
Pathogenesis and Current Treatment Strategies of Hepatocellular Carcinoma
 ,
,
Abstract
:1. Introduction
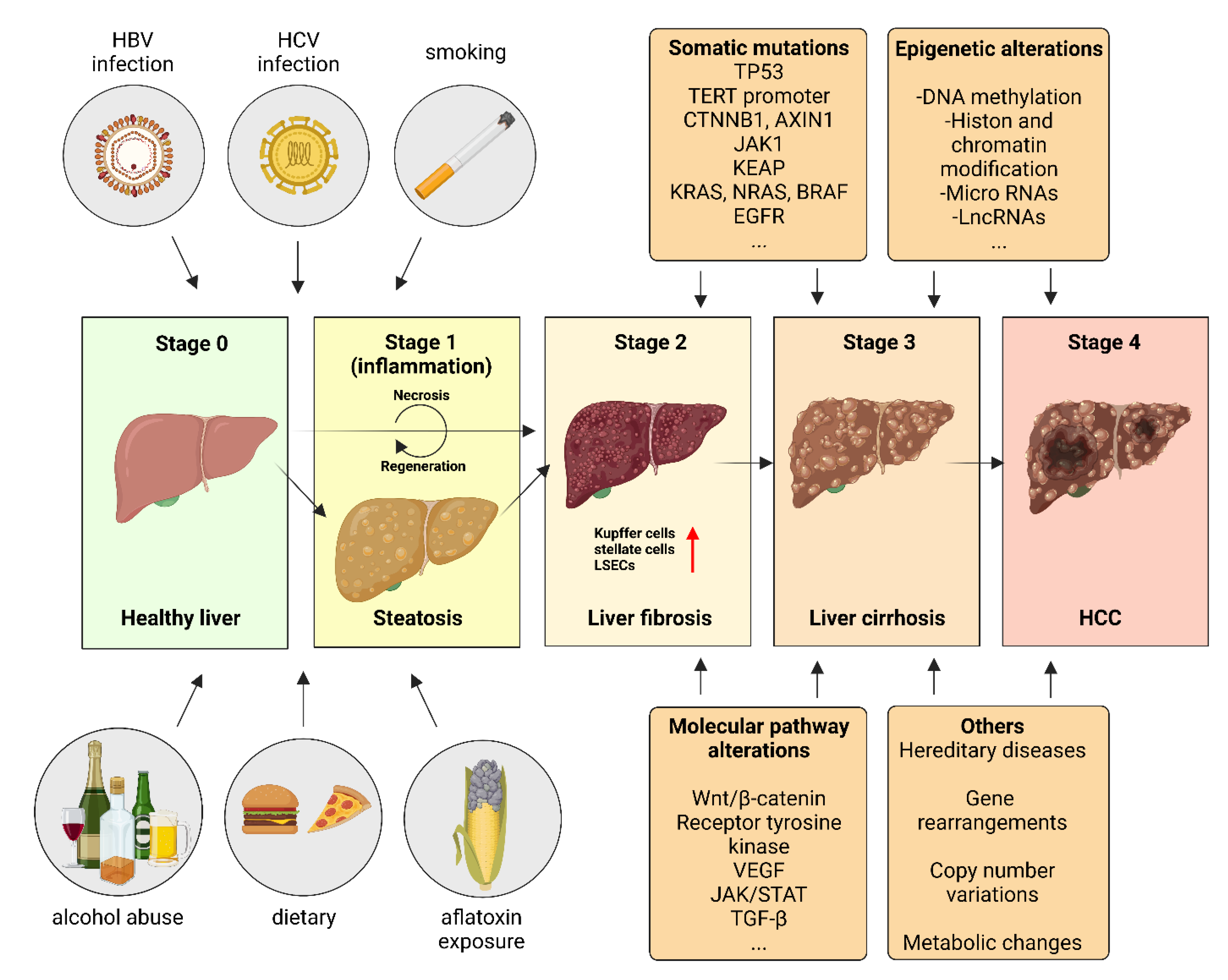
2. Etiology and Risk Factors in HCC Pathophysiology
2.1. Viral Risk Factors
2.2. Aflatoxin
2.3. Alcohol Consumption
2.4. Metabolic Syndrome, Non-Alcoholic Fatty Liver Disease (NAFLD) and Non-Alcoholic Steatohepatitis (NASH)
3. Somatic Mutations in HCC Pathogenesis
3.1. The Tumor Suppressor p53

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Gene/Pathway | Incidence | Reference |
|---|---|---|---|
| Somatic mutations | Tumor suppressor p53 | AFB1 induced R249S | [58,59,60] |
| AFB1 induced V157F | [58,60,61] | ||
| HBV promoted Mdm2 promoter polymorphism (SNP309, rs2279744) | [61,62] | ||
| ZNF498 | [51] | ||
| SLC7A11 | [52] | ||
| GPX4 | [52] | ||
| ATM | [63] | ||
| RPS6KA3 | [63] | ||
| Telomerase promoter | TERT promoter C to T transition (−228 to −250) | [64,65] | |
| HBV-promoted | HBV-DNA integration in TERT | [66,67,68] | |
| MLL4, KMT2B | [69,70] | ||
| CCNE1, CCNA2 | [69,70] | ||
| SENP5 | [71] | ||
| RARβ | [72] | ||
| ROCK1, ARHGEF12 | [69,70] | ||
| FN1 | [73] | ||
| CYP2C8 | [73] | ||
| PHACTR4 | [73] | ||
| SMAD5 | [73] | ||
| Others | CTNNB1, AXIN1 | [74] | |
| mTORC1, ARID1A, ARID2 | [74,75] | ||
| TTN | [74,75] | ||
| JAK1 | [74,75] | ||
| KEAP1 | [74,75] | ||
| NRF2, NFE2L2 | [76,77] | ||
| KRAS, NRAS, BRAF | [78] | ||
| EGFR | [78] | ||
| IDH1, IDH2 | [78] | ||
| Epigenetic changes | DNA methylation | genes involved in Ras/Raf/ERK and Wnt/β-catenin pathway | [79] |
| GSTP1 | [80] | ||
| FANCB | [81] | ||
| KIF15 | [81] | ||
| KIF4A | [81] | ||
| ERCC6L | [81] | ||
| UBE2C | [81] | ||
| CDKN2A | [82] | ||
| p16 | [83] | ||
| DNMT1, DNMT3A, DNMT3A2 | [84] | ||
| Histon and chromatin modification | H3K3me3, H3K27ac, H3K9ac, H3K4me2 | [85] | |
| H3Kme27, H3K4me3 | [85,86] | ||
| Ash2 | [87] | ||
| LSD1 | [87] | ||
| MMP1/MMP3 | [88] | ||
| VASH2 | [87,88] | ||
| EZH2 | [89] | ||
| SMARCD1 | [90] | ||
| ARID2 | [91] | ||
| Hbx-antigen interaction with CREB-binding protein/p300 | [92] | ||
| Micro RNAs | miR-1/-122/-124/-132/-148/-200/-205 | [93,94] | |
| miR-210-3p | [95] | ||
| miR224 | [96] | ||
| miR-196a and miR-196b | [97] | ||
| miRNA-1468 | [96,97] | ||
| miRNA144 | [98] | ||
| miR-342-3p | [99] | ||
| miR-1/-124/-214/-34-A/-449 | [100] | ||
| Long-non-coding RNAs | LINC01234 | [101] | |
| PTTG3P | [102] | ||
| HULC | [103] | ||
| HEIH | [104] | ||
| MVIH | [103,104,105,106,107] | ||
| MAIT | [102] | ||
| MIR31HG | [108] | ||
| Pathway dysregulations | Wnt/β-catenin | CTNNB1 | [78] |
| AXIN1 | [109,110] | ||
| AXIN2 | [109,110] | ||
| APC | [109,110] | ||
| UBE2T | [111] | ||
| TFAP4 | [112] | ||
| DDX39 | [113] | ||
| circ_0004018 | [114] | ||
| circ_0003418 | [115] | ||
| Receptor tyrosine kinase | mTORC1, mTORC2 | [116] | |
| PTEN | [117,118] | ||
| LZTS2 | [119] | ||
| HJURP | [120] | ||
| RSK2 | [121] | ||
| miR155-5p/-494/-493/-519a | [122] | ||
| VEGF | HIF-1, HIF-2 | [123] | |
| ERO1α | [124] | ||
| lncRNA PAARH | [125] | ||
| JAK/STAT | IL-6 | [126] | |
| GP130 | [127] | ||
| JAK1 | [128] | ||
| IL6R | [128] | ||
| IL6ST | [128] | ||
| TGF-β | SMAD2/3 | [129,130] | |
| PTPRε | [129] | ||
| exosomes | [131] |
3.2. The Telomerase Promoter
3.3. Hepatitis B Virus Integration Mediated Mutations
3.4. Other Somatic Mutations
4. Epigenetic Regulation in HCC
4.1. DNA Methylation
4.2. Histon Modification and Chromatin Remodeling
4.3. Micro RNAs
4.4. Long Non-Coding RNAs
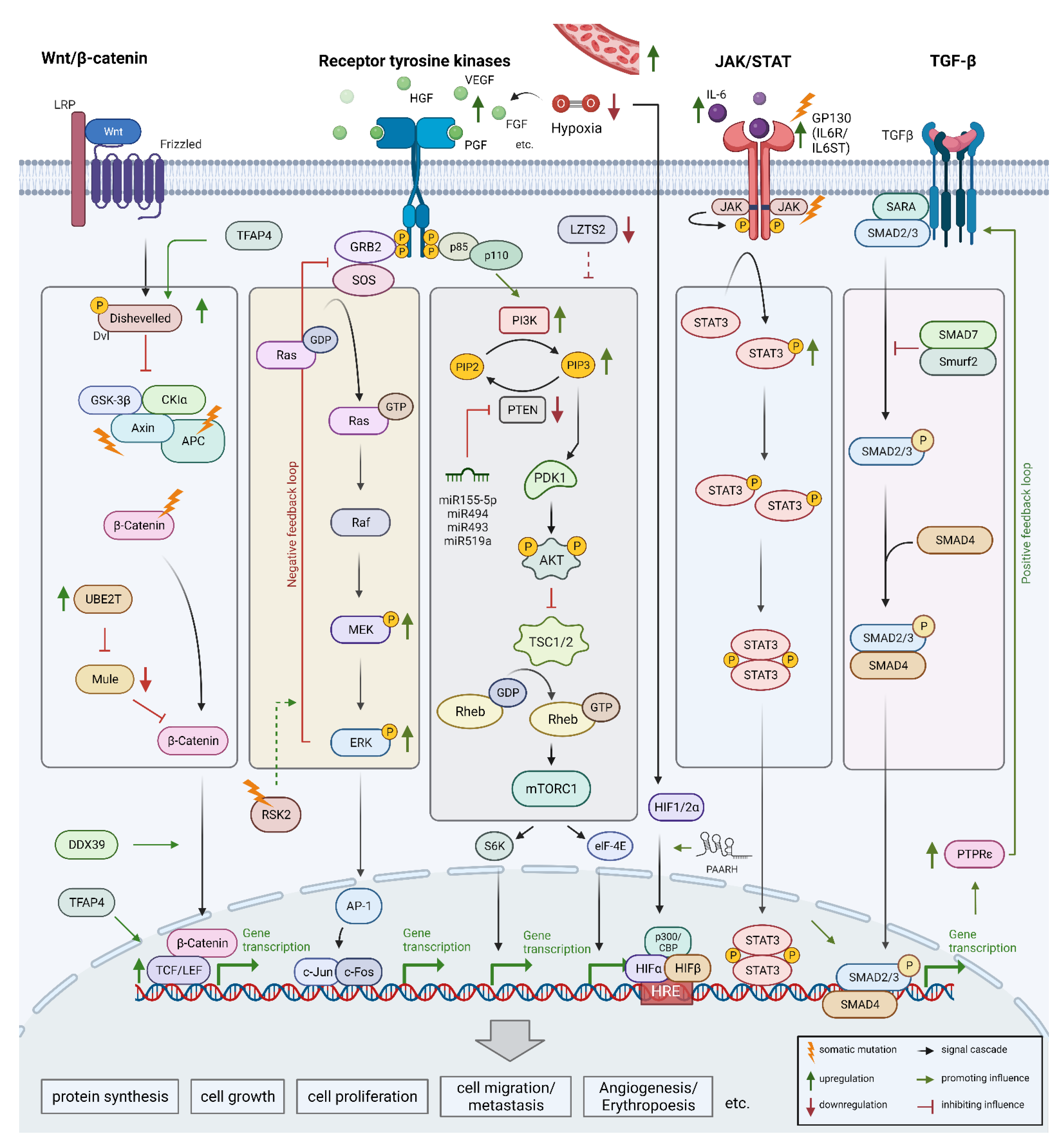
5. Dysregulation of Key Signaling Pathways
5.1. Wnt/β-Catenin Signaling Pathway
5.2. Receptor Tyrosine Kinase Pathway
5.3. Vascular Endothelial Growth Factor and Further Signaling Pathways
5.4. JAK/STAT Pathway
5.5. Transforming Growth Factor-Beta Pathway
6. Current Treatment Strategies
6.1. Transarterial Chemoembolization (TACE) for BCLC Stage B
6.1.1. Conventional TACE (cTACE)
6.1.2. Drug-Eluting-Bed TACE (DEB-TACE)
6.1.3. Combination of TACE with Systemic Therapy
6.1.4. Current Knowledge: Combination of TACE + Immunotherapy or TKI
6.1.5. Current Therapy Recommendations for TACE in HCC
6.2. Combination of TACE and RFA (Radiofrequency Ablation)
Current Therapy Recommendation for TACE and RFA in HCC
7. Systemic Therapy for HCC, BCLC C
7.1. Immunotherapeutic Strategies in HCC Treatment
7.1.1. IO combination Therapies in 1st-Line Therapy of HCC
7.1.2. Clinical Data for Other IO-Based Treatment Regimens in Later Therapy Lines
7.2. Tyrosine Kinase Inhibitors
7.2.1. Sorafenib in 1st Line Therapy
7.2.2. Lenvatinib in 1st-Line Therapy
7.2.3. Regorafenib in 2nd Line Therapy after Sorafenib
7.2.4. Cabozantinib in 2nd Line Therapy
7.2.5. Ramucirumab in 2nd Line Therapy in AFP-Overexpressing HCC
8. Future Directions
8.1. Immunotherapeutic Strategies
8.2. Adoptive Cell Transfer
8.3. Therapeutic Vaccination
8.4. Locoregional Therapy Enhancement
8.5. Novel Therapeutic Targets in HCC-Related Signaling Pathways
8.6. Non-Coding RNA-Based Therapies
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Petrick, J.L.; Florio, A.A.; Znaor, A.; Ruggieri, D.; Laversanne, M.; Alvarez, C.S.; Ferlay, J.; Valery, P.C.; Bray, F.; McGlynn, K.A. International Trends in Hepatocellular Carcinoma Incidence, 1978–2012. Int. J. Cancer 2020, 147, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Massarweh, N.N.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma and Intrahepatic Cholangiocarcinoma. Cancer Control J. Moffitt Cancer Cent. 2017, 24, 1073274817729245. [Google Scholar] [CrossRef] [PubMed]
- Altekruse, S.F.; Devesa, S.S.; Dickie, L.A.; McGlynn, K.A.; Kleiner, D.E. Histological Classification of Liver and Intrahepatic Bile Duct Cancers in SEER Registries. J. Regist. Manag. 2011, 38, 201–205. [Google Scholar]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular Carcinoma: Epidemiology and Molecular Carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef]
- Jemal, A.; Ward, E.M.; Johnson, C.J.; Cronin, K.A.; Ma, J.; Ryerson, B.; Mariotto, A.; Lake, A.J.; Wilson, R.; Sherman, R.L.; et al. Annual Report to the Nation on the Status of Cancer, 1975–2014, Featuring Survival. J. Natl. Cancer Inst. 2017, 109, djx030. [Google Scholar] [CrossRef] [Green Version]
- Sangiovanni, A.; Prati, G.M.; Fasani, P.; Ronchi, G.; Romeo, R.; Manini, M.; Del Ninno, E.; Morabito, A.; Colombo, M. The Natural History of Compensated Cirrhosis Due to Hepatitis C Virus: A 17-Year Cohort Study of 214 Patients. Hepatology 2006, 43, 1303–1310. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Splan, M.F.; Weiss, N.S.; McDonald, G.B.; Beretta, L.; Lee, S.P. Incidence and Predictors of Hepatocellular Carcinoma in Patients with Cirrhosis. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2007, 5, 938–945.e4. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.R.; Asch, S.M.; Cao, Y.; Li, L.; El-Serag, H.B. Long-Term Risk of Hepatocellular Carcinoma in HCV Patients Treated With Direct Acting Antiviral Agents. Hepatology 2020, 71, 44–55. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of Hepatocellular Carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [Green Version]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of Hepatocellular Cancer in Patients With Non-Alcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1828–1837.e2. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- Sidali, S.; Trépo, E.; Sutter, O.; Nault, J.-C. New Concepts in the Treatment of Hepatocellular Carcinoma. United Eur. Gastroenterol. J. 2022, 10, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Reig, M.; Forner, A.; Rimola, J.; Ferrer-Fàbrega, J.; Burrel, M.; Garcia-Criado, Á.; Kelley, R.K.; Galle, P.R.; Mazzaferro, V.; Salem, R.; et al. BCLC Strategy for Prognosis Prediction and Treatment Recommendation: The 2022 Update. J. Hepatol. 2022, 76, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.M.; Meyer, T.; Nault, J.C.; Neumann, U.; Ricke, J.; Sangro, B.; et al. Hepatocellular carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018, 29 (Suppl. S4), iv238–iv255, Erratum in: Ann Oncol. 2019, 30, 871–873. [Google Scholar] [CrossRef]
- Cheng, A.-L.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Lim, H.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. Updated Efficacy and Safety Data from IMbrave150: Atezolizumab plus Bevacizumab vs. Sorafenib for Unresectable Hepatocellular Carcinoma. J. Hepatol. 2022, 76, 862–873. [Google Scholar] [CrossRef]
- Cao, W.; Chen, H.-D.; Yu, Y.-W.; Li, N.; Chen, W.-Q. Changing Profiles of Cancer Burden Worldwide and in China: A Secondary Analysis of the Global Cancer Statistics 2020. Chin. Med. J. 2021, 134, 783–791. [Google Scholar] [CrossRef]
- Ghouri, Y.A.; Mian, I.; Rowe, J.H. Review of Hepatocellular Carcinoma: Epidemiology, Etiology, and Carcinogenesis. J. Carcinog. 2017, 16, 1. [Google Scholar] [CrossRef]
- Kuper, H.; Tzonou, A.; Kaklamani, E.; Hsieh, C.C.; Lagiou, P.; Adami, H.O.; Trichopoulos, D.; Stuver, S.O. Tobacco Smoking, Alcohol Consumption and Their Interaction in the Causation of Hepatocellular Carcinoma. Int. J. Cancer 2000, 85, 498–502. [Google Scholar] [CrossRef]
- Wang, L.-Y.; You, S.-L.; Lu, S.-N.; Ho, H.-C.; Wu, M.-H.; Sun, C.-A.; Yang, H.-I.; Chien-Jen, C. Risk of Hepatocellular Carcinoma and Habits of Alcohol Drinking, Betel Quid Chewing and Cigarette Smoking: A Cohort of 2416 HBsAg-Seropositive and 9421 HBsAg-Seronegative Male Residents in Taiwan. Cancer Causes Control CCC 2003, 14, 241–250. [Google Scholar] [CrossRef]
- Schütze, M.; Boeing, H.; Pischon, T.; Rehm, J.; Kehoe, T.; Gmel, G.; Olsen, A.; Tjønneland, A.M.; Dahm, C.C.; Overvad, K.; et al. Alcohol Attributable Burden of Incidence of Cancer in Eight European Countries Based on Results from Prospective Cohort Study. BMJ 2011, 342, d1584. [Google Scholar] [CrossRef]
