

Oncolytic Adenovirus, a New Treatment Strategy for Prostate Cancer

Abstract
:1. Introduction
2. Characteristics of Oncolytic Adenoviruses
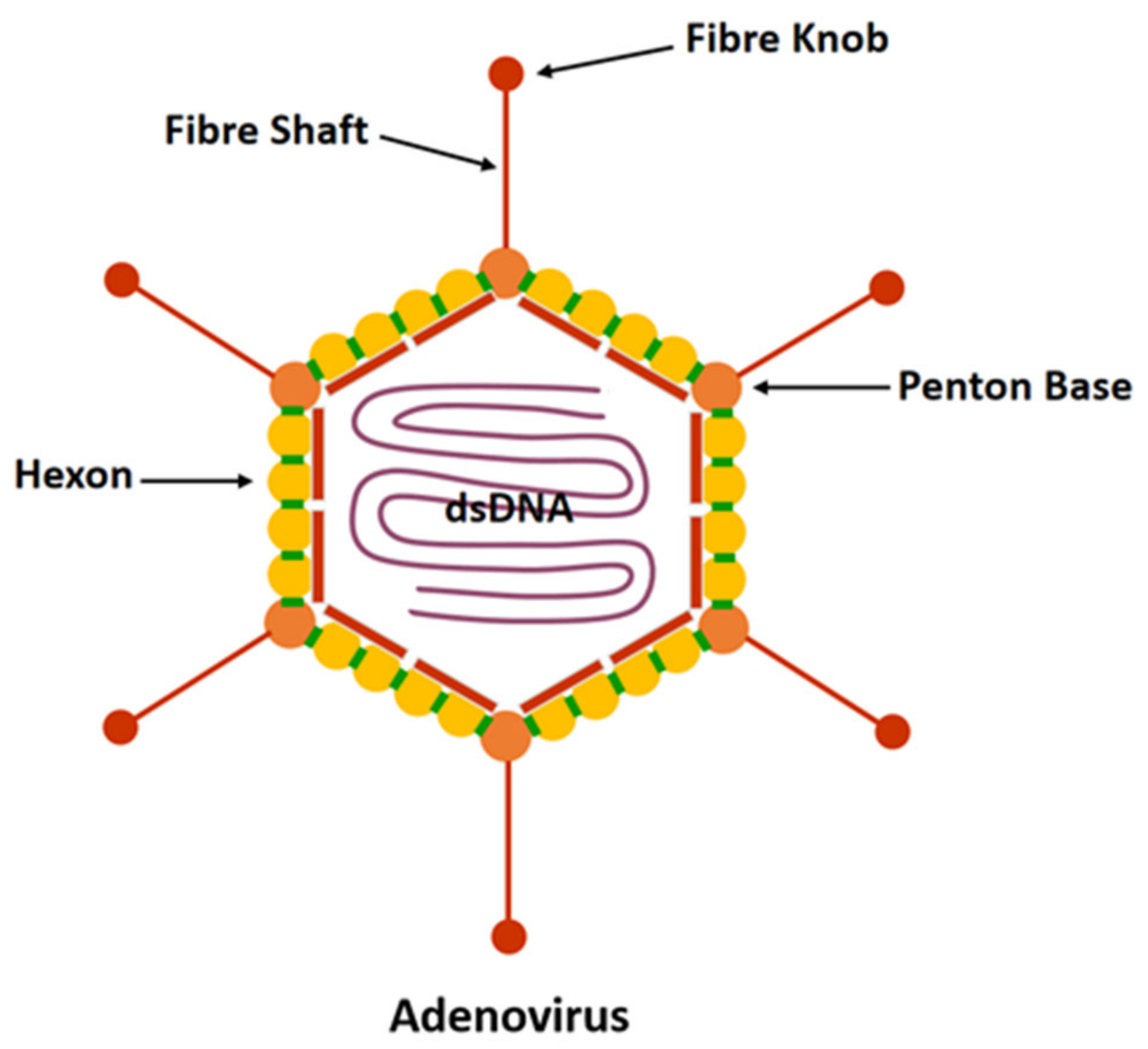
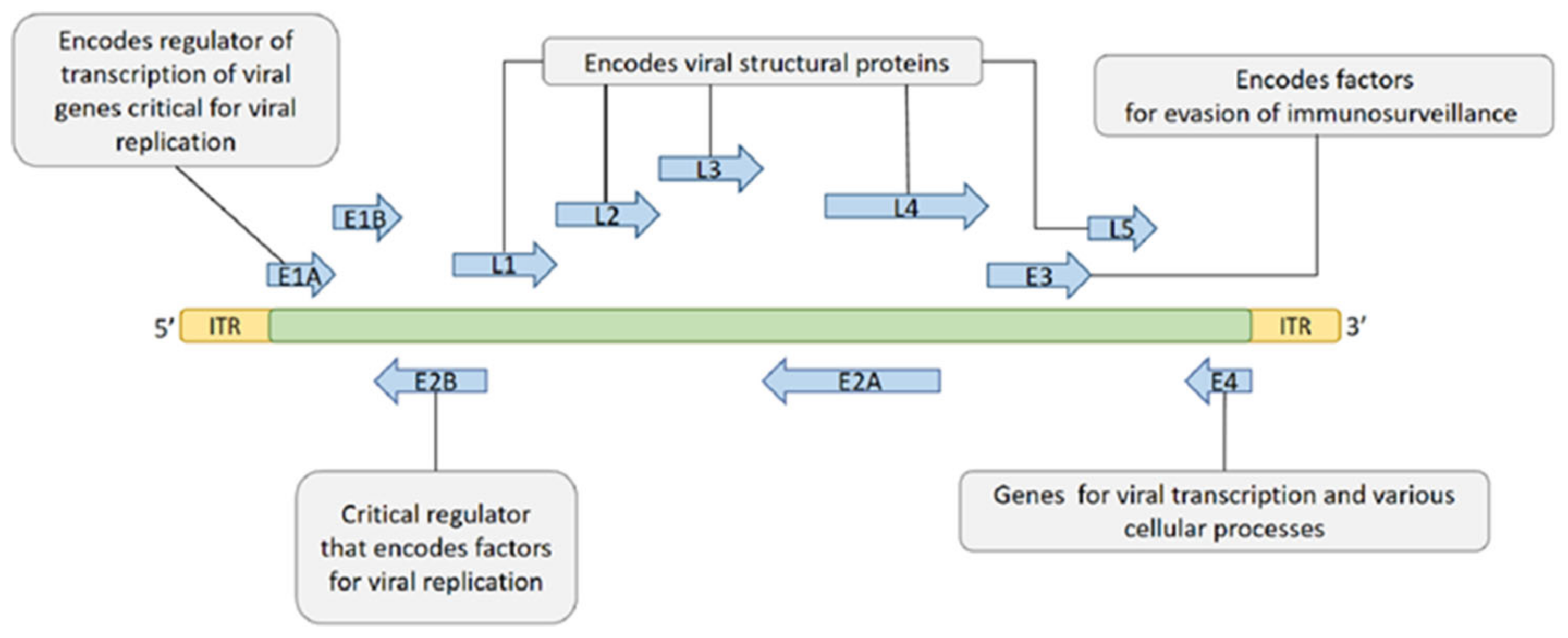
2.1. Structure, Proteins, Genome, and Classification of HAdV
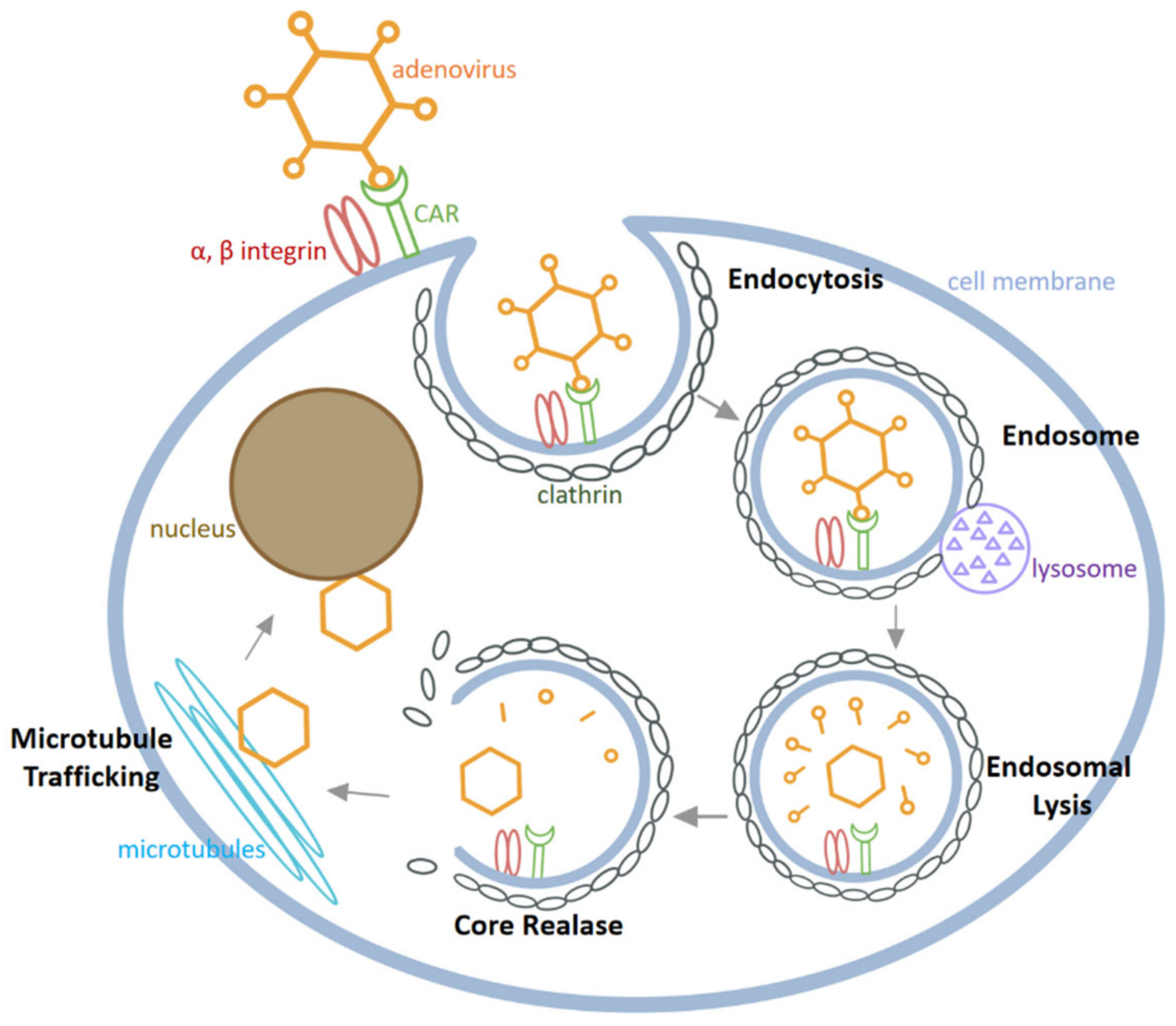
2.2. HAdV Infection
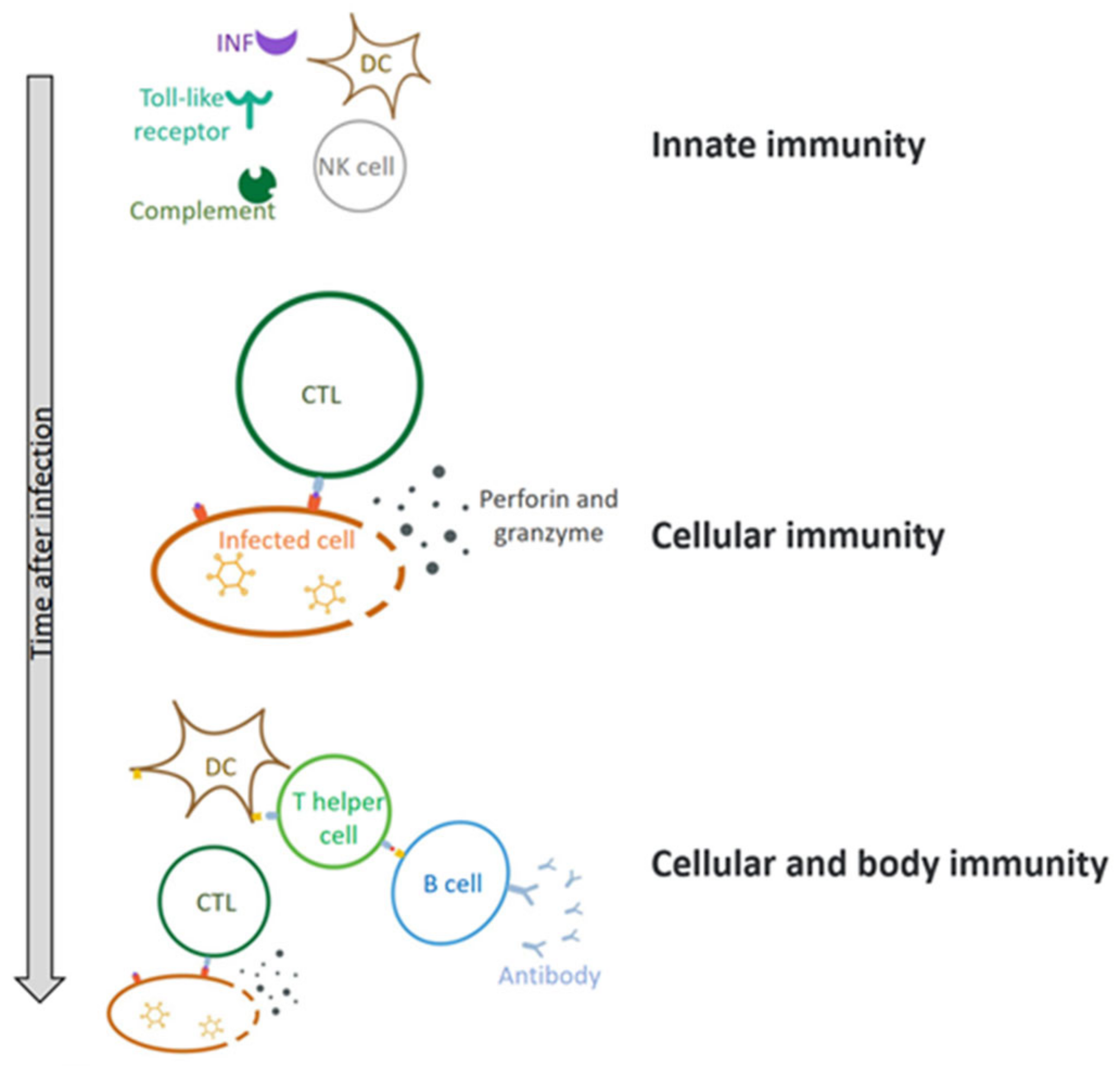
2.3. Adenovirus Causes an Immune Response When It Enters Cells
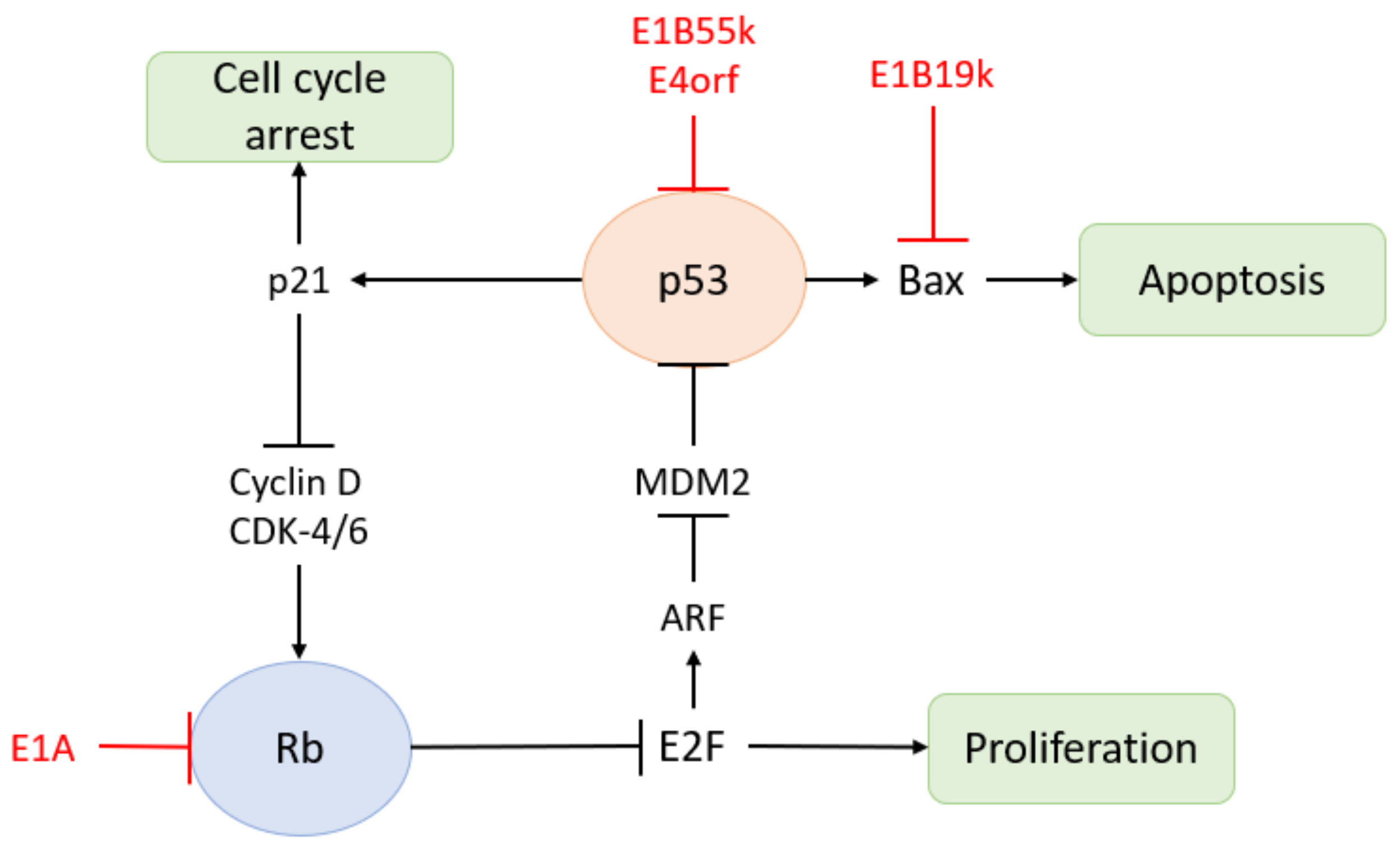
2.4. Immune Escape of HAdV
2.5. Advantages of Adenovirus as Oncolytic Virus
2.6. Genetic Modifications of Oncolytic Adenovirus
2.6.1. Modification Based on Targeting Mechanisms
- (1)
- Genetic modifications for specific infections
- (2)
- Genetic modifications for selective replication
2.6.2. Genetic Modifications with Anti-Tumour Molecules
2.6.3. Using Carriers
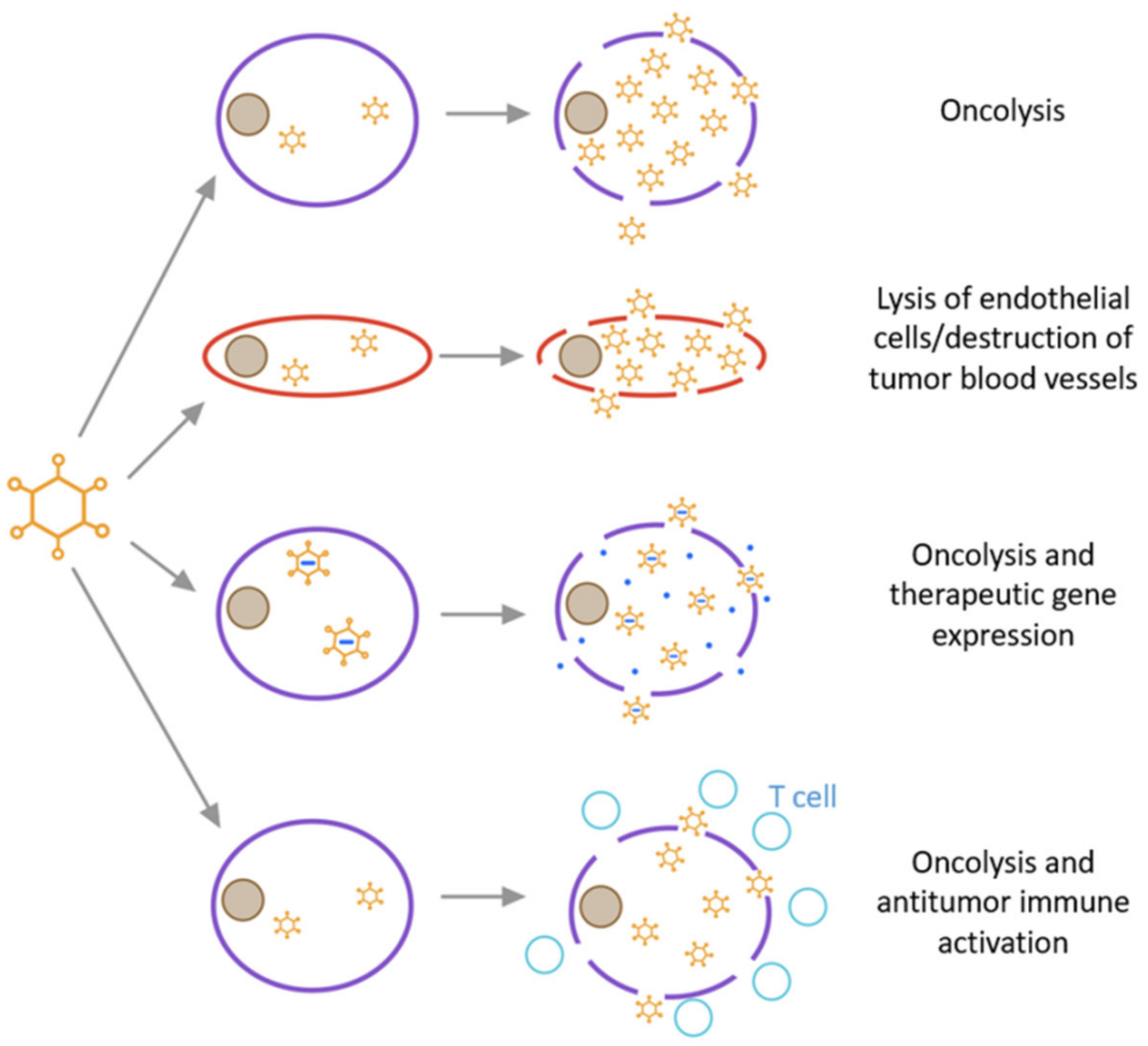
2.7. The Mechanism Underlying the Action of Oncolytic Adenovirus
2.7.1. Virus-Mediated Tumour-Killing Mechanism
2.7.2. The Anti-Tumour Immune Response Mechanism
3. Oncolytic Adenovirus in Clinical Trials
3.1. CV706 (CN706)
3.2. CG7870 (CV787)
3.3. Ad5-CD/TKrep (FGR)
3.4. Ad5-yCD/mutTKSR39rep-ADP
4. Existing Limitations and Future Directions
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Russell, S.J.; Peng, K.-W. Viruses as anticancer drugs. Trends Pharmacol. Sci. 2007, 28, 326–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galanis, E.; Atherton, P.J.; Maurer, M.J.; Knutson, K.L.; Dowdy, S.C.; Cliby, W.A.; Haluska, P.; Long, H.J.; Oberg, A.; Aderca, I.; et al. Oncolytic Measles Virus Expressing the Sodium Iodide Symporter to Treat Drug-Resistant Ovarian Cancer. Cancer Res. 2015, 75, 22–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, J.C.; McFadden, G. Editorial overview: Oncolytic viruses—Replicating virus therapeutics for the treatment of cancer. Curr. Opin. Virol. 2015, 13, viii–ix. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, M.; Sun, F.; Zhang, J.; Liu, F. Enhancing the immune effect of oHSV-1 therapy through TLR3 signaling in uveal melanoma. J. Cancer Res. Clin. Oncol. 2022. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Obaid, Q.A.; Al-Shammari, A.M.; Khudair, K.K. Glucose Deprivation Induced by Acarbose and Oncolytic Newcastle Disease Virus Promote Metabolic Oxidative Stress and Cell Death in a Breast Cancer Model. Front. Mol. Biosci. 2022, 9, 816510. [Google Scholar] [CrossRef]
- Singh, H.M.; Leber, M.F.; Bossow, S.; Engeland, C.E.; Dessila, J.; Grossardt, C.; Zaoui, K.; Bell, J.C.; Jäger, D.; von Kalle, C.; et al. MicroRNA-sensitive oncolytic measles virus for chemovirotherapy of pancreatic cancer. Mol. Ther. Oncolytics 2021, 21, 340–355. [Google Scholar] [CrossRef]
- Ammour, Y.I.; Ryabaya, O.O.; Milovanova, A.V.; Sidorov, A.V.; Shohin, I.E.; Zverev, V.V.; Nasedkina, T.V. Oncolytic Properties of a Mumps Virus Vaccine Strain in Human Melanoma Cell Lines. Mol. Biol. 2018, 52, 570–576. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, Z.; Zhang, C.; Zhang, N.; Wang, P.; Chu, Y.; Dunmall, L.S.C.; Lemoine, N.R.; Wang, Y. An effective therapeutic regime for treatment of glioma using oncolytic vaccinia virus expressing IL-21 in combination with immune checkpoint inhibition. Mol. Ther.-Oncolytics 2022, 26, 105–119. [Google Scholar] [CrossRef]
- Zhang, Y.; Nagalo, B.M. Immunovirotherapy Based on Recombinant Vesicular Stomatitis Virus: Where Are We? Front. Immunol. 2022, 13, 898631. [Google Scholar] [CrossRef]
- Garber, K. China approves world’s first oncolytic virus therapy for cancer treatment. J. Natl. Cancer Inst. 2006, 98, 298–300. [Google Scholar] [CrossRef]
- Cheng, G.; Dong, H.; Yang, C.; Liu, Y.; Wu, Y.; Zhu, L.; Tong, X.; Wang, S. A review on the advances and challenges of immunotherapy for head and neck cancer. Cancer Cell Int. 2021, 21, 406. [Google Scholar] [CrossRef]
