del(20)(q11.2q13.1)[ | 25 metaphases were analysed: 1 metaphase had karyotype 46,XY 24 metaphases had karyotype 42,XY,-3,der(5)t(3;5)(q21;q15), -7, der(12)t(7;12)(q11.2;p11.2~13), -16,-17, -8,add(18)(p11.3),+?21[24]/46,xx [1] | Monosomy 7 and structurally abnormal chromosome 19p (pre-conditioning BM negative for monosomy 7 and mutations associated with myeloid disorders |
| Transformation to AML | Transformation to AML after 34 days | Not reported | Transformation to AML after 2 years | none | no transformation | Transformation to AML during initial MDS treatment |
| Treatment | Induction chemotherapy with idarubicin and cytarabine, then high cytarabine in anticipation of bone-marrow transplant-ation | Not reported | Induction chemotherapy with cytosine arabinoside and etoposide | Conditioning regimen including busulfan, fludarabine, and HSCT graft | corticosteroids and ciclosporin | 5-Azacytadine and decitabine, then after AML diagnosis, induction chemotherapy of idarubicin/cytarabine, followed by reinduction with cladribine, high-dose cytarabine, and granulocyte colony-stimulating factor, then myeloablative doses of melphalan, fludarabine, and total-body irradiation, followed by an HLA-haploidentical HSCT and cyclophosphamide posttransplant. |
| Outcome | Pancyto-penia, sepsis, subarach-noidal haemorrhage, death | Not reported | Disease progression with CNS involvement, death | Alive after 21 months | Death 3 months after diagnosis | Relapse 6 months after HSCT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flevari, P.; Voskaridou, E.; Galactéros, F.; Cannas, G.; Loko, G.; Joseph, L.; Bartolucci, P.; Gellen-Dautremer, J.; Bernit, E.; Charneau, C.; et al. Case Report of Myelodysplastic Syndrome in a Sickle-Cell Disease Patient Treated with Hydroxyurea and Literature Review. Biomedicines 2022, 10, 3201. https://doi.org/10.3390/biomedicines10123201
Flevari P, Voskaridou E, Galactéros F, Cannas G, Loko G, Joseph L, Bartolucci P, Gellen-Dautremer J, Bernit E, Charneau C, et al. Case Report of Myelodysplastic Syndrome in a Sickle-Cell Disease Patient Treated with Hydroxyurea and Literature Review. Biomedicines. 2022; 10(12):3201. https://doi.org/10.3390/biomedicines10123201
Chicago/Turabian StyleFlevari, Pagona, Ersi Voskaridou, Frédéric Galactéros, Giovanna Cannas, Gylna Loko, Laure Joseph, Pablo Bartolucci, Justine Gellen-Dautremer, Emmanuelle Bernit, Corine Charneau, and et al. 2022. "Case Report of Myelodysplastic Syndrome in a Sickle-Cell Disease Patient Treated with Hydroxyurea and Literature Review" Biomedicines 10, no. 12: 3201. https://doi.org/10.3390/biomedicines10123201
APA StyleFlevari, P., Voskaridou, E., Galactéros, F., Cannas, G., Loko, G., Joseph, L., Bartolucci, P., Gellen-Dautremer, J., Bernit, E., Charneau, C., & Habibi, A. (2022). Case Report of Myelodysplastic Syndrome in a Sickle-Cell Disease Patient Treated with Hydroxyurea and Literature Review. Biomedicines, 10(12), 3201. https://doi.org/10.3390/biomedicines10123201