- Global Burden of Disease Liver Cancer Collaboration; Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzman, N.; Amoako, Y.; et al. The Burden of Primary Liver Cancer and Underlying Etiologies From 1990 to 2015 at the Global, Regional, and National Level: Results From the Global Burden of Disease Study 2015. JAMA Oncol. 2017, 3, 1683–1691. [Google Scholar] [CrossRef]
- Heimbach, J.K.; Kulik, L.M.; Finn, R.S.; Sirlin, C.B.; Abecassis, M.M.; Roberts, L.R.; Zhu, A.X.; Murad, M.H.; Marrero, J.A. AASLD Guidelines for the Treatment of Hepatocellular Carcinoma. Hepatology 2018, 67, 358–380. [Google Scholar] [CrossRef] [Green Version]
- Indolfi, G.; Easterbrook, P.; Dusheiko, G.; Siberry, G.; Chang, M.-H.; Thorne, C.; Bulterys, M.; Chan, P.-L.; El-Sayed, M.H.; Giaquinto, C.; et al. Hepatitis B Virus Infection in Children and Adolescents. Lancet Gastroenterol. Hepatol. 2019, 4, 466–476. [Google Scholar] [CrossRef]
- Chang, M.H.; Chen, C.J.; Lai, M.S.; Hsu, H.M.; Wu, T.C.; Kong, M.S.; Liang, D.C.; Shau, W.Y.; Chen, D.S. Universal Hepatitis B Vaccination in Taiwan and the Incidence of Hepatocellular Carcinoma in Children. Taiwan Childhood Hepatoma Study Group. N. Engl. J. Med. 1997, 336, 1855–1859. [Google Scholar] [CrossRef] [Green Version]
- El-Serag, H.B.; Kanwal, F.; Richardson, P.; Kramer, J. Risk of Hepatocellular Carcinoma after Sustained Virological Response in Veterans with Hepatitis C Virus Infection. Hepatology 2016, 64, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, G.N.; Beste, L.A.; Green, P.K.; Singal, A.G.; Tapper, E.B.; Waljee, A.K.; Sterling, R.K.; Feld, J.J.; Kaplan, D.E.; Taddei, T.H.; et al. Increased Risk for Hepatocellular Carcinoma Persists Up to 10 Years After HCV Eradication in Patients With Baseline Cirrhosis or High FIB-4 Scores. Gastroenterology 2019, 157, 1264–1278.e4. [Google Scholar] [CrossRef] [Green Version]
- McGlynn, K.A.; London, W.T. The Global Epidemiology of Hepatocellular Carcinoma: Present and Future. Clin. Liver Dis. 2011, 15, 223–243. [Google Scholar] [CrossRef] [Green Version]
- Vandenbulcke, H.; Moreno, C.; Colle, I.; Knebel, J.-F.; Francque, S.; Sersté, T.; George, C.; de Galocsy, C.; Laleman, W.; Delwaide, J.; et al. Alcohol Intake Increases the Risk of HCC in Hepatitis C Virus-Related Compensated Cirrhosis: A Prospective Study. J. Hepatol. 2016, 65, 543–551. [Google Scholar] [CrossRef] [Green Version]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans Is Associated With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2016, 14, 124–131.e1. [Google Scholar] [CrossRef] [Green Version]
- Loomba, R.; Lim, J.K.; Patton, H.; El-Serag, H.B. AGA Clinical Practice Update on Screening and Surveillance for Hepatocellular Carcinoma in Patients With Nonalcoholic Fatty Liver Disease: Expert Review. Gastroenterology 2020, 158, 1822–1830. [Google Scholar] [CrossRef] [PubMed]
- Petrick, J.L.; Thistle, J.E.; Zeleniuch-Jacquotte, A.; Zhang, X.; Wactawski-Wende, J.; Van Dyke, A.L.; Stampfer, M.J.; Sinha, R.; Sesso, H.D.; Schairer, C.; et al. Body Mass Index, Diabetes and Intrahepatic Cholangiocarcinoma Risk: The Liver Cancer Pooling Project and Meta-Analysis. Am. J. Gastroenterol. 2018, 113, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Marrero, J.A.; Kulik, L.M.; Sirlin, C.B.; Zhu, A.X.; Finn, R.S.; Abecassis, M.M.; Roberts, L.R.; Heimbach, J.K. Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2018, 68, 723–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omata, M.; Cheng, A.-L.; Kokudo, N.; Kudo, M.; Lee, J.M.; Jia, J.; Tateishi, R.; Han, K.-H.; Chawla, Y.K.; Shiina, S.; et al. Asia-Pacific Clinical Practice Guidelines on the Management of Hepatocellular Carcinoma: A 2017 Update. Hepatol. Int. 2017, 11, 317–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, L. Glycogen Storage Disease I and Hepatocellular Tumours. Eur. J. Pediatr. 1993, 152 (Suppl. S1), S63–S70. [Google Scholar] [CrossRef]
- de Fost, M.; Vom Dahl, S.; Weverling, G.J.; Brill, N.; Brett, S.; Häussinger, D.; Hollak, C.E.M. Increased Incidence of Cancer in Adult Gaucher Disease in Western Europe. Blood Cells Mol. Dis. 2006, 36, 53–58. [Google Scholar] [CrossRef]
- Bartlett, D.C.; Lloyd, C.; McKiernan, P.J.; Newsome, P.N. Early Nitisinone Treatment Reduces the Need for Liver Transplantation in Children with Tyrosinaemia Type 1 and Improves Post-Transplant Renal Function. J. Inherit. Metab. Dis. 2014, 37, 745–752. [Google Scholar] [CrossRef]
- Li, S.; Mao, M. Next Generation Sequencing Reveals Genetic Landscape of Hepatocellular Carcinomas. Cancer Lett. 2013, 340, 247–253. [Google Scholar] [CrossRef]
- Hao, D.; Wang, L.; Di, L. Distinct Mutation Accumulation Rates among Tissues Determine the Variation in Cancer Risk. Sci. Rep. 2016, 6, 19458. [Google Scholar] [CrossRef] [Green Version]
- Craig, A.J.; von Felden, J.; Garcia-Lezana, T.; Sarcognato, S.; Villanueva, A. Tumour Evolution in Hepatocellular Carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 139–152. [Google Scholar] [CrossRef]
- Mendiratta, G.; Ke, E.; Aziz, M.; Liarakos, D.; Tong, M.; Stites, E.C. Cancer Gene Mutation Frequencies for the U.S. Population. Nat. Commun. 2021, 12, 5961. [Google Scholar] [CrossRef]
- Zhu, G.; Pan, C.; Bei, J.-X.; Li, B.; Liang, C.; Xu, Y.; Fu, X. Mutant P53 in Cancer Progression and Targeted Therapies. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Müller, M.; Strand, S.; Hug, H.; Heinemann, E.M.; Walczak, H.; Hofmann, W.J.; Stremmel, W.; Krammer, P.H.; Galle, P.R. Drug-Induced Apoptosis in Hepatoma Cells Is Mediated by the CD95 (APO-1/Fas) Receptor/Ligand System and Involves Activation of Wild-Type P53. J. Clin. Investig. 1997, 99, 403–413. [Google Scholar] [CrossRef]
- Müller, M.; Wilder, S.; Bannasch, D.; Israeli, D.; Lehlbach, K.; Li-Weber, M.; Friedman, S.L.; Galle, P.R.; Stremmel, W.; Oren, M.; et al. P53 Activates the CD95 (APO-1/Fas) Gene in Response to DNA Damage by Anticancer Drugs. J. Exp. Med. 1998, 188, 2033–2045. [Google Scholar] [CrossRef] [Green Version]
- Müller, M.; Scaffidi, C.A.; Galle, P.R.; Stremmel, W.; Krammer, P.H. The Role of P53 and the CD95 (APO-1/Fas) Death System in Chemotherapy-Induced Apoptosis. Eur. Cytokine Netw. 1998, 9, 685–686. [Google Scholar]
- Pitolli, C.; Wang, Y.; Candi, E.; Shi, Y.; Melino, G.; Amelio, I. P53-Mediated Tumor Suppression: DNA-Damage Response and Alternative Mechanisms. Cancers 2019, 11, 1983. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-H.; Ho, T.L.F.; Hariharan, A.; Goh, H.C.; Wong, Y.L.; Verkaik, N.S.; Lee, M.Y.; Tam, W.L.; van Gent, D.C.; Venkitaraman, A.R.; et al. Rapid Recruitment of P53 to DNA Damage Sites Directs DNA Repair Choice and Integrity. Proc. Natl. Acad. Sci. USA 2022, 119, e2113233119. [Google Scholar] [CrossRef]
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of Transcriptional Regulation by P53. Cell Death Differ. 2018, 25, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How Does P53 Induce Apoptosis and How Does This Relate to P53-Mediated Tumour Suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Pflaum, J.; Schlosser, S.; Müller, M. P53 Family and Cellular Stress Responses in Cancer. Front. Oncol. 2014, 4, 285. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zheng, Q.; Yue, X.; Yuan, Z.; Ling, J.; Yuan, Y.; Liang, Y.; Sun, A.; Liu, Y.; Li, H.; et al. ZNF498 Promotes Hepatocellular Carcinogenesis by Suppressing P53-Mediated Apoptosis and Ferroptosis via the Attenuation of P53 Ser46 Phosphorylation. J. Exp. Clin. Cancer Res. 2022, 41, 79. [Google Scholar] [CrossRef] [PubMed]
- Gnanapradeepan, K.; Basu, S.; Barnoud, T.; Budina-Kolomets, A.; Kung, C.-P.; Murphy, M.E. The P53 Tumor Suppressor in the Control of Metabolism and Ferroptosis. Front. Endocrinol. 2018, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Schleithoff, E.S.; Stremmel, W.; Melino, G.; Krammer, P.H.; Schilling, T. One, Two, Three–P53, P63, P73 and Chemosensitivity. Drug Resist. Updat. 2006, 9, 288–306. [Google Scholar] [CrossRef] [PubMed]
- Gressner, O.; Schilling, T.; Lorenz, K.; Schulze Schleithoff, E.; Koch, A.; Schulze-Bergkamen, H.; Lena, A.M.; Candi, E.; Terrinoni, A.; Catani, M.V.; et al. TAp63alpha Induces Apoptosis by Activating Signaling via Death Receptors and Mitochondria. EMBO J. 2005, 24, 2458–2471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, M.; Schilling, T.; Sayan, A.E.; Kairat, A.; Lorenz, K.; Schulze-Bergkamen, H.; Oren, M.; Koch, A.; Tannapfel, A.; Stremmel, W.; et al. TAp73/ΔNp73 Influences Apoptotic Response, Chemosensitivity and Prognosis in Hepatocellular Carcinoma. Cell Death Differ. 2005, 12, 1564–1577. [Google Scholar] [CrossRef]
- Candi, E.; Dinsdale, D.; Rufini, A.; Salomoni, P.; Knight, R.A.; Mueller, M.; Krammer, P.H.; Melino, G. TAp63 and DeltaNp63 in Cancer and Epidermal Development. Cell Cycle 2007, 6, 274–285. [Google Scholar] [CrossRef] [Green Version]
- Cai, B.-H.; Hsu, Y.-C.; Yeh, F.-Y.; Lin, Y.-R.; Lu, R.-Y.; Yu, S.-J.; Shaw, J.-F.; Wu, M.-H.; Tsai, Y.-Z.; Lin, Y.-C.; et al. P63 and P73 Activation in Cancers with P53 Mutation. Biomedicines 2022, 10, 1490. [Google Scholar] [CrossRef]
- Truong, D.-J.J.; Phlairaharn, T.; Eßwein, B.; Gruber, C.; Tümen, D.; Baligács, E.; Armbrust, N.; Vaccaro, F.L.; Lederer, E.-M.; Beck, E.M.; et al. Non-Invasive and High-Throughput Interrogation of Exon-Specific Isoform Expression. Nat. Cell Biol. 2021, 23, 652–663. [Google Scholar] [CrossRef]
- Müller, M.; Bird, T.G.; Nault, J.-C. The Landscape of Gene Mutations in Cirrhosis and Hepatocellular Carcinoma. J. Hepatol. 2020, 72, 990–1002. [Google Scholar] [CrossRef] [Green Version]