- Coffin, R. Interview with Robert Coffin, inventor of T-VEC: The first oncolytic immunotherapy approved for the treatment of cancer. Immunotherapy 2016, 8, 103–106. [Google Scholar] [CrossRef] [Green Version]
- Mao, L.J.; Kan, Y.; Li, B.H.; Ma, S.; Liu, Y.; Yang, D.L.; Yang, C. Combination Therapy of Prostate Cancer by Oncolytic Adenovirus Harboring Interleukin 24 and Ionizing Radiation. Front. Oncol. 2020, 10, 421. [Google Scholar] [CrossRef]
- Deweese, T.L.; Van Der Poel, H.; Li, S.; Mikhak, B.; Drew, R.; Goemann, M.; Hamper, U.; DeJong, R.; Detorie, N.; Rodriguez, R.; et al. A phase I trial of CV706, a replication-competent, PSA selective oncolytic adenovirus, for the treatment of locally recurrent prostate cancer following radiation therapy. Cancer Res. 2001, 61, 7464–7472. [Google Scholar]
- Small, E.J.; Carducci, M.A.; Burke, J.M.; Rodriguez, R.; Fong, L.; van Ummersen, L.; Yu, D.C.; Aimi, J.; Ando, D.; Working, P.; et al. A phase I trial of intravenous CG7870, a replication-selective, prostate-specific antigen-targeted oncolytic adenovirus, for the treatment of hormone-refractory, metastatic prostate cancer. Mol. Ther. 2006, 14, 107–117. [Google Scholar] [CrossRef]
- Hallden, G.; Sweeney, K. Oncolytic adenovirus-mediated therapy for prostate cancer. Oncolytic Virother. 2016, 5, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Freytag, S.O.; Rogulski, K.R.; Paielli, D.L.; Gilbert, J.D.; Kim, J.H. A novel three-pronged approach to kill cancer cells selectively: Concomitant viral, double suicide gene, and radiotherapy. Hum. Gene Ther. 1998, 9, 1323–1333. [Google Scholar] [CrossRef]
- Freytag, S.O.; Khil, M.; Stricker, H.; Peabody, J.; Menon, M.; DePeralta-Venturina, M.; Nafziger, D.; Pegg, J.; Paielli, D.; Brown, S.; et al. Phase I study of replication-competent adenovirus-mediated double suicide gene therapy for the treatment of locally recurrent prostate cancer. Cancer Res. 2002, 62, 4968–4976. [Google Scholar]
- Freytag, S.O.; Stricker, H.; Peabody, J.; Pegg, J.; Paielli, D.; Movsas, B.; Barton, K.N.; Brown, S.L.; Lu, M.; Kim, J.H. Five-year follow-up of trial of replication-competent adenovirus-mediated suicide gene therapy for treatment of prostate cancer. Mol. Ther. 2007, 15, 636–642. [Google Scholar] [CrossRef]
- Freytag, S.O.; Stricker, H.; Pegg, J.; Paielli, D.; Pradhan, D.G.; Peabody, J.; DePeralta-Venturina, M.; Xia, X.; Brown, S.; Lu, M.; et al. Phase I study of replication-competent adenovirus-mediated double-suicide gene therapy in combination with conventional-dose three-dimensional conformal radiation therapy for the treatment of newly diagnosed, intermediate- to high-risk prostate cancer. Cancer Res. 2003, 63, 7497–7506. [Google Scholar]
- Freytag, S.O.; Movsas, B.; Aref, I.; Stricker, H.; Peabody, J.; Pegg, J.; Zhang, Y.; Barton, K.N.; Brown, S.L.; Lu, M.; et al. Phase I Trial of Replication-competent Adenovirus-mediated Suicide Gene Therapy Combined with IMRT for Prostate Cancer. Mol. Ther. 2007, 15, 1016–1023. [Google Scholar] [CrossRef]
- Freytag, S.O.; Stricker, H.; Lu, M.; Elshaikh, M.; Aref, I.; Pradhan, D.; Levin, K.; Kim, J.H.; Peabody, J.; Siddiqui, F.; et al. Prospective Randomized Phase 2 Trial of Intensity Modulated Radiation Therapy With or Without Oncolytic Adenovirus-Mediated Cytotoxic Gene Therapy in Intermediate-Risk Prostate Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2014, 89, 268–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djavan, B.; Moul, J.W.; Zlotta, A.; Remzi, M.; Ravery, V. PSA progression following radical prostatectomy and radiation therapy: New standards in the new Millennium. Eur Urol. 2003, 43, 12–27. [Google Scholar] [CrossRef]
- Nandana, S.; Chung, L.W. Prostate cancer progression and metastasis: Potential regulatory pathways for therapeutic targeting. Am. J. Clin. Exp. Urol. 2014, 2, 92–101. [Google Scholar]
- Lorente, D.; Mateo, J.; Perez-Lopez, R.; de Bono, J.S.; Attard, G. Sequencing of agents in castration-resistant prostate cancer. Lancet Oncol. 2015, 16, e279–e292. [Google Scholar] [CrossRef]
- Harris, W.P.; Mostaghel, E.A.; Nelson, P.S.; Montgomery, B. Androgen deprivation therapy: Progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol. 2009, 6, 76–85. [Google Scholar] [CrossRef]
- Akaza, H.; Procopio, G.; Pripatnanont, C.; Facchini, G.; Fava, S.; Wheatley, D.; Leung, K.C.; Butt, M.; Silva, A.; Castillo, L.; et al. Metastatic Castration-Resistant Prostate Cancer Previously Treated With Docetaxel-Based Chemotherapy: Treatment Patterns From the PROXIMA Prospective Registry. J. Glob. Oncol. 2018, 4, 1–12. [Google Scholar] [CrossRef]
- Cannata, D.H.; Kirschenbaum, A.; Levine, A.C. Androgen Deprivation Therapy as Primary Treatment for Prostate Cancer. J. Clin. Endocrinol. Metab. 2012, 97, 360–365. [Google Scholar] [CrossRef] [Green Version]
- Van der Zande, K.; Oyen, W.J.G.; Zwart, W.; Bergman, A.M. Radium-223 Treatment of Patients with Metastatic Castration Resistant Prostate Cancer: Biomarkers for Stratification and Response Evaluation. Cancers 2021, 13, 4346. [Google Scholar] [CrossRef]
- Fallah, J.; Agrawal, S.; Gittleman, H.; Fiero, M.H.; Subramaniam, S.; John, C.; Chen, W.; Ricks, T.K.; Niu, G.; Fotenos, A.; et al. FDA Approval Summary: Lutetium Lu 177 vipivotide tetraxetan for patients with metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2022. online ahead of print. [Google Scholar] [CrossRef]
- De Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B., Jr.; Saad, F.; et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.-E.; Sternberg, C.N.; Miller, K.; De Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Ekblad, M.; Halldén, G. Adenovirus-based therapy for prostate cancer. Curr. Opin. Mol. Ther. 2010, 12, 421–431. [Google Scholar]
- Rux, J.J.; Burnett, R.M. Adenovirus structure. Hum. Gene Ther. 2004, 15, 1167–1176. [Google Scholar] [CrossRef]
- Stasiak, A.C.; Stehle, T. Human adenovirus binding to host cell receptors: A structural view. Med. Microbiol. Immunol. 2020, 209, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Eager, R.M.; Nemunaitis, J. Clinical development directions in oncolytic viral therapy. Cancer Gene Ther. 2011, 18, 305–317. [Google Scholar] [CrossRef] [Green Version]
- Arnberg, N. Adenovirus receptors: Implications for targeting of viral vectors. Trends Pharmacol. Sci. 2012, 33, 442–448. [Google Scholar] [CrossRef]
- Gao, J.; Zhang, W.; Ehrhardt, A. Expanding the Spectrum of Adenoviral Vectors for Cancer Therapy. Cancers 2020, 12, 1139. [Google Scholar] [CrossRef]
- Diana, G.; Hearing, P. Adenovirus replication. In Adenoviral Vectors for Gene Therapy; Academic Press: Cambridge, MA, USA, 2016; pp. 59–84. [Google Scholar]
- Afkhami, S.; Yao, Y.; Xing, Z. Methods and clinical development of adenovirus-vectored vaccines against mucosal pathogens. Mol. Ther. -Methods Clin. Dev. 2016, 3, 16030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waye, M.M.Y.; Sing, C.W. Anti-viral drugs for human adenoviruses. Pharmaceuticals 2010, 3, 3343–3354. [Google Scholar] [CrossRef]
- Bil-Lula, I.; Ussowicz, M.; Woniak, M. Adenoviral Infection—Common Complication Following Hematopoietic Stem Cell Transplantation. In New Advances in Stem Cell Transplantation; IntechOpen: London, UK, 2012; pp. 533–556. [Google Scholar]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009, 388, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Capasso, C.; Garofalo, M.; Hirvinen, M.; Cerullo, V. The Evolution of Adenoviral Vectors through Genetic and Chemical Surface Modifications. Viruses 2014, 6, 832–855. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Upton, J.W.; Mocarski, E.S. Viral modulation of programmed necrosis. Curr. Opin. Virol. 2013, 3, 296–306. [Google Scholar] [CrossRef] [Green Version]