- Jiao, J.; Niu, W.; Wang, Y.; Baggerly, K.; Ye, Y.; Wu, X.; Davenport, D.; Almeda, J.L.; Betancourt-Garcia, M.M.; Forse, R.A.; et al. Prevalence of Aflatoxin-Associated TP53R249S Mutation in Hepatocellular Carcinoma in Hispanics in South Texas. Cancer Prev. Res. 2017, 11, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, F.; Harris, C.C.; Sun, T.; Hollstein, M.; Cerutti, P. Geographic Variation of P53 Mutational Profile in Nonmalignant Human Liver. Science 1994, 264, 1317–1319. [Google Scholar] [CrossRef] [PubMed]
- Kunst, C.; Haderer, M.; Heckel, S.; Schlosser, S.; Müller, M. The p53 family in hepatocellular carcinoma. Transl. Cancer Res. 2016, 5, 632–638. [Google Scholar] [CrossRef]
- Madden, C.R.; Finegold, M.J.; Slagle, B.L. Altered DNA Mutation Spectrum in Aflatoxin B1-Treated Transgenic Mice That Express the Hepatitis B Virus x Protein. J. Virol. 2002, 76, 11770–11774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, T.; Li, S.-F.; Wang, J.-L.; Zhang, T.; Zhang, S.; Chen, H.-T.; Xiao, Q.-Y.; Ren, W.-H.; Liu, C.; Peng, B.; et al. TP53 R249S Mutation Detected in Circulating Tumour DNA Is Associated with Prognosis of Hepatocellular Carcinoma Patients with or without Hepatectomy. Liver Int. 2020, 40, 2834–2847. [Google Scholar] [CrossRef] [PubMed]
- An, P.; Xu, J.; Yu, Y.; Winkler, C.A. Host and Viral Genetic Variation in HBV-Related Hepatocellular Carcinoma. Front. Genet. 2018, 9, 261. [Google Scholar] [CrossRef]
- Dharel, N.; Kato, N.; Muroyama, R.; Moriyama, M.; Shao, R.-X.; Kawabe, T.; Omata, M. MDM2 Promoter SNP309 Is Associated with the Risk of Hepatocellular Carcinoma in Patients with Chronic Hepatitis C. Clin. Cancer Res. 2006, 12, 4867–4871. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Franklin, D.A.; Dong, J.; Zhang, Y. MDM2–P53 Pathway in Hepatocellular Carcinoma. Cancer Res. 2014, 74, 7161–7167. [Google Scholar] [CrossRef] [Green Version]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-Ancestry Mutational Landscape of Hepatocellular Carcinoma Genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef]
- Shampay, J.; Szostak, J.W.; Blackburn, E.H. DNA Sequences of Telomeres Maintained in Yeast. Nature 1984, 310, 154–157. [Google Scholar] [CrossRef]
- In der Stroth, L.; Tharehalli, U.; Günes, C.; Lechel, A. Telomeres and Telomerase in the Development of Liver Cancer. Cancers 2020, 12, 2048. [Google Scholar] [CrossRef]
- Paradis, V.; Youssef, N.; Dargère, D.; Bâ, N.; Bonvoust, F.; Deschatrette, J.; Bedossa, P. Replicative Senescence in Normal Liver, Chronic Hepatitis C, and Hepatocellular Carcinomas. Hum. Pathol. 2001, 32, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Wiemann, S.U.; Satyanarayana, A.; Tsahuridu, M.; Tillmann, H.L.; Zender, L.; Klempnauer, J.; Flemming, P.; Franco, S.; Blasco, M.A.; Manns, M.P.; et al. Hepatocyte Telomere Shortening and Senescence Are General Markers of Human Liver Cirrhosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 935–942. [Google Scholar] [CrossRef]
- Urabe, Y.; Nouso, K.; Higashi, T.; Nakatsukasa, H.; Hino, N.; Ashida, K.; Kinugasa, N.; Yoshida, K.; Uematsu, S.; Tsuji, T. Telomere Length in Human Liver Diseases. Liver 1996, 16, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Ningarhari, M.; Caruso, S.; Hirsch, T.Z.; Bayard, Q.; Franconi, A.; Védie, A.-L.; Noblet, B.; Blanc, J.-F.; Amaddeo, G.; Ganne, N.; et al. Telomere Length Is Key to Hepatocellular Carcinoma Diversity and Telomerase Addiction Is an Actionable Therapeutic Target. J. Hepatol. 2021, 74, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Larsson, C.; Xu, D. Mechanisms Underlying the Activation of TERT Transcription and Telomerase Activity in Human Cancer: Old Actors and New Players. Oncogene 2019, 38, 6172–6183. [Google Scholar] [CrossRef] [Green Version]
- Kwa, W.T.; Effendi, K.; Yamazaki, K.; Kubota, N.; Hatano, M.; Ueno, A.; Masugi, Y.; Sakamoto, M. Telomerase Reverse Transcriptase (TERT) Promoter Mutation Correlated with Intratumoral Heterogeneity in Hepatocellular Carcinoma. Pathol. Int. 2020, 70, 624–632. [Google Scholar] [CrossRef]
- Ma, Z.-X.; Yang, C.-M.; Li, M.-G.; Tu, H. Telomerase Reverse Transcriptase Promoter Mutations in Hepatocellular Carcinogenesis. Hepatoma Res. 2019, 5, 8. [Google Scholar] [CrossRef]
- Wan, S.; Liu, X.; Hua, W.; Xi, M.; Zhou, Y.; Wan, Y. The Role of Telomerase Reverse Transcriptase (TERT) Promoter Mutations in Prognosis in Bladder Cancer. Bioengineered 2021, 12, 1495–1504. [Google Scholar] [CrossRef]
- Furuta, M.; Tanaka, H.; Shiraishi, Y.; Unida, T.; Imamura, M.; Fujimoto, A.; Fujita, M.; Sasaki-Oku, A.; Maejima, K.; Nakano, K.; et al. Characterization of HBV Integration Patterns and Timing in Liver Cancer and HBV-Infected Livers. Oncotarget 2018, 9, 25075–25088. [Google Scholar] [CrossRef] [Green Version]
- Trung, N.T.; Hoan, N.X.; Trung, P.Q.; Binh, M.T.; van Tong, H.; Toan, N.L.; Bang, M.H.; Le Song, H. Clinical Significance of Combined Circulating TERT Promoter Mutations and MiR-122 Expression for Screening HBV-Related Hepatocellular Carcinoma. Sci. Rep. 2020, 10, 8181. [Google Scholar] [CrossRef]
- Mathkar, P.P.; Chen, X.; Sulovari, A.; Li, D. Characterization of Hepatitis B Virus Integrations Identified in Hepatocellular Carcinoma Genomes. Viruses 2021, 13, 245. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Yoo, J.E.; Jeon, Y.; Chong, J.U.; Choi, G.H.; Song, D.-G.; Jung, S.H.; Oh, B.-K.; Park, Y.N. Suppression of PROX1-Mediated TERT Expression in Hepatitis B Viral Hepatocellular Carcinoma. Int. J. Cancer 2018, 143, 3155–3168. [Google Scholar] [CrossRef]
- Li, W.; Yan, Y.; Zheng, Z.; Zhu, Q.; Long, Q.; Sui, S.; Luo, M.; Chen, M.; Li, Y.; Hua, Y.; et al. Targeting the NCOA3-SP1-TERT Axis for Tumor Growth in Hepatocellular Carcinoma. Cell Death Dis. 2020, 11, 1011. [Google Scholar] [CrossRef]
- Yu, J.; Yuan, X.; Sjöholm, L.; Liu, T.; Kong, F.; Ekström, T.J.; Björkholm, M.; Xu, D. Telomerase Reverse Transcriptase Regulates DNMT3B Expression/Aberrant DNA Methylation Phenotype and AKT Activation in Hepatocellular Carcinoma. Cancer Lett. 2018, 434, 33–41. [Google Scholar] [CrossRef]
- Liang, T.J. Hepatitis B: The Virus and Disease. Hepatology 2009, 49, S13–S21. [Google Scholar] [CrossRef] [Green Version]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef]
- Jin, Z.-L.; Pei, H.; Xu, Y.-H.; Yu, J.; Deng, T. The SUMO-Specific Protease SENP5 Controls DNA Damage Response and Promotes Tumorigenesis in Hepatocellular Carcinoma. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 3566–3573. [Google Scholar]
- Shimizu, M.; Shirakami, Y.; Imai, K.; Takai, K.; Moriwaki, H. Acyclic Retinoid in Chemoprevention of Hepatocellular Carcinoma: Targeting Phosphorylated Retinoid X Receptor-α for Prevention of Liver Carcinogenesis. J. Carcinog. 2012, 11, 11. [Google Scholar] [CrossRef]
- Sung, W.-K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-Wide Survey of Recurrent HBV Integration in Hepatocellular Carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef]
- Tatsuno, K.; Midorikawa, Y.; Takayama, T.; Yamamoto, S.; Nagae, G.; Moriyama, M.; Nakagawa, H.; Koike, K.; Moriya, K.; Aburatani, H. Impact of AAV2 and Hepatitis B Virus Integration Into Genome on Development of Hepatocellular Carcinoma in Patients with Prior Hepatitis B Virus Infection. Clin. Cancer Res. 2019, 25, 6217–6227. [Google Scholar] [CrossRef]
- Midorikawa, Y.; Tatsuno, K.; Moriyama, M. Genome-Wide Analysis of Hepatitis B Virus Integration in Hepatocellular Carcinoma: Insights next Generation Sequencing. Hepatobiliary Surg. Nutr. 2021, 10, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Lou, X.; Hua, D.; Yu, W.; Li, L.; Wang, J.; Gao, F.; Zhao, N.; Ren, G.; Li, L.; et al. Recurrent Targeted Genes of Hepatitis B Virus in the Liver Cancer Genomes Identified by a Next-Generation Sequencing–Based Approach. PLoS Genet. 2012, 8, e1003065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hino, O.; Kajino, K.; Umeda, T.; Arakawa, Y. Understanding the Hypercarcinogenic State in Chronic Hepatitis: A Clue to the Prevention of Human Hepatocellular Carcinoma. J. Gastroenterol. 2002, 37, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Howell, J.; Atkinson, S.R.; Pinato, D.J.; Knapp, S.; Ward, C.; Minisini, R.; Burlone, M.E.; Leutner, M.; Pirisi, M.; Büttner, R.; et al. Identification of Mutations in Circulating Cell-Free Tumour DNA as a Biomarker in Hepatocellular Carcinoma. Eur. J. Cancer 2019, 116, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, Y.-F.; Cao, J.; Burley, S.K.; Wang, H.-Y.; Zheng, X.F.S. MTORC1 Promotes ARID1A Degradation and Oncogenic Chromatin Remodeling in Hepatocellular Carcinoma. Cancer Res. 2021, 81, 5652–5665. [Google Scholar] [CrossRef]
- Loesch, R.; Chenane, L.; Colnot, S. ARID2 Chromatin Remodeler in Hepatocellular Carcinoma. Cells 2020, 9, 2152. [Google Scholar] [CrossRef]
- Raghunath, A.; Sundarraj, K.; Arfuso, F.; Sethi, G.; Perumal, E. Dysregulation of Nrf2 in Hepatocellular Carcinoma: Role in Cancer Progression and Chemoresistance. Cancers 2018, 10, 481. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.; Singh, S.; Liu, S.; Bell, A.; Chen, X.; Monga, S.P. NFE2L2 Synergizes with Beta-Catenin Gene Mutations to Induce HCC in Patients and Mice. FASEB J. 2019, 33, 126.12. [Google Scholar] [CrossRef]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated Analysis of Somatic Mutations and Focal Copy-Number Changes Identifies Key Genes and Pathways in Hepatocellular Carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef]
- Bollati, V.; Baccarelli, A. Environmental Epigenetics. Heredity 2010, 105, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Wei, D.; Ji, Y.; Chen, L.; Yang, L.; Li, G.; Wu, L.; Hou, T.; Xie, L.; Ding, G.; et al. Integrative Analysis of DNA Methylation and Gene Expression Reveals Hepatocellular Carcinoma-Specific Diagnostic Biomarkers. Genome Med. 2018, 10, 42. [Google Scholar] [CrossRef]