- Burgert, H.G.; Ruzsics, Z.; Obermeier, S.; Hilgendorf, A.; Windheim, M.; Elsing, A. Subversion of host defense mechanisms by adenoviruses. Curr. Top. Microbiol. Immunol. 2002, 269, 273–318. [Google Scholar]
- Cook, J.L.; Walker, T.A.; Worthen, G.S.; Radke, J.R. Role of the E1A Rb-binding domain in repression of the NF-kappa B-dependent defense against tumor necrosis factor-alpha. Proc. Natl. Acad. Sci. USA 2002, 99, 9966–9971. [Google Scholar] [CrossRef] [Green Version]
- Berhane, S.; Aresté, C.; Ablack, J.N.; Ryan, G.B.; Blackbourn, D.J.; Mymryk, J.S.; Turnell, A.S.; Steele, J.C.; Grand, R.J. Adenovirus E1A interacts directly with, and regulates the level of expression of, the immunoproteasome component MECL1. Virology 2011, 421, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Ries, S.; Korn, W.M. ONYX-015: Mechanisms of action and clinical potential of a replication-selective adenovirus. Br. J. Cancer 2002, 86, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Ferreon, A.C.M.; Ferreon, J.C.; Wright, P.E.; Deniz, A.A. Modulation of allostery by protein intrinsic disorder. Nature 2013, 498, 390–394. [Google Scholar] [CrossRef] [Green Version]
- Schreiner, S.; Bürck, C.; Glass, M.; Groitl, P.; Wimmer, P.; Kinkley, S.; Mund, A.; Everett, R.D.; Dobner, T. Control of human adenovirus type 5 gene expression by cellular Daxx/ATRX chromatin-associated complexes. Nucleic Acids Res. 2013, 41, 3532–3550. [Google Scholar] [CrossRef] [Green Version]
- Flint, S.J.; Gonzalez, R.A. Regulation of mRNA Production by the Adenoviral E1B 55-kDa and E4 Orf6 Proteins. Curr. Top. Microbiol. Immunol. 2003, 272, 287–330. [Google Scholar]
- Carson, C.T.; Orazio, N.I.; Lee, D.V.; Suh, J.; Bekker-Jensen, S.; Araujo, F.D.; Lakdawala, S.S.; Lilley, C.E.; Bartek, J.; Lukas, J.; et al. Mislocalization of the MRN complex prevents ATR signaling during adenovirus infection. EMBO J. 2009, 28, 652–662. [Google Scholar] [CrossRef] [Green Version]
- McSharry, B.P.; Burgert, H.G.; Owen, D.P.; Stanton, R.J.; Prod’homme, V.; Sester, M.; Koebernick, K.; Groh, V.; Spies, T.; Cox, S.; et al. Adenovirus E3/19K promotes evasion of NK cell recognition by intracellular sequestration of the NKG2D ligands major histocompatibility complex class I chain-related proteins A and B. J. Virol. 2008, 82, 4585–4594. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.; Gawanbacht, A.; Habjan, M.; Rang, A.; Borner, C.; Schmidt, A.M.; Veitinger, S.; Jacob, R.; Devignot, S.; Kochs, G.; et al. Incoming RNA Virus Nucleocapsids Containing a 5′-Triphosphorylated Genome Activate RIG-I and Antiviral Signaling. Cell Host Microbe 2013, 13, 336–346. [Google Scholar] [CrossRef] [Green Version]
- McNeish, I.; Bell, S.J.; Lemoine, N.R. Gene Therapy Progress and Prospects: Cancer gene therapy using tumour suppressor genes. Gene Ther. 2004, 11, 497–503. [Google Scholar] [CrossRef]
- Sharma, A.; Tandon, M.; Bangari, D.S.; Mittal, S.K. Adenoviral vector-based strategies for cancer therapy. Curr. Drug Ther. 2009, 4, 117–138. [Google Scholar] [CrossRef] [Green Version]
- Kanerva, A.; Hemminki, A. Adenoviruses for treatment of cancer. Ann. Med. 2005, 37, 33–43. [Google Scholar] [CrossRef]
- Yamamoto, M.; Curiel, D.T. Current Issues and Future Directions of Oncolytic Adenoviruses. Mol. Ther. 2010, 18, 243–250. [Google Scholar] [CrossRef]
- Choi, J.-W.; Lee, J.-S.; Kim, S.W.; Yun, C.-O. Evolution of oncolytic adenovirus for cancer treatment. Adv. Drug Deliv. Rev. 2012, 64, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Ji, W.; Hu, H.; Ma, J.; Li, X.; Mei, W.; Xu, Y.; Hu, H.-Z.; Yan, Y.; Song, Q.; et al. Survivin promoter-regulated oncolytic adenovirus with Hsp70 gene exerts effective antitumor efficacy in gastric cancer immunotherapy. Oncotarget 2013, 5, 150–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, S.; Xu, J.; Wu, Y.; Ding, C. Targeting autophagy to enhance oncolytic virus-based cancer therapy. Expert Opin. Biol. Ther. 2013, 13, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pong, R.C.; Bergelson, J.M.; Hall, M.C.; Sagalowsky, A.I.; Tseng, C.P.; Wang, Z.; Hsieh, J.T. Loss of adenoviral receptor expression in human bladder cancer cells: A potential impact on the efficacy of gene therapy. Cancer Res. 1999, 59, 325–330. [Google Scholar]
- Sounni, N.E.; Noel, A. Targeting the Tumor Microenvironment for Cancer Therapy. Clin. Chem. 2013, 59, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Shashkova, E.V.; May, S.M.; Doronin, K.; Barry, M.A. Expanded anticancer therapeutic window of hexon-modified oncolytic adenovirus. Mol. Ther. 2009, 17, 2121–2130. [Google Scholar] [CrossRef]
- Van der Poel, H.G.; Molenaar, B.; van Beusechem, V.W.; Haisma, H.J.; Rodriguez, R.; Curiel, D.T.; Gerritsen, W.R. Epidermal growth factor receptor targeting of replication competent adenovirus enhances cytotoxicity in bladder cancer. J. Urol. 2002, 168, 266–272. [Google Scholar] [CrossRef]
- Cripe, T.P.; Dunphy, E.J.; Holub, A.D.; Saini, A.; Vasi, N.H.; Mahller, Y.Y.; Collins, M.H.; Snyder, J.D.; Krasnykh, V.; Curiel, D.T.; et al. Fiber knob modifications overcome low, heterogeneous expression of the coxsackievirus-adenovirus receptor that limits adenovirus gene transfer and oncolysis for human rhabdomyosarcoma cells. Cancer Res. 2001, 61, 2953–2960. [Google Scholar]
- Mok, W.; Boucher, Y.; Jain, R.K. Matrix Metalloproteinases-1 and -8 Improve the Distribution and Efficacy of an Oncolytic Virus. Cancer Res. 2007, 67, 10664–10668. [Google Scholar] [CrossRef] [Green Version]
- Chiocca, E.A.; Rabkin, S.D. Oncolytic Viruses and Their Application to Cancer Immunotherapy. Cancer Immunol. Res. 2014, 2, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Baird, S.K.; Aerts, J.L.; Eddaoudi, A.; Lockley, M.; Lemoine, N.R.; McNeish, I.A. Oncolytic adenoviral mutants induce a novel mode of programmed cell death in ovarian cancer. Oncogene 2007, 27, 3081–3090. [Google Scholar] [CrossRef] [Green Version]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Kuppuswamy, M.; Spencer, J.F.; Doronin, K.; Tollefson, A.E.; Wold, W.S.M.; Tóth, K. Oncolytic adenovirus that overproduces ADP and replicates selectively in tumors due to hTERT promoter-regulated E4 gene expression. Gene Ther. 2005, 12, 1608–1617. [Google Scholar] [CrossRef] [Green Version]
- Hodzic, J.; Sie, D.; Vermeulen, A.; Van Beusechem, V.W. Functional Screening Identifies Human miRNAs that Modulate Adenovirus Propagation in Prostate Cancer Cells. Hum. Gene Ther. 2017, 28, 766–780. [Google Scholar] [CrossRef]
- Ding, S.-W.; Voinnet, O. Antiviral Immunity Directed by Small RNAs. Cell 2007, 130, 413–426. [Google Scholar] [CrossRef] [Green Version]
- Lecellier, C.-H.; Dunoyer, P.; Arar, K.; Lehmann-Che, J.; Eyquem, S.; Himber, C.; Saïb, A.; Voinnet, O. A Cellular MicroRNA Mediates Antiviral Defense in Human Cells. Science 2005, 308, 557–560. [Google Scholar] [CrossRef] [Green Version]
- Li, H.-C.; Yang, C.-H.; Lo, S.-Y. Roles of microRNAs in Hepatitis C Virus Replication and Pathogenesis. Viruses 2022, 14, 1776. [Google Scholar] [CrossRef]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of Hepatitis C Virus RNA Abundance by a Liver-Specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [Green Version]