- Deng, Y.-B.; Nagae, G.; Midorikawa, Y.; Yagi, K.; Tsutsumi, S.; Yamamoto, S.; Hasegawa, K.; Kokudo, N.; Aburatani, H.; Kaneda, A. Identification of Genes Preferentially Methylated in Hepatitis C Virus-Related Hepatocellular Carcinoma. Cancer Sci. 2010, 101, 1501–1510. [Google Scholar] [CrossRef]
- Su, P.-F.; Lee, T.-C.; Lin, P.-J.; Lee, P.-H.; Jeng, Y.-M.; Chen, C.-H.; Liang, J.-D.; Chiou, L.-L.; Huang, G.-T.; Lee, H.-S. Differential DNA Methylation Associated with Hepatitis B Virus Infection in Hepatocellular Carcinoma. Int. J. Cancer 2007, 121, 1257–1264. [Google Scholar] [CrossRef]
- Matsushita, J.; Suzuki, T.; Okamura, K.; Ichihara, G.; Nohara, K. Identification by TCGA Database Search of Five Genes That Are Aberrantly Expressed and Involved in Hepatocellular Carcinoma Potentially via DNA Methylation Changes. Environ. Health Prev. Med. 2020, 25, 31. [Google Scholar] [CrossRef]
- Zheng, Y.-F.; Lu, X.; Zhang, X.-Y.; Guan, B.-G. The Landscape of DNA Methylation in Hepatocellular Carcinoma. J. Cell. Physiol. 2019, 234, 2631–2638. [Google Scholar] [CrossRef]
- Zang, J.-J.; Xie, F.; Xu, J.-F.; Qin, Y.-Y.; Shen, R.-X.; Yang, J.-M.; He, J. P16 Gene Hypermethylation and Hepatocellular Carcinoma: A Systematic Review and Meta-Analysis. World J. Gastroenterol. WJG 2011, 17, 3043–3048. [Google Scholar] [CrossRef]
- Zhang, C.; Li, J.; Huang, T.; Duan, S.; Dai, D.; Jiang, D.; Sui, X.; Li, D.; Chen, Y.; Ding, F.; et al. Meta-Analysis of DNA Methylation Biomarkers in Hepatocellular Carcinoma. Oncotarget 2016, 7, 81255–81267. [Google Scholar] [CrossRef] [Green Version]
- Arechederra, M.; Recalde, M.; Gárate-Rascón, M.; Fernández-Barrena, M.G.; Ávila, M.A.; Berasain, C. Epigenetic Biomarkers for the Diagnosis and Treatment of Liver Disease. Cancers 2021, 13, 1265. [Google Scholar] [CrossRef]
- Liu, Y.-X.; Li, Q.-Z.; Cao, Y.-N.; Zhang, L.-Q. Identification of Key Genes and Important Histone Modifications in Hepatocellular Carcinoma. Comput. Struct. Biotechnol. J. 2020, 18, 2657–2669. [Google Scholar] [CrossRef]
- Li, D.; Zeng, Z. Epigenetic Regulation of Histone H3 in the Process of Hepatocellular Tumorigenesis. Biosci. Rep. 2019, 39, BSR20191815. [Google Scholar] [CrossRef] [Green Version]
- Cai, M.-Y.; Hou, J.-H.; Rao, H.-L.; Luo, R.-Z.; Li, M.; Pei, X.-Q.; Lin, M.C.; Guan, X.-Y.; Kung, H.-F.; Zeng, Y.-X.; et al. High Expression of H3K27me3 in Human Hepatocellular Carcinomas Correlates Closely with Vascular Invasion and Predicts Worse Prognosis in Patients. Mol. Med. 2011, 17, 12–20. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Xu, J.; Zhang, J.; Xie, D.; Ye, H.; Xiao, Z.; Cai, M.; Xu, K.; Zeng, Y.; Li, H.; et al. High Expression of Trimethylated Histone H3 Lysine 4 Is Associated with Poor Prognosis in Hepatocellular Carcinoma. Hum. Pathol. 2012, 43, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Hayashi Concurrent Activation of Acetylation and Tri-Methylation of H3K27 in a Subset of Hepatocellular Carcinoma with Aggressive Behavior. Available online: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0091330 (accessed on 3 April 2022).
- Magerl, C.; Ellinger, J.; Braunschweig, T.; Kremmer, E.; Koch, L.K.; Höller, T.; Büttner, R.; Lüscher, B.; Gütgemann, I. H3K4 Dimethylation in Hepatocellular Carcinoma Is Rare Compared with Other Hepatobiliary and Gastrointestinal Carcinomas and Correlates with Expression of the Methylase Ash2 and the Demethylase LSD1. Hum. Pathol. 2010, 41, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Liu, H.; Sasagawa, S.; Dong, Y.; Trainor, P.A.; Cheng, E.H.; Hsieh, J.J. HGF-MET Signals via the MLL-ETS2 Complex in Hepatocellular Carcinoma. J. Clin. Investig. 2013, 123, 3154–3165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, X.; Gao, W.; Sun, B.; Xu, Y.; Han, B.; Wang, F.; Zhang, Y.; Sun, J.; Wei, J.; Lu, Z.; et al. Vasohibin 2 Is Transcriptionally Activated and Promotes Angiogenesis in Hepatocellular Carcinoma. Oncogene 2013, 32, 1724–1734. [Google Scholar] [CrossRef]
- Zhao, P.; Malik, S.; Xing, S. Epigenetic Mechanisms Involved in HCV-Induced Hepatocellular Carcinoma (HCC). Front. Oncol. 2021, 11, 677926. [Google Scholar] [CrossRef]
- Jancewicz, I.; Siedlecki, J.A.; Sarnowski, T.J.; Sarnowska, E. BRM: The Core ATPase Subunit of SWI/SNF Chromatin-Remodelling Complex—A Tumour Suppressor or Tumour-Promoting Factor? Epigenet. Chromatin 2019, 12, 68. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, Q.; Tao, L.; Chen, Y.; Shu, Y.; Wu, Z.; Lu, C.; Shi, Y.; Bu, H. Enhanced SMARCD1, a Subunit of the SWI/SNF Complex, Promotes Liver Cancer Growth through the MTOR Pathway. Clin. Sci. 2020, 134, 1457–1472. [Google Scholar] [CrossRef]
- Li, M.; Zhao, H.; Zhang, X.; Wood, L.D.; Anders, R.A.; Choti, M.A.; Pawlik, T.M.; Daniel, H.D.; Kannangai, R.; Offerhaus, G.J.A.; et al. Inactivating Mutations of the Chromatin Remodeling Gene ARID2 in Hepatocellular Carcinoma. Nat. Genet. 2011, 43, 828–829. [Google Scholar] [CrossRef] [Green Version]
- Cougot, D.; Wu, Y.; Cairo, S.; Caramel, J.; Renard, C.-A.; Lévy, L.; Buendia, M.A.; Neuveut, C. The Hepatitis B Virus X Protein Functionally Interacts with CREB-Binding Protein/P300 in the Regulation of CREB-Mediated Transcription. J. Biol. Chem. 2007, 282, 4277–4287. [Google Scholar] [CrossRef] [Green Version]
- Vidigal, J.A.; Ventura, A. The Biological Functions of MiRNAs: Lessons from in Vivo Studies. Trends Cell Biol. 2015, 25, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Sartorius, K.; An, P.; Winkler, C.; Chuturgoon, A.; Li, X.; Makarova, J.; Kramvis, A. The Epigenetic Modulation of Cancer and Immune Pathways in Hepatitis B Virus-Associated Hepatocellular Carcinoma: The Influence of HBx and MiRNA Dysregulation. Front. Immunol. 2021, 12, 661204. [Google Scholar] [CrossRef]
- Wei, X.; Xiang, T.; Ren, G.; Tan, C.; Liu, R.; Xu, X.; Wu, Z. MiR-101 Is down-Regulated by the Hepatitis B Virus x Protein and Induces Aberrant DNA Methylation by Targeting DNA Methyltransferase 3A. Cell. Signal. 2013, 25, 439–446. [Google Scholar] [CrossRef]
- Wei, X.; Tan, C.; Tang, C.; Ren, G.; Xiang, T.; Qiu, Z.; Liu, R.; Wu, Z. Epigenetic Repression of MiR-132 Expression by the Hepatitis B Virus x Protein in Hepatitis B Virus-Related Hepatocellular Carcinoma. Cell. Signal. 2013, 25, 1037–1043. [Google Scholar] [CrossRef]
- Lin, X.-J.; Fang, J.-H.; Yang, X.-J.; Zhang, C.; Yuan, Y.; Zheng, L.; Zhuang, S.-M. Hepatocellular Carcinoma Cell-Secreted Exosomal MicroRNA-210 Promotes Angiogenesis In Vitro and In Vivo. Mol. Ther.-Nucleic Acids 2018, 11, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Xu, H.-F.; Liu, M.-Y.; Xu, Y.-J.; He, J.-C.; Zhou, Y.; Cang, S.-D. Mechanism of Exosomal MicroRNA-224 in Development of Hepatocellular Carcinoma and Its Diagnostic and Prognostic Value. World J. Gastroenterol. 2019, 25, 1890–1898. [Google Scholar] [CrossRef]
- Ren, W.; Wu, S.; Wu, Y.; Liu, T.; Zhao, X.; Li, Y. MicroRNA-196a/-196b Regulate the Progression of Hepatocellular Carcinoma through Modulating the JAK/STAT Pathway via Targeting SOCS2. Cell Death Dis. 2019, 10, 333. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Lui, V.W.Y.; Yeo, W. Targeting the PI3K/Akt/MTOR Pathway in Hepatocellular Carcinoma. Future Oncol. 2011, 7, 1149–1167. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, Y.; Dou, C.; Sun, L.; Li, Q.; Wang, L.; Xu, Q.; Yang, W.; Liu, Q.; Tu, K. MicroRNA-1468 Promotes Tumor Progression by Activating PPAR-γ-Mediated AKT Signaling in Human Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 49. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Liu, X.; Li, J.; He, Y. MicroRNA-144 Inhibits Cell Proliferation, Migration and Invasion in Human Hepatocellular Carcinoma by Targeting CCNB1. Cancer Cell Int. 2019, 19, 15. [Google Scholar] [CrossRef] [Green Version]
- Komoll, R.-M.; Hu, Q.; Olarewaju, O.; von Döhlen, L.; Yuan, Q.; Xie, Y.; Tsay, H.-C.; Daon, J.; Qin, R.; Manns, M.P.; et al. MicroRNA-342-3p Is a Potent Tumour Suppressor in Hepatocellular Carcinoma. J. Hepatol. 2021, 74, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Lu, H.; Wang, X.; Jin, H. MicroRNAs in Hepatocellular Carcinoma: Regulation, Function, and Clinical Implications. Sci. World J. 2013, 2013, e924206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preethi, K.A.; Selvakumar, S.C.; Ross, K.; Jayaraman, S.; Tusubira, D.; Sekar, D. Liquid Biopsy: Exosomal MicroRNAs as Novel Diagnostic and Prognostic Biomarkers in Cancer. Mol. Cancer 2022, 21, 54. [Google Scholar] [CrossRef] [PubMed]
- Drula, R.; Ott, L.F.; Berindan-Neagoe, I.; Pantel, K.; Calin, G.A. MicroRNAs from Liquid Biopsy Derived Extracellular Vesicles: Recent Advances in Detection and Characterization Methods. Cancers 2020, 12, 2009. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Shu, Z.; Ye, T.; Zhang, M. Identification and Analysis of the Blood LncRNA Signature for Liver Cirrhosis and Hepatocellular Carcinoma. Front. Genet. 2020, 11, 595699. [Google Scholar] [CrossRef]
- Jiang, M.-C.; Ni, J.-J.; Cui, W.-Y.; Wang, B.-Y.; Zhuo, W. Emerging Roles of LncRNA in Cancer and Therapeutic Opportunities. Am. J. Cancer Res. 2019, 9, 1354–1366. [Google Scholar]
- He, Q.; Long, J.; Yin, Y.; Li, Y.; Lei, X.; Li, Z.; Zhu, W. Emerging Roles of LncRNAs in the Formation and Progression of Colorectal Cancer. Front. Oncol. 2020, 9, 1542. [Google Scholar]
- Huang, Z.; Zhou, J.-K.; Peng, Y.; He, W.; Huang, C. The Role of Long Noncoding RNAs in Hepatocellular Carcinoma. Mol. Cancer 2020, 19, 77. [Google Scholar] [CrossRef] [Green Version]