- Herzog, K.; Bandiera, S.; Pernot, S.; Fauvelle, C.; Jühling, F.; Weiss, A.; Bull, A.; Durand, S.C.; Chane-Woon-Ming, B.; Pfeffer, S.; et al. Functional microRNA screen uncovers O-linked N-acetylglucosamine transferase as a host factor modulating hepatitis C virus morphogenesis and infectivity. Gut 2019, 69, 380–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, K.; Liu, H.; Rice, A.P. miR-132 enhances HIV-1 replication. Virology 2013, 438, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanerva, A.; Nokisalmi, P.; Diaconu, I.; Koski, A.; Cerullo, V.; Liikanen, I.; Tähtinen, S.; Oksanen, M.; Heiskanen, R.; Pesonen, S.; et al. Antiviral and Antitumor T-cell Immunity in Patients Treated with GM-CSF–Coding Oncolytic Adenovirus. Clin. Cancer Res. 2013, 19, 2734–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varghese, S.; Rabkin, S.D.; Liu, R.; Nielsen, P.G.; Ipe, T.; Martuza, R.L. Enhanced therapeutic efficacy of IL-12, but not GM-CSF, expressing oncolytic herpes simplex virus for transgenic mouse derived prostate cancers. Cancer Gene Ther. 2006, 13, 253–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.L.; Robinson, M.; Han, Z.-Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [Green Version]
- Atherton, M.J.; Lichty, B.D. Evolution of oncolytic viruses: Novel strategies for cancer treatment. Immunotherapy 2013, 5, 1191–1206. [Google Scholar] [CrossRef]
- Mao, L.-J.; Ding, M.; Xu, K.; Pan, J.; Yu, H.; Yang, C. Oncolytic Adenovirus Harboring Interleukin-24 Improves Chemotherapy for Advanced Prostate Cancer. J. Cancer 2018, 9, 4391–4397. [Google Scholar] [CrossRef]
- Clubb, J.H.A.; Kudling, T.V.; Heiniö, C.; Basnet, S.; Pakola, S.; Carrascón, V.C.; Santos, J.M.; Quixabeira, D.C.A.; Havunen, R.; Sorsa, S.; et al. Adenovirus Encoding Tumor Necrosis Factor Alpha and Interleukin 2 Induces a Tertiary Lymphoid Structure Signature in Immune Checkpoint Inhibitor Refractory Head and Neck Cancer. Front. Immunol. 2022, 13, 794251. [Google Scholar] [CrossRef]
- Miller, K.E.; Cassady, K.A.; Roth, J.C.; Clements, J.; Schieffer, K.M.; Leraas, K.; Miller, A.R.; Prasad, N.; Leavenworth, J.W.; Aban, I.B.; et al. Immune Activity and Response Differences of Oncolytic Viral Therapy in Recurrent Glioblastoma: Gene Expression Analyses of a Phase IB Study. Clin. Cancer Res. 2022, 28, 498–506. [Google Scholar] [CrossRef]
- Hulou, M.M.; Cho, C.F.; Chiocca, E.A.; Bjerkvig, R. Experimental therapies: Gene therapies and oncolytic viruses. Handb. Clin. Neurol. 2016, 134, 183–197. [Google Scholar]
- Cowen, R.L.; Williams, J.C.; Emery, S.; Blakey, D.; Darling, J.L.; Lowenstein, P.R.; Castro, M.G. Adenovirus vector–mediated delivery of the prodrug-converting enzyme carboxypeptidase G2 in a secreted or GPI-anchored form: High-level expression of this active conditional cytotoxic enzyme at the plasma membrane. Cancer Gene Ther. 2002, 9, 897–907. [Google Scholar] [CrossRef] [Green Version]
- Platonov, M.E.; Borovjagin, A.V.; Kaverina, N.; Xiao, T.; Kadagidze, Z.; Lesniak, M.; Baryshnikova, M.; Ulasov, I.V. KISS1 tumor suppressor restricts angiogenesis of breast cancer brain metastases and sensitizes them to oncolytic virotherapy in vitro. Cancer Lett. 2018, 417, 75–88. [Google Scholar] [CrossRef]
- Li, Y.; Shang, C.; Liu, Z.; Han, J.; Li, W.; Xiao, P.; Li, N.; Li, S.; Xiu, Z.; Song, G.; et al. Apoptin mediates mitophagy and endogenous apoptosis by regulating the level of ROS in hepatocellular carcinoma. Cell Commun. Signal. 2022, 20, 134. [Google Scholar] [CrossRef]
- Li, M.; Zhu, Y.; Bai, B.; Fang, J.; Yao, W.; Li, Y.; Li, S.; Li, X.; Jin, N.; Jiang, R. Suppression effect of a dual cancer-specific oncolytic adenovirus on luciferase-labeled human melanoma cells in vitro and in vivo. Cancer Biomark. 2021, 32, 251–262. [Google Scholar] [CrossRef]
- Cáceres, B.; Ramirez, A.; Carrillo, E.; Jimenez, G.; Griñán-Lisón, C.; López-Ruiz, E.; Jiménez-Martínez, Y.; Marchal, J.A.; Boulaiz, H. Deciphering the Mechanism of Action Involved in Enhanced Suicide Gene Colon Cancer Cell Killer Effect Mediated by Gef and Apoptin. Cancers 2019, 11, 264. [Google Scholar] [CrossRef] [Green Version]
- Song, G.; Fang, J.; Shang, C.; Li, Y.; Zhu, Y.; Xiu, Z.; Sun, L.; Jin, N.; Li, X. Ad-apoptin inhibits glycolysis, migration and invasion in lung cancer cells targeting AMPK/mTOR signaling pathway. Exp. Cell Res. 2021, 409, 112926. [Google Scholar] [CrossRef]
- Cui, C.-X.; Li, Y.-Q.; Sun, Y.-J.; Zhu, Y.-L.; Fang, J.-B.; Bai, B.; Li, W.-J.; Li, S.-Z.; Ma, Y.-Z.; Li, X.; et al. Antitumor effect of a dual cancer-specific oncolytic adenovirus on prostate cancer PC-3 cells. Urol. Oncol. Semin. Orig. Investig. 2019, 37, 352.e1–352.e18. [Google Scholar] [CrossRef]
- Li, X.; Liu, Y.; Wen, Z.; Li, C.; Lu, H.; Tian, M.; Jin, K.; Sun, L.; Gao, P.; Yang, E.; et al. Potent anti-tumor effects of a dual specific oncolytic adenovirus expressing apoptin in vitro and in vivo. Mol. Cancer 2010, 9, 10. [Google Scholar]
- Kanerva, A.; Koski, A.; Liikanen, I.; Oksanen, M.; Joensuu, T.; Hemminki, O.; Palmgren, J.; Hemminki, K.; Hemminki, A. Case–Control Estimation of the Impact of Oncolytic Adenovirus on the Survival of Patients With Refractory Solid Tumors. Mol. Ther. 2015, 23, 321–329. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.; Heiniö, C.; Quixabeira, D.; Zafar, S.; Clubb, J.; Pakola, S.; Cervera-Carrascon, V.; Havunen, R.; Kanerva, A.; Hemminki, A. Systemic Delivery of Oncolytic Adenovirus to Tumors Using Tumor-Infiltrating Lymphocytes as Carriers. Cells 2021, 10, 978. [Google Scholar] [CrossRef]
- Shimizu, Y.; Gumin, J.; Gao, F.; Hossain, A.; Shpall, E.J.; Kondo, A.; Kerrigan, B.C.P.; Yang, J.; Ledbetter, D.; Fueyo, J.; et al. Characterization of patient-derived bone marrow human mesenchymal stem cells as oncolytic virus carriers for the treatment of glioblastoma. J. Neurosurg. 2022, 136, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.-T.; Wu, M.-H.; Chen, M.-J.; Lin, S.-P.; Yen, Y.-T.; Hung, S.-C. Combination of Mesenchymal Stem Cell-Delivered Oncolytic Virus with Prodrug Activation Increases Efficacy and Safety of Colorectal Cancer Therapy. Biomedicines 2021, 9, 548. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.; Zhang, P.-B.; Feng, Q.; Pan, X.-Y.; Song, S.-L.; Cui, J.; Yang, J.-L. Cytokine-induced killer cells carrying recombinant oncolytic adenovirus expressing p21Ras scFv inhibited liver cancer. J. Cancer 2021, 12, 2768–2776. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Zhang, Z.; Liao, X.; Huang, L.; Liao, Y.; Deng, W. Combination of oncolytic adenovirus ZD55 harboring TRAIL-IETD-MnSOD and cytokine-induced killer cells against lung cancer. Ann. Transl. Med. 2021, 9, 1527. [Google Scholar] [CrossRef] [PubMed]
- Parker Kerrigan, B.C.; Shimizu, Y.; Andreeff, M.; Lang, F.F. Mesenchymal stromal cells for the delivery of oncolytic viruses in gliomas. Cytotherapy 2017, 19, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Iscaro, A.; Jones, C.; Forbes, N.; Mughal, A.; Howard, F.N.; Al Janabi, H.; Demiral, S.; Perrie, Y.; Essand, M.; Weglarz, A.; et al. Targeting circulating monocytes with CCL2-loaded liposomes armed with an oncolytic adenovirus. Nanomedicine 2021, 40, 102506. [Google Scholar] [CrossRef]
- Man, Y.K.S.; Aguirre-Hernandez, C.; Fernandez, A.; Martin-Duque, P.; González-Pastor, R.; Halldén, G. Complexing the Oncolytic Adenoviruses Ad∆∆ and Ad-3∆-A20T with Cationic Nanoparticles Enhances Viral Infection and Spread in Prostate and Pancreatic Cancer Models. Int. J. Mol. Sci. 2022, 23, 8884. [Google Scholar] [CrossRef]
- Tong, A.W.; Senzer, N.; Cerullo, V.; Templeton, N.S.; Hemminki, A.; Nemunaitis, J. Oncolytic viruses for induction of anti-tumor immunity. Curr. Pharm. Biotechnol. 2012, 13, 1750–1760. [Google Scholar] [CrossRef]
- Ungerechts, G.; Bossow, S.; Leuchs, B.; Holm, P.S.; Rommelaere, J.; Coffey, M.; Coffin, R.; Bell, J.; Nettelbeck, D.M. Moving oncolytic viruses into the clinic: Clinical-grade production, purification, and characterization of diverse oncolytic viruses. Mol. Ther. Methods Clin. Dev. 2016, 3, 16018. [Google Scholar] [CrossRef] [Green Version]
- Sato-Dahlman, M.; Miura, Y.; Huang, J.L.; Hajeri, P.; Jacobsen, K.; Davydova, J.; Yamamoto, M. CD133-targeted oncolytic adenovirus demonstrates anti-tumor effect in colorectal cancer. Oncotarget 2017, 8, 76044–76056. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, D.L.; Liu, Z.; Sathaiah, M.; Ravindranathan, R.; Guo, Z.; He, Y.; Guo, Z.S. Oncolytic viruses as therapeutic cancer vaccines. Mol. Cancer 2013, 12, 103. [Google Scholar] [CrossRef]