- Dhanasekaran, R.; Bandoh, S.; Roberts, L.R. Molecular Pathogenesis of Hepatocellular Carcinoma and Impact of Therapeutic Advances. F1000Research 2016, 5, 879. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, H.; Li, H.; Feng, X.; Tang, H.; Qiu, C.; Zhang, J.; Fu, B. LINC01234/MicroRNA-31-5p/MAGEA3 Axis Mediates the Proliferation and Chemoresistance of Hepatocellular Carcinoma Cells. Mol. Ther.-Nucleic Acids 2020, 19, 168–178. [Google Scholar] [CrossRef]
- Huang, J.; Cao, S.; Ou, Q.; Yang, B.; Zheng, S.; Tang, J.; Chen, J.; Hu, Y.; Zheng, L.; Wang, Q. The Long Non-Coding RNA PTTG3P Promotes Cell Growth and Metastasis via up-Regulating PTTG1 and Activating PI3K/AKT Signaling in Hepatocellular Carcinoma. Mol. Cancer 2018, 17, 93. [Google Scholar] [CrossRef]
- Panzitt, K.; Tschernatsch, M.M.O.; Guelly, C.; Moustafa, T.; Stradner, M.; Strohmaier, H.M.; Buck, C.R.; Denk, H.; Schroeder, R.; Trauner, M.; et al. Characterization of HULC, a Novel Gene With Striking Up-Regulation in Hepatocellular Carcinoma, as Noncoding RNA. Gastroenterology 2007, 132, 330–342. [Google Scholar] [CrossRef]
- Du, Y.; Kong, G.; You, X.; Zhang, S.; Zhang, T.; Gao, Y.; Ye, L.; Zhang, X. Elevation of Highly Up-Regulated in Liver Cancer (HULC) by Hepatitis B Virus X Protein Promotes Hepatoma Cell Proliferation via Down-Regulating P18. J. Biol. Chem. 2012, 287, 26302–26311. [Google Scholar] [CrossRef] [Green Version]
- Hirai, H.; Roussel, M.F.; Kato, J.Y.; Ashmun, R.A.; Sherr, C.J. Novel INK4 Proteins, P19 and P18, Are Specific Inhibitors of the Cyclin D-Dependent Kinases CDK4 and CDK6. Mol. Cell. Biol. 1995, 15, 2672–2681. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Zhang, L.; Huo, X.; Yuan, J.; Xu, D.; Yuan, S.; Zhu, N.; Zhou, W.; Yang, G.; Wang, Y.; et al. Long Noncoding RNA High Expression in Hepatocellular Carcinoma Facilitates Tumor Growth through Enhancer of Zeste Homolog 2 in Humans. Hepatology 2011, 54, 1679–1689. [Google Scholar] [CrossRef]
- Lay, A.J.; Jiang, X.-M.; Kisker, O.; Flynn, E.; Underwood, A.; Condron, R.; Hogg, P.J. Phosphoglycerate Kinase Acts in Tumour Angiogenesis as a Disulphide Reductase. Nature 2000, 408, 869–873. [Google Scholar] [CrossRef]
- He, Y.; Luo, Y.; Zhang, D.; Wang, X.; Zhang, P.; Li, H.; Ejaz, S.; Liang, S. PGK1-Mediated Cancer Progression and Drug Resistance. Am. J. Cancer Res. 2019, 9, 2280–2302. [Google Scholar]
- Feng, J.; Li, J.; Wu, L.; Yu, Q.; Ji, J.; Wu, J.; Dai, W.; Guo, C. Emerging Roles and the Regulation of Aerobic Glycolysis in Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. 2020, 39, 126. [Google Scholar] [CrossRef]
- Bao, M.H.-R.; Wong, C.C.-L. Hypoxia, Metabolic Reprogramming, and Drug Resistance in Liver Cancer. Cells 2021, 10, 1715. [Google Scholar] [CrossRef]
- Cassim, S.; Raymond, V.-A.; Dehbidi-Assadzadeh, L.; Lapierre, P.; Bilodeau, M. Metabolic Reprogramming Enables Hepatocarcinoma Cells to Efficiently Adapt and Survive to a Nutrient-Restricted Microenvironment. Cell Cycle 2018, 17, 903–916. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. We Need to Talk about the Warburg Effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.; Tang, Z.; Chen, K.; Liu, Y.; Yu, G.; Chen, Q.; Dang, H.; Chen, F.; Ling, J.; Zhu, L.; et al. Long Noncoding RNA MIR31HG Inhibits Hepatocellular Carcinoma Proliferation and Metastasis by Sponging MicroRNA-575 to Modulate ST7L Expression. J. Exp. Clin. Cancer Res. 2018, 37, 214. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Xiong, D.; He, R.; Yang, X.; Lai, Z.; Liu, L.; Huang, Z.; Wu, H.; Yang, L.; Ma, J.; et al. MIR22HG As A Tumor Suppressive LncRNA In HCC: A Comprehensive Analysis Integrating RT-QPCR, MRNA-Seq, And Microarrays. OncoTargets Ther. 2019, 12, 9827–9848. [Google Scholar] [CrossRef] [Green Version]
- Khalaf, A.M.; Fuentes, D.; Morshid, A.I.; Burke, M.R.; Kaseb, A.O.; Hassan, M.; Hazle, J.D.; Elsayes, K.M. Role of Wnt/β-Catenin Signaling in Hepatocellular Carcinoma, Pathogenesis, and Clinical Significance. J. Hepatocell. Carcinoma 2018, 5, 61–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bévant, K.; Coulouarn, C. Landscape of Genomic Alterations in Hepatocellular Carcinoma: Current Knowledge and Perspectives for Targeted Therapies. Hepatobiliary Surg. Nutr. 2017, 6, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Ho, N.P.Y.; Leung, C.O.N.; Wong, T.L.; Lau, E.Y.T.; Lei, M.M.L.; Mok, E.H.K.; Leung, H.W.; Tong, M.; Ng, I.O.L.; Yun, J.P.; et al. The Interplay of UBE2T and Mule in Regulating Wnt/β-Catenin Activation to Promote Hepatocellular Carcinoma Progression. Cell Death Dis. 2021, 12, 148. [Google Scholar] [CrossRef]
- Song, J.; Xie, C.; Jiang, L.; Wu, G.; Zhu, J.; Zhang, S.; Tang, M.; Song, L.; Li, J. Transcription Factor AP-4 Promotes Tumorigenic Capability and Activates the Wnt/β-Catenin Pathway in Hepatocellular Carcinoma. Theranostics 2018, 8, 3571–3583. [Google Scholar] [CrossRef]
- Zhang, T.; Ma, Z.; Liu, L.; Sun, J.; Tang, H.; Zhang, B.; Zou, Y.; Li, H. DDX39 Promotes Hepatocellular Carcinoma Growth and Metastasis through Activating Wnt/β-Catenin Pathway. Cell Death Dis. 2018, 9, 657. [Google Scholar] [CrossRef] [Green Version]
- Zhu, P.; Liang, H.; Huang, X.; Zeng, Q.; Liu, Y.; Lv, J.; Ming, L. Circular RNA Hsa_circ_0004018 Inhibits Wnt/β-Catenin Signaling Pathway by Targeting MicroRNA-626/DKK3 in Hepatocellular Carcinoma. OncoTargets Ther. 2020, 13, 9351–9364. [Google Scholar] [CrossRef]
- Chen, H.; Liu, S.; Li, M.; Huang, P.; Li, X. Circ_0003418 Inhibits Tumorigenesis And Cisplatin Chemoresistance Through Wnt/β-Catenin Pathway In Hepatocellular Carcinoma. OncoTargets Ther. 2019, 12, 9539–9549. [Google Scholar] [CrossRef] [Green Version]
- Ferrín, G.; Guerrero, M.; Amado, V.; Rodríguez-Perálvarez, M.; De la Mata, M. Activation of MTOR Signaling Pathway in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 1266. [Google Scholar] [CrossRef] [Green Version]
- Rahmani, F.; Ziaeemehr, A.; Shahidsales, S.; Gharib, M.; Khazaei, M.; Ferns, G.A.; Ryzhikov, M.; Avan, A.; Hassanian, S.M. Role of Regulatory MiRNAs of the PI3K/AKT/MTOR Signaling in the Pathogenesis of Hepatocellular Carcinoma. J. Cell. Physiol. 2020, 235, 4146–4152. [Google Scholar] [CrossRef]
- Wang, L.; Wang, W.-L.; Zhang, Y.; Guo, S.-P.; Zhang, J.; Li, Q.-L. Epigenetic and Genetic Alterations of PTEN in Hepatocellular Carcinoma. Hepatol. Res. 2007, 37, 389–396. [Google Scholar] [CrossRef]
- Lu, Y.; Li, X.; Liu, H.; Xue, J.; Zeng, Z.; Dong, X.; Zhang, T.; Wu, G.; Yang, K.; Xu, S. β-Trcp and CK1δ-Mediated Degradation of LZTS2 Activates PI3K/AKT Signaling to Drive Tumorigenesis and Metastasis in Hepatocellular Carcinoma. Oncogene 2021, 40, 1269–1283. [Google Scholar] [CrossRef]
- Chen, T.; Huang, H.; Zhou, Y.; Geng, L.; Shen, T.; Yin, S.; Zhou, L.; Zheng, S. HJURP Promotes Hepatocellular Carcinoma Proliferation by Destabilizing P21 via the MAPK/ERK1/2 and AKT/GSK3β Signaling Pathways. J. Exp. Clin. Cancer Res. 2018, 37, 193. [Google Scholar] [CrossRef] [Green Version]
- Chan, L.-K.; Ho, D.W.-H.; Kam, C.S.; Chiu, E.Y.-T.; Lo, I.L.-O.; Yau, D.T.-W.; Cheung, E.T.-Y.; Tang, C.-N.; Tang, V.W.-L.; Lee, T.K.-W.; et al. RSK2-Inactivating Mutations Potentiate MAPK Signaling and Support Cholesterol Metabolism in Hepatocellular Carcinoma. J. Hepatol. 2021, 74, 360–371. [Google Scholar] [CrossRef]
- Fu, X.; Wen, H.; Jing, L.; Yang, Y.; Wang, W.; Liang, X.; Nan, K.; Yao, Y.; Tian, T. MicroRNA-155-5p Promotes Hepatocellular Carcinoma Progression by Suppressing PTEN through the PI3K/Akt Pathway. Cancer Sci. 2017, 108, 620–631. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zhang, Y.; Qin, X.; Geng, H.; Zuo, D.; Zhao, Q. PI3K/AKT/MTOR Pathway-Related Long Non-Coding RNAs: Roles and Mechanisms in Hepatocellular Carcinoma. Pharmacol. Res. 2020, 160, 105195. [Google Scholar] [CrossRef]
- Yang, S.; Yang, C.; Yu, F.; Ding, W.; Hu, Y.; Cheng, F.; Zhang, F.; Guan, B.; Wang, X.; Lu, L.; et al. Endoplasmic Reticulum Resident Oxidase ERO1-Lalpha Promotes Hepatocellular Carcinoma Metastasis and Angiogenesis through the S1PR1/STAT3/VEGF-A Pathway. Cell Death Dis. 2018, 9, 1105. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Xu, Z.; Chen, L.; Wei, Q.; Huang, Z.; Liu, G.; Li, W.; Wang, J.; Tang, Q.; Pu, J. Long Non-Coding RNA PAARH Promotes Hepatocellular Carcinoma Progression and Angiogenesis via Upregulating HOTTIP and Activating HIF-1α/VEGF Signaling. Cell Death Dis. 2022, 13, 102. [Google Scholar] [CrossRef]
- Brooks, A.J.; Putoczki, T. JAK-STAT Signalling Pathway in Cancer. Cancers 2020, 12, 1971. [Google Scholar] [CrossRef] [PubMed]
- Lokau, J.; Schoeder, V.; Haybaeck, J.; Garbers, C. Jak-Stat Signaling Induced by Interleukin-6 Family Cytokines in Hepatocellular Carcinoma. Cancers 2019, 11, 1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, A.; Llovet, J.M. Mutational Landscape of HCC—The End of the Beginning. Nat. Rev. Clin. Oncol. 2014, 11, 73–74. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Chen, L.; Zhang, X.; Zhang, H.; Tan, X.; Dong, K.; Lu, X.; Zhu, H.; Liu, Q.; Zhang, Z.; et al. PTPRε Acts as a Metastatic Promoter in Hepatocellular Carcinoma by Facilitating Recruitment of SMAD3 to TGF-β Receptor 1. Hepatology 2020, 72, 997–1012. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-Induced Epithelial to Mesenchymal Transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Qu, Z.; Feng, J.; Pan, H.; Jiang, Y.; Duan, Y.; Fa, Z. Exosomes Derived from HCC Cells with Different Invasion Characteristics Mediated EMT through TGF-β/Smad Signaling Pathway. OncoTargets Ther. 2019, 12, 6897–6905. [Google Scholar] [CrossRef]
- Garcia-Lezana, T.; Lopez-Canovas, J.L.; Villanueva, A. Signaling pathways in hepatocellular carcinoma. Adv. Cancer Res. 2021, 149, 63–101. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Dimri, M.; Satyanarayana, A. Molecular Signaling Pathways and Therapeutic Targets in Hepatocellular Carcinoma. Cancers 2020, 12, 491. [Google Scholar] [CrossRef] [Green Version]
- Farzaneh, Z.; Vosough, M.; Agarwal, T.; Farzaneh, M. Critical Signaling Pathways Governing Hepatocellular Carcinoma Behavior; Small Molecule-Based Approaches. Cancer Cell Int. 2021, 21, 208. [Google Scholar] [CrossRef]
- Parsons, M.J.; Tammela, T.; Dow, L.E. WNT as a Driver and Dependency in Cancer. Cancer Discov. 2021, 11, 2413–2429. [Google Scholar] [CrossRef]
- Perugorria, M.J.; Olaizola, P.; Labiano, I.; Esparza-Baquer, A.; Marzioni, M.; Marin, J.J.G.; Bujanda, L.; Banales, J.M. Wnt–β-Catenin Signalling in Liver Development, Health and Disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 121–136. [Google Scholar] [CrossRef]
- Shi, J.; Chi, S.; Xue, J.; Yang, J.; Li, F.; Liu, X. Emerging Role and Therapeutic Implication of Wnt Signaling Pathways in Autoimmune Diseases. J. Immunol. Res. 2016, 2016, e9392132. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Xu, Z.; Zhang, Y.; Evert, M.; Calvisi, D.F.; Chen, X. β-Catenin Signaling in Hepatocellular Carcinoma. J. Clin. Investig. 2022, 132, e154515. [Google Scholar] [CrossRef]
- Xiao, X.; Mo, H.; Tu, K. CTNNB1 Mutation Suppresses Infiltration of Immune Cells in Hepatocellular Carcinoma through MiRNA-Mediated Regulation of Chemokine Expression. Int. Immunopharmacol. 2020, 89, 107043. [Google Scholar] [CrossRef]
- Diniz, P.H.C.; Silva, S.D.C.; Vidigal, P.V.T.; Xavier, M.A.P.; Lima, C.X.; Faria, L.C.; Ferrari, T.C.A. Expression of MAPK and PI3K/AKT/MTOR Proteins According to the Chronic Liver Disease Etiology in Hepatocellular Carcinoma. J. Oncol. 2020, 2020, 4609360. [Google Scholar] [CrossRef]
- Moon, H.; Ro, S.W. MAPK/ERK Signaling Pathway in Hepatocellular Carcinoma. Cancers 2021, 13, 3026. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK Signal Pathways in the Regulation of Cell Proliferation in Mammalian Cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Roberts, L.R.; Gores, G.J. Hepatocellular Carcinoma: Molecular Pathways and New Therapeutic Targets. Semin. Liver Dis. 2005, 25, 212–225. [Google Scholar] [CrossRef] [Green Version]
- Delire, B.; Stärkel, P. The Ras/MAPK Pathway and Hepatocarcinoma: Pathogenesis and Therapeutic Implications. Eur. J. Clin. Investig. 2015, 45, 609–623. [Google Scholar] [CrossRef]
- Benetatos, L.; Voulgaris, E.; Vartholomatos, G. The Crosstalk between Long Non-Coding RNAs and PI3K in Cancer. Med. Oncol. 2017, 34, 39. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in Cancer: Mechanisms and Advances in Clinical Trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semela, D.; Dufour, J.-F. Angiogenesis and Hepatocellular Carcinoma. J. Hepatol. 2004, 41, 864–880. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-Z.; Xie, G.-R.; Chen, D. Hypoxia and Hepatocellular Carcinoma: The Therapeutic Target for Hepatocellular Carcinoma. J. Gastroenterol. Hepatol. 2007, 22, 1178–1182. [Google Scholar] [CrossRef] [PubMed]
- Cannito, S.; Turato, C.; Paternostro, C.; Biasiolo, A.; Colombatto, S.; Cambieri, I.; Quarta, S.; Novo, E.; Morello, E.; Villano, G.; et al. Hypoxia Up-Regulates SERPINB3 through HIF-2α in Human Liver Cancer Cells. Oncotarget 2014, 6, 2206–2221. [Google Scholar] [CrossRef] [Green Version]
- Longo, R.; Sarmiento, R.; Fanelli, M.; Capaccetti, B.; Gattuso, D.; Gasparini, G. Anti-Angiogenic Therapy: Rationale, Challenges and Clinical Studies. Angiogenesis 2002, 5, 237–256. [Google Scholar] [CrossRef]
- Tang, J.J.H.; Thng, D.K.H.; Lim, J.J.; Toh, T.B. JAK/STAT Signaling in Hepatocellular Carcinoma. Hepatic Oncol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Tzavlaki, K.; Moustakas, A. TGF-β Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef] [Green Version]
- Dituri, F.; Mancarella, S.; Cigliano, A.; Chieti, A.; Giannelli, G. TGF-β as Multifaceted Orchestrator in HCC Progression: Signaling, EMT, Immune Microenvironment, and Novel Therapeutic Perspectives. Semin. Liver Dis. 2019, 39, 53–69. [Google Scholar] [CrossRef]
- Raoul, J.-L.; Forner, A.; Bolondi, L.; Cheung, T.T.; Kloeckner, R.; de Baere, T. Updated Use of TACE for Hepatocellular Carcinoma Treatment: How and When to Use It Based on Clinical Evidence. Cancer Treat. Rev. 2019, 72, 28–36. [Google Scholar] [CrossRef]
- O’Leary, C.; Mahler, M.; Soulen, M.C. Liver-Directed Therapy for Hepatocellular Carcinoma. Chin. Clin. Oncol. 2021, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, M.; Saisho, H.; Ebara, M.; Iijima, T.; Iwama, S.; Endo, F.; Kimura, M.; Shimamura, Y.; Suzuki, Y.; Nakano, T. A Randomized Trial of Intrahepatic Arterial Infusion of 4′-Epidoxorubicin with Lipiodol versus 4′-Epidoxorubicin Alone in the Treatment of Hepatocellular Carcinoma. Cancer Chemother. Pharmacol. 1994, 33, S149–S152. [Google Scholar] [CrossRef] [PubMed]
- Takayasu, K.; Shima, Y.; Muramatsu, Y.; Moriyama, N.; Yamada, T.; Makuuchi, M.; Hasegawa, H.; Hirohashi, S. Hepatocellular Carcinoma: Treatment with Intraarterial Iodized Oil with and without Chemotherapeutic Agents. Radiology 1987, 163, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Song, J.E.; Kim, D.Y. Conventional vs Drug-Eluting Beads Transarterial Chemoembolization for Hepatocellular Carcinoma. World J. Hepatol. 2017, 9, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Suh, J.H.; Yoo, H.S.; Lee, J.T.; Kim, D.I. Evaluation of Intrahepatic I-131 Ethiodol on a Patient with Hepatocellular Carcinoma. Therapeutic Feasibility Study. Clin. Nucl. Med. 1986, 11, 514–517. [Google Scholar] [CrossRef]
- Park, C.; Choi, S.I.; Kim, H.; Yoo, H.S.; Lee, Y.B. Distribution of Lipiodol in Hepatocellular Carcinoma. Liver 1990, 10, 72–78. [Google Scholar] [CrossRef]
- Marelli, L.; Stigliano, R.; Triantos, C.; Senzolo, M.; Cholongitas, E.; Davies, N.; Tibballs, J.; Meyer, T.; Patch, D.W.; Burroughs, A.K. Transarterial Therapy for Hepatocellular Carcinoma: Which Technique Is More Effective? A Systematic Review of Cohort and Randomized Studies. Cardiovasc. Interv. Radiol. 2007, 30, 6–25. [Google Scholar] [CrossRef]
- Lencioni, R. Loco-Regional Treatment of Hepatocellular Carcinoma. Hepatology 2010, 52, 762–773. [Google Scholar] [CrossRef]
- Liu, Z.; Tu, K.; Wang, Y.; Yao, B.; Li, Q.; Wang, L.; Dou, C.; Liu, Q.; Zheng, X. Hypoxia Accelerates Aggressiveness of Hepatocellular Carcinoma Cells Involving Oxidative Stress, Epithelial-Mesenchymal Transition and Non-Canonical Hedgehog Signaling. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 44, 1856–1868. [Google Scholar] [CrossRef] [Green Version]
- Kirchhoff, T.D.; Rudolph, K.L.; Layer, G.; Chavan, A.; Greten, T.F.; Rosenthal, H.; Kubicka, S.; Galanski, M.; Manns, M.P.; Schild, H.; et al. Chemoocclusion vs Chemoperfusion for Treatment of Advanced Hepatocellular Carcinoma: A Randomised Trial. Eur. J. Surg. Oncol. J. Eur. Soc. Surg. Oncol. Br. Assoc. Surg. Oncol. 2006, 32, 201–207. [Google Scholar] [CrossRef]
- Okusaka, T.; Kasugai, H.; Shioyama, Y.; Tanaka, K.; Kudo, M.; Saisho, H.; Osaki, Y.; Sata, M.; Fujiyama, S.; Kumada, T.; et al. Transarterial Chemotherapy Alone versus Transarterial Chemoembolization for Hepatocellular Carcinoma: A Randomized Phase III Trial. J. Hepatol. 2009, 51, 1030–1036. [Google Scholar] [CrossRef]
- Llovet, J.M.; Bruix, J. Systematic Review of Randomized Trials for Unresectable Hepatocellular Carcinoma: Chemoembolization Improves Survival. Hepatology 2003, 37, 429–442. [Google Scholar] [CrossRef] [Green Version]
- de Baere, T.; Plotkin, S.; Yu, R.; Sutter, A.; Wu, Y.; Cruise, G.M. An In Vitro Evaluation of Four Types of Drug-Eluting Microspheres Loaded with Doxorubicin. J. Vasc. Interv. Radiol. JVIR 2016, 27, 1425–1431. [Google Scholar] [CrossRef] [Green Version]
- Jordan, O.; Denys, A.; De Baere, T.; Boulens, N.; Doelker, E. Comparative Study of Chemoembolization Loadable Beads: In Vitro Drug Release and Physical Properties of DC Bead and Hepasphere Loaded with Doxorubicin and Irinotecan. J. Vasc. Interv. Radiol. JVIR 2010, 21, 1084–1090. [Google Scholar] [CrossRef]
- Lewis, A.L.; Taylor, R.R.; Hall, B.; Gonzalez, M.V.; Willis, S.L.; Stratford, P.W. Pharmacokinetic and Safety Study of Doxorubicin-Eluting Beads in a Porcine Model of Hepatic Arterial Embolization. J. Vasc. Interv. Radiol. JVIR 2006, 17, 1335–1343. [Google Scholar] [CrossRef]
- Facciorusso, A. Drug-Eluting Beads Transarterial Chemoembolization for Hepatocellular Carcinoma: Current State of the Art. World J. Gastroenterol. 2018, 24, 161–169. [Google Scholar] [CrossRef]
- de Baere, T.; Arai, Y.; Lencioni, R.; Geschwind, J.-F.; Rilling, W.; Salem, R.; Matsui, O.; Soulen, M.C. Treatment of Liver Tumors with Lipiodol TACE: Technical Recommendations from Experts Opinion. Cardiovasc. Intervent. Radiol. 2016, 39, 334–343. [Google Scholar] [CrossRef]
- Golfieri, R.; Giampalma, E.; Renzulli, M.; Cioni, R.; Bargellini, I.; Bartolozzi, C.; Breatta, A.D.; Gandini, G.; Nani, R.; Gasparini, D.; et al. Randomised Controlled Trial of Doxorubicin-Eluting Beads vs. Conventional Chemoembolisation for Hepatocellular Carcinoma. Br. J. Cancer 2014, 111, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Lammer, J.; Malagari, K.; Vogl, T.; Pilleul, F.; Denys, A.; Watkinson, A.; Pitton, M.; Sergent, G.; Pfammatter, T.; Terraz, S.; et al. Prospective Randomized Study of Doxorubicin-Eluting-Bead Embolization in the Treatment of Hepatocellular Carcinoma: Results of the PRECISION V Study. Cardiovasc. Intervent. Radiol. 2010, 33, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [Green Version]
- Schaaf, M.B.; Garg, A.D.; Agostinis, P. Defining the Role of the Tumor Vasculature in Antitumor Immunity and Immunotherapy. Cell Death Dis. 2018, 9, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morse, M.A.; Sun, W.; Kim, R.; He, A.R.; Abada, P.B.; Mynderse, M.; Finn, R.S. The Role of Angiogenesis in Hepatocellular Carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 912–920. [Google Scholar] [CrossRef] [Green Version]