- Wang, Y.; Hallden, G.; Hill, R.; Anand, A.; Liu, T.-C.; Francis, J.; Brooks, G.A.; Lemoine, N.R.; Kirn, D.H. E3 gene manipulations affect oncolytic adenovirus activity in immunocompetent tumor models. Nat. Biotechnol. 2003, 21, 1328–1335. [Google Scholar] [CrossRef]
- Hutzen, B.; Bid, H.K.; Houghton, P.J.; Pierson, C.R.; Powell, K.; Bratasz, A.; Raffel, C.; Studebaker, A.W. Treatment of medulloblastoma with oncolytic measles viruses expressing the angiogenesis inhibitors endostatin and angiostatin. BMC Cancer 2014, 14, 206. [Google Scholar] [CrossRef]
- Breitbach, C.J.; Arulanandam, R.; De Silva, N.; Thorne, S.H.; Patt, R.; Daneshmand, M.; Moon, A.; Ilkow, C.; Burke, J.; Hwang, T.H.; et al. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res. 2013, 73, 1265–1275. [Google Scholar] [CrossRef] [Green Version]
- Tyynelä, K.; Sandmair, A.-M.; Turunen, M.; Vanninen, R.; Vainio, P.; Kauppinen, R.; Johansson, R.; Vapalahti, M.; Ylä-Herttuala, S. Adenovirus-mediated herpes simplex virus thymidine kinase gene therapy in BT4C rat glioma model. Cancer Gene Ther. 2002, 9, 917–924. [Google Scholar] [CrossRef]
- Lemay, C.G.; Rintoul, J.L.; Kus, A.; Paterson, J.M.; Garcia, V.; Falls, T.J.; Ferreira, L.; Bridle, B.W.; Conrad, D.P.; Tang, V.A.; et al. Harnessing Oncolytic Virus-mediated Antitumor Immunity in an Infected Cell Vaccine. Mol. Ther. 2012, 20, 1791–1799. [Google Scholar] [CrossRef] [Green Version]
- Gujar, S.A.; Lee, P.W.K. Oncolytic Virus-Mediated Reversal of Impaired Tumor Antigen Presentation. Front. Oncol. 2014, 4, 77. [Google Scholar] [CrossRef] [Green Version]
- Woller, N.; Gürlevik, E.; Fleischmann-Mundt, B.; Schumacher, A.; Knocke, S.; Kloos, A.M.; Saborowski, M.; Geffers, R.; Manns, M.P.; Wirth, T.C.; et al. Viral Infection of Tumors Overcomes Resistance to PD-1-immunotherapy by Broadening Neoantigenome-directed T-cell Responses. Mol. Ther. 2015, 23, 1630–1640. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://clinicaltrials.gov/ct2/show/NCT01931046?term=NCT01931046&draw=2&rank=1 (accessed on 11 December 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT03514836?term=NCT03514836&draw=2&rank=1 (accessed on 11 December 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT04374240?term=NCT04374240&draw=2&rank=1 (accessed on 11 December 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT02555397?term=NCT02555397&draw=2&rank=1 (accessed on 11 December 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT04097002?term=NCT04097002&draw=1&rank=1 (accessed on 11 December 2022).
- Yang, Y.; Nunes, F.A.; Berencsi, K.; Furth, E.E.; Gönczöl, E.; Wilson, J.M. Cellular immunity to viral antigens limits E1-deleted adenoviruses for gene therapy. Proc. Natl. Acad. Sci. USA 1994, 91, 4407–4411. [Google Scholar] [CrossRef] [Green Version]
- Lieber, A.; He, C.Y.; Meuse, L.; Schowalter, D.; Kirillova, I.; Winther, B.; Kay, M.A. The role of Kupffer cell activation and viral gene expression in early liver toxicity after infusion of recombinant adenovirus vectors. J. Virol. 1997, 71, 8798–8807. [Google Scholar] [CrossRef] [Green Version]
- Ura, T.; Okuda, K.; Shimada, M. Developments in Viral Vector-Based Vaccines. Vaccines 2014, 2, 624–641. [Google Scholar] [CrossRef] [PubMed]
- Melcher, A.; Parato, K.; Rooney, C.M.; Bell, J.C. Thunder and Lightning: Immunotherapy and Oncolytic Viruses Collide. Mol. Ther. 2011, 19, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.H.; Lemoine, N.R.; Wang, Y. Oncolytic Viruses for Cancer Therapy: Overcoming the Obstacles. Viruses 2010, 2, 78–106. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subclasses | Serotypes | Identified Receptor(s) | Major Site(s) of