- Kudo, M. Immuno-Oncology Therapy for Hepatocellular Carcinoma: Current Status and Ongoing Trials. Liver Cancer 2019, 8, 221–238. [Google Scholar] [CrossRef]
- Kudo, M. Immune Checkpoint Inhibition in Hepatocellular Carcinoma: Basics and Ongoing Clinical Trials. Oncology 2017, 92 (Suppl 1), 50–62. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Ueshima, K.; Ikeda, M.; Torimura, T.; Tanabe, N.; Aikata, H.; Izumi, N.; Yamasaki, T.; Nojiri, S.; Hino, K.; et al. Randomised, Multicentre Prospective Trial of Transarterial Chemoembolisation (TACE) plus Sorafenib as Compared with TACE Alone in Patients with Hepatocellular Carcinoma: TACTICS Trial. Gut 2020, 69, 1492–1501. [Google Scholar] [CrossRef]
- Fu, Z.; Li, X.; Zhong, J.; Chen, X.; Cao, K.; Ding, N.; Liu, L.; Zhang, X.; Zhai, J.; Qu, Z. Lenvatinib in Combination with Transarterial Chemoembolization for Treatment of Unresectable Hepatocellular Carcinoma (UHCC): A Retrospective Controlled Study. Hepatol. Int. 2021, 15, 663–675. [Google Scholar] [CrossRef]
- Lencioni, R.; Llovet, J.M.; Han, G.; Tak, W.Y.; Yang, J.; Guglielmi, A.; Paik, S.W.; Reig, M.; Kim, D.Y.; Chau, G.-Y.; et al. Sorafenib or Placebo plus TACE with Doxorubicin-Eluting Beads for Intermediate Stage HCC: The SPACE Trial. J. Hepatol. 2016, 64, 1090–1098. [Google Scholar] [CrossRef] [Green Version]
- Meyer, T.; Fox, R.; Ma, Y.T.; Ross, P.J.; James, M.W.; Sturgess, R.; Stubbs, C.; Stocken, D.D.; Wall, L.; Watkinson, A.; et al. Sorafenib in Combination with Transarterial Chemoembolisation in Patients with Unresectable Hepatocellular Carcinoma (TACE 2): A Randomised Placebo-Controlled, Double-Blind, Phase 3 Trial. Lancet Gastroenterol. Hepatol. 2017, 2, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Kudo, M.; Han, G.; Finn, R.S.; Poon, R.T.P.; Blanc, J.-F.; Yan, L.; Yang, J.; Lu, L.; Tak, W.-Y.; Yu, X.; et al. Brivanib as Adjuvant Therapy to Transarterial Chemoembolization in Patients with Hepatocellular Carcinoma: A Randomized Phase III Trial. Hepatology 2014, 60, 1697–1707. [Google Scholar] [CrossRef]
- Kudo, M.; Cheng, A.-L.; Park, J.-W.; Park, J.H.; Liang, P.-C.; Hidaka, H.; Izumi, N.; Heo, J.; Lee, Y.J.; Sheen, I.-S.; et al. Orantinib versus Placebo Combined with Transcatheter Arterial Chemoembolisation in Patients with Unresectable Hepatocellular Carcinoma (ORIENTAL): A Randomised, Double-Blind, Placebo-Controlled, Multicentre, Phase 3 Study. Lancet Gastroenterol. Hepatol. 2018, 3, 37–46. [Google Scholar] [CrossRef]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; ElGindi, M.; et al. Tremelimumab in Combination with Ablation in Patients with Advanced Hepatocellular Carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef] [Green Version]
- Kloeckner, R.; Galle, P.R.; Bruix, J. Local and Regional Therapies for Hepatocellular Carcinoma. Hepatology. 2021, 73 (Suppl. S1), 137–149. [Google Scholar] [CrossRef] [PubMed]
- Fulgenzi, C.A.M.; Cortellini, A.; D’Alessio, A.; Thomas, R.; Tait, P.; Ross, P.J.; Young, A.-M.; Talbot, T.; Goldin, R.; Ward, C.; et al. A Phase Ib Study of Pembrolizumab Following Trans-Arterial Chemoembolization (TACE) in Hepatocellular Carcinoma (HCC): PETAL. J. Clin. Oncol. 2022, 40, e16195. [Google Scholar] [CrossRef]
- Llovet, J.M.; Vogel, A.; Madoff, D.C.; Finn, R.S.; Ogasawara, S.; Ren, Z.; Mody, K.; Li, J.J.; Siegel, A.B.; Dubrovsky, L.; et al. Randomized Phase 3 LEAP-012 Study: Transarterial Chemoembolization With or Without Lenvatinib Plus Pembrolizumab for Intermediate-Stage Hepatocellular Carcinoma Not Amenable to Curative Treatment. Cardiovasc. Intervent. Radiol. 2022, 45, 405–412. [Google Scholar] [CrossRef]
- Peng, Z.-W.; Zhang, Y.-J.; Liang, H.-H.; Lin, X.-J.; Guo, R.-P.; Chen, M.-S. Recurrent Hepatocellular Carcinoma Treated with Sequential Transcatheter Arterial Chemoembolization and RF Ablation versus RF Ablation Alone: A Prospective Randomized Trial. Radiology 2012, 262, 689–700. [Google Scholar] [CrossRef]
- Peng, Z.-W.; Zhang, Y.-J.; Chen, M.-S.; Xu, L.; Liang, H.-H.; Lin, X.-J.; Guo, R.-P.; Zhang, Y.-Q.; Lau, W.Y. Radiofrequency Ablation with or without Transcatheter Arterial Chemoembolization in the Treatment of Hepatocellular Carcinoma: A Prospective Randomized Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 426–432. [Google Scholar] [CrossRef]
- Lee, M.S.; Ryoo, B.-Y.; Hsu, C.-H.; Numata, K.; Stein, S.; Verret, W.; Hack, S.P.; Spahn, J.; Liu, B.; Abdullah, H.; et al. Atezolizumab with or without Bevacizumab in Unresectable Hepatocellular Carcinoma (GO30140): An Open-Label, Multicentre, Phase 1b Study. Lancet Oncol. 2020, 21, 808–820. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Kudo, M. Durvalumab plus Tremelimumab in Unresectable Hepatocellular Carcinoma. Hepatobiliary Surg. Nutr. 2022, 11, 592–596. [Google Scholar] [CrossRef]
- Nawaz, K. ESMO 2022: Ten Key Takeaways on Europe’s Top Oncology Event; Clarivate: London, UK, 2022. [Google Scholar]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Qin, S.; Kudo, M.; Meyer, T.; Finn, R.S.; Vogel, A.; Bai, Y.; Guo, Y.; Meng, Z.; Zhang, T.; Satoh, T.; et al. LBA36 Final Analysis of RATIONALE-301: Randomized, Phase III Study of Tislelizumab versus Sorafenib as First-Line Treatment for Unresectable Hepatocellular Carcinoma. Ann. Oncol. 2022, 33, S1402–S1403. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in Patients with Advanced Hepatocellular Carcinoma (CheckMate 040): An Open-Label, Non-Comparative, Phase 1/2 Dose Escalation and Expansion Trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Park, J.-W.; Finn, R.S.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus Sorafenib in Advanced Hepatocellular Carcinoma (CheckMate 459): A Randomised, Multicentre, Open-Label, Phase 3 Trial. Lancet Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Yau, T.; Kang, Y.-K.; Kim, T.-Y.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Nivolumab (NIVO) plus Ipilimumab (IPI) Combination Therapy in Patients (Pts) with Advanced Hepatocellular Carcinoma (AHCC): Long-Term Results from CheckMate 040. J. Clin. Oncol. 2021, 39, 269. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib (KEYNOTE-224): A Non-Randomised, Open-Label Phase 2 Trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.H.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Updated Efficacy and Safety of KEYNOTE-224: A Phase II Study of Pembrolizumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib. Eur. J. Cancer 2022, 167, 1–12. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Qin, S.; Chen, Z.; Fang, W.; Ren, Z.; Xu, R.; Ryoo, B.-Y.; Meng, Z.; Bai, Y.; Chen, X.; Liu, X.; et al. Pembrolizumab plus Best Supportive Care versus Placebo plus Best Supportive Care as Second-Line Therapy in Patients in Asia with Advanced Hepatocellular Carcinoma (HCC): Phase 3 KEYNOTE-394 Study. J. Clin. Oncol. 2022, 40, 383. [Google Scholar] [CrossRef]
- Galle, P.R.; Dufour, J.-F.; Peck-Radosavljevic, M.; Trojan, J.; Vogel, A. Systemic Therapy of Advanced Hepatocellular Carcinoma. Future Oncol. 2021, 17, 1237–1251. [Google Scholar] [CrossRef]
- Fernández, M.; Semela, D.; Bruix, J.; Colle, I.; Pinzani, M.; Bosch, J. Angiogenesis in Liver Disease. J. Hepatol. 2009, 50, 604–620. [Google Scholar] [CrossRef]
- Villanueva, A.; Llovet, J.M. Targeted Therapies for Hepatocellular Carcinoma. Gastroenterology 2011, 140, 1410–1426. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, A.; Newell, P.; Chiang, D.Y.; Friedman, S.L.; Llovet, J.M. Genomics and Signaling Pathways in Hepatocellular Carcinoma. Semin. Liver Dis. 2007, 27, 55–76. [Google Scholar] [CrossRef]
- Cervello, M.; Bachvarov, D.; Lampiasi, N.; Cusimano, A.; Azzolina, A.; McCubrey, J.A.; Montalto, G. Molecular Mechanisms of Sorafenib Action in Liver Cancer Cells. Cell Cycle 2012, 11, 2843–2855. [Google Scholar] [CrossRef] [Green Version]
- Stotz, M.; Gerger, A.; Haybaeck, J.; Kiesslich, T.; Bullock, M.D.; Pichler, M. Molecular Targeted Therapies in Hepatocellular Carcinoma: Past, Present and Future. Anticancer Res. 2015, 35, 5737–5744. [Google Scholar]
- Méndez-Sánchez, N.; Vásquez-Fernández, F.; Zamora-Valdés, D.; Uribe, M. Sorafenib, a Systemic Therapy for Hepatocellular Carcinoma. Ann. Hepatol. 2008, 7, 46–51. [Google Scholar] [CrossRef]
- Marrero, J.A.; Kudo, M.; Venook, A.P.; Ye, S.-L.; Bronowicki, J.-P.; Chen, X.-P.; Dagher, L.; Furuse, J.; Geschwind, J.-F.H.; de Guevara, L.L.; et al. Observational Registry of Sorafenib Use in Clinical Practice across Child-Pugh Subgroups: The GIDEON Study. J. Hepatol. 2016, 65, 1140–1147. [Google Scholar] [CrossRef]
- Bruix, J.; Takayama, T.; Mazzaferro, V.; Chau, G.-Y.; Yang, J.; Kudo, M.; Cai, J.; Poon, R.T.; Han, K.-H.; Tak, W.Y.; et al. Adjuvant Sorafenib for Hepatocellular Carcinoma after Resection or Ablation (STORM): A Phase 3, Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Oncol. 2015, 16, 1344–1354. [Google Scholar] [CrossRef]
- Tohyama, O.; Matsui, J.; Kodama, K.; Hata-Sugi, N.; Kimura, T.; Okamoto, K.; Minoshima, Y.; Iwata, M.; Funahashi, Y. Antitumor Activity of Lenvatinib (E7080): An Angiogenesis Inhibitor That Targets Multiple Receptor Tyrosine Kinases in Preclinical Human Thyroid Cancer Models. J. Thyroid Res. 2014, 2014, 638747. [Google Scholar] [CrossRef]
- Matsuki, M.; Hoshi, T.; Yamamoto, Y.; Ikemori-Kawada, M.; Minoshima, Y.; Funahashi, Y.; Matsui, J. Lenvatinib Inhibits Angiogenesis and Tumor Fibroblast Growth Factor Signaling Pathways in Human Hepatocellular Carcinoma Models. Cancer Med. 2018, 7, 2641–2653. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus Sorafenib in First-Line Treatment of Patients with Unresectable Hepatocellular Carcinoma: A Randomised Phase 3 Non-Inferiority Trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Rimini, M.; Rimassa, L.; Ueshima, K.; Burgio, V.; Shigeo, S.; Tada, T.; Suda, G.; Yoo, C.; Cheon, J.; Pinato, D.J.; et al. Atezolizumab plus Bevacizumab versus Lenvatinib or Sorafenib in Non-Viral Unresectable Hepatocellular Carcinoma: An International Propensity Score Matching Analysis. ESMO Open 2022, 7, 100591. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH Limits Anti-Tumour Surveillance in Immunotherapy-Treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Rimassa, L.; Cheng, A.-L.; Kaseb, A.; Qin, S.; Zhu, A.X.; Chan, S.L.; Melkadze, T.; Sukeepaisarnjaroen, W.; Breder, V.; et al. Cabozantinib plus Atezolizumab versus Sorafenib for Advanced Hepatocellular Carcinoma (COSMIC-312): A Multicentre, Open-Label, Randomised, Phase 3 Trial. Lancet Oncol. 2022, 23, 995–1008. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Kang, Y.-K.; Yen, C.-J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after Sorafenib in Patients with Advanced Hepatocellular Carcinoma and Increased α-Fetoprotein Concentrations (REACH-2): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Flecken, T.; Schmidt, N.; Hild, S.; Gostick, E.; Drognitz, O.; Zeiser, R.; Schemmer, P.; Bruns, H.; Eiermann, T.; Price, D.A.; et al. Immunodominance and Functional Alterations of Tumor-Associated Antigen-Specific CD8+ T-Cell Responses in Hepatocellular Carcinoma. Hepatology 2014, 59, 1415–1426. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Johnson, B.A.; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting Neoantigens to Augment Antitumour Immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef]
- He, X.; Xu, C. Immune Checkpoint Signaling and Cancer Immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- Crispe, I.N. The Liver as a Lymphoid Organ. Annu. Rev. Immunol. 2009, 27, 147–163. [Google Scholar] [CrossRef]
- Krenkel, O.; Tacke, F. Liver Macrophages in Tissue Homeostasis and Disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Dunham, R.M.; Thapa, M.; Velazquez, V.M.; Elrod, E.J.; Denning, T.L.; Pulendran, B.; Grakoui, A. Hepatic Stellate Cells Preferentially Induce Foxp3+ Regulatory T Cells by Production of Retinoic Acid. J. Immunol. 2013, 190, 2009–2016. [Google Scholar] [CrossRef] [Green Version]
- Bruix, J.; Chan, S.L.; Galle, P.R.; Rimassa, L.; Sangro, B. Systemic Treatment of Hepatocellular Carcinoma: An EASL Position Paper. J. Hepatol. 2021, 75, 960–974. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Han, K.-H.; Harding, J.J.; Merle, P.; et al. LBA38_PR—CheckMate 459: A Randomized, Multi-Center Phase III Study of Nivolumab (NIVO) vs Sorafenib (SOR) as First-Line (1L) Treatment in Patients (Pts) with Advanced Hepatocellular Carcinoma (AHCC). Ann. Oncol. 2019, 30, v874–v875. [Google Scholar] [CrossRef]
- Cabibbo, G.; Reig, M.; Celsa, C.; Torres, F.; Battaglia, S.; Enea, M.; Rizzo, G.E.M.; Petta, S.; Calvaruso, V.; Marco, V.D.; et al. First-Line Immune Checkpoint Inhibitor-Based Sequential Therapies for Advanced Hepatocellular Carcinoma: Rationale for Future Trials. Liver Cancer 2022, 11, 75–84. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive Cell Transfer as Personalized Immunotherapy for Human Cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Sangro, B.; Sarobe, P.; Hervás-Stubbs, S.; Melero, I. Advances in Immunotherapy for Hepatocellular Carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 525–543. [Google Scholar] [CrossRef]
- Gao, H.; Li, K.; Tu, H.; Pan, X.; Jiang, H.; Shi, B.; Kong, J.; Wang, H.; Yang, S.; Gu, J.; et al. Development of T Cells Redirected to Glypican-3 for the Treatment of Hepatocellular Carcinoma. Clin. Cancer Res. 2014, 20, 6418–6428. [Google Scholar] [CrossRef]
- Sun, B. Clinical Study of GPC3-Targeted Chimeric Antigen Receptor T Cells Fo Treating Advanced Hepatocellular Carcinoma; ClinicalTrials: Bethesda, MD, USA, 2021. [Google Scholar]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards Personalized, Tumour-Specific, Therapeutic Vaccines for Cancer. Nat. Rev. Immunol. 2018, 18, 168–182. [Google Scholar] [CrossRef]
- Tagliamonte, M.; Petrizzo, A.; Mauriello, A.; Tornesello, M.L.; Buonaguro, F.M.; Buonaguro, L. Potentiating Cancer Vaccine Efficacy in Liver Cancer. OncoImmunology 2018, 7, e1488564. [Google Scholar] [CrossRef]
- Rammensee, H.-G.; Singh-Jasuja, H. HLA Ligandome Tumor Antigen Discovery for Personalized Vaccine Approach. Expert Rev. Vaccines 2013, 12, 1211–1217. [Google Scholar] [CrossRef] [Green Version]
- Buonaguro, L. HEPAVAC Consortium Developments in Cancer Vaccines for Hepatocellular Carcinoma. Cancer Immunol. Immunother. 2016, 65, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic Cell Death in Cancer and Infectious Disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Hoffmann, J.A.; Galluzzi, L. Pharmacological Modulation of Nucleic Acid Sensors—Therapeutic Potential and Persisting Obstacles. Nat. Rev. Drug Discov. 2019, 18, 845–867. [Google Scholar] [CrossRef] [PubMed]
- Tenneti, P.; Borad, M.J.; Babiker, H.M. Exploring the Role of Oncolytic Viruses in Hepatobiliary Cancers. Immunotherapy 2018, 10, 971–986. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Gato, M.; Shekarian, T.; Aznar, A.; Valsesia-Wittmann, S.; Caux, C.; Etxeberrria, I.; Teijeira, A.; Marabelle, A. Repurposing Infectious Disease Vaccines for Intratumoral Immunotherapy. J. Immunother. Cancer 2020, 8, e000443. [Google Scholar] [CrossRef]
- Vacante, F.; Senesi, P.; Montesano, A.; Paini, S.; Luzi, L.; Terruzzi, I. Metformin Counteracts HCC Progression and Metastasis Enhancing KLF6/P21 Expression and Downregulating the IGF Axis. Int. J. Endocrinol. 2019, 2019, 7570146. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Lin, X.; Yan, Q.; Gu, C.; Li, M.; Pan, L.; Meng, Y.; Zhao, X.; Liu, S.; Li, A. PBLD Inhibits Angiogenesis via Impeding VEGF/VEGFR2-Mediated Microenvironmental Cross-Talk between HCC Cells and Endothelial Cells. Oncogene 2022, 41, 1851–1865. [Google Scholar] [CrossRef]
- Su, Q.; Fan, M.; Wang, J.; Ullah, A.; Ghauri, M.A.; Dai, B.; Zhan, Y.; Zhang, D.; Zhang, Y. Sanguinarine Inhibits Epithelial–Mesenchymal Transition via Targeting HIF-1α/TGF-β Feed-Forward Loop in Hepatocellular Carcinoma. Cell Death Dis. 2019, 10, 939. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Xu, W.; Xu, H.; Zheng, J.; Gu, Y. Overexpression of HDAC6 Suppresses Tumor Cell Proliferation and Metastasis by Inhibition of the Canonical Wnt/Β-catenin Signaling Pathway in Hepatocellular Carcinoma. Oncol. Lett. 2018, 16, 7082–7090. [Google Scholar] [CrossRef] [Green Version]
- Streubel, G.; Schrepfer, S.; Kallus, H.; Parnitzke, U.; Wulff, T.; Hermann, F.; Borgmann, M.; Hamm, S. Histone Deacetylase Inhibitor Resminostat in Combination with Sorafenib Counteracts Platelet-Mediated pro-Tumoral Effects in Hepatocellular Carcinoma. Sci. Rep. 2021, 11, 9587. [Google Scholar] [CrossRef]
- Garbo, S.; Tripodi, M.; Battistelli, C. A Novel RNA-Based Approach to Counteract EMT. Oncoscience 2021, 8, 53–54. [Google Scholar] [CrossRef]
- Kimura, T.; Aikata, H.; Takahashi, S.; Takahashi, I.; Nishibuchi, I.; Doi, Y.; Kenjo, M.; Murakami, Y.; Honda, Y.; Kakizawa, H.; et al. Stereotactic Body Radiotherapy for Patients with Small Hepatocellular Carcinoma Ineligible for Resection or Ablation Therapies. Hepatol. Res. 2015, 45, 378–386. [Google Scholar] [CrossRef]
- Takahashi, H.; Carninci, P. Widespread Genome Transcription: New Possibilities for RNA Therapies. Biochem. Biophys. Res. Commun. 2014, 452, 294–301. [Google Scholar] [CrossRef] [Green Version]
- Parasramka, M.A.; Maji, S.; Matsuda, A.; Yan, I.K.; Patel, T. Long Non-Coding RNAs as Novel Targets for Therapy in Hepatocellular Carcinoma. Pharmacol. Ther. 2016, 161, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Battistelli, C.; Cicchini, C.; Santangelo, L.; Tramontano, A.; Grassi, L.; Gonzalez, F.J.; de Nonno, V.; Grassi, G.; Amicone, L.; Tripodi, M. The Snail Repressor Recruits EZH2 to Specific Genomic Sites through the Enrollment of the LncRNA HOTAIR in Epithelial-to-Mesenchymal Transition. Oncogene 2017, 36, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Cirillo, D.; Blanco, M.; Armaos, A.; Buness, A.; Avner, P.; Guttman, M.; Cerase, A.; Tartaglia, G.G. Quantitative Predictions of Protein Interactions with Long Noncoding RNAs. Nat. Methods 2017, 14, 5–6. [Google Scholar] [CrossRef]
- Battistelli, C.; Garbo, S.; Riccioni, V.; Montaldo, C.; Santangelo, L.; Vandelli, A.; Strippoli, R.; Tartaglia, G.G.; Tripodi, M.; Cicchini, C. Design and Functional Validation of a Mutant Variant of the LncRNA HOTAIR to Counteract Snail Function in Epithelial-to-Mesenchymal Transition. Cancer Res. 2021, 81, 103–113. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tümen, D.; Heumann, P.; Gülow, K.; Demirci, C.-N.; Cosma, L.-S.; Müller, M.; Kandulski, A. Pathogenesis and Current Treatment Strategies of Hepatocellular Carcinoma. Biomedicines 2022, 10, 3202. https://doi.org/10.3390/biomedicines10123202
Tümen D, Heumann P, Gülow K, Demirci C-N, Cosma L-S, Müller M, Kandulski A. Pathogenesis and Current Treatment Strategies of Hepatocellular Carcinoma. Biomedicines. 2022; 10(12):3202. https://doi.org/10.3390/biomedicines10123202
Chicago/Turabian StyleTümen, Deniz, Philipp Heumann, Karsten Gülow, Cagla-Nur Demirci, Lidia-Sabina Cosma, Martina Müller, and Arne Kandulski. 2022. "Pathogenesis and Current Treatment Strategies of Hepatocellular Carcinoma" Biomedicines 10, no. 12: 3202. https://doi.org/10.3390/biomedicines10123202
APA StyleTümen, D., Heumann, P., Gülow, K., Demirci, C. -N., Cosma, L. -S., Müller, M., & Kandulski, A. (2022). Pathogenesis and Current Treatment Strategies of Hepatocellular Carcinoma. Biomedicines, 10(12), 3202. https://doi.org/10.3390/biomedicines10123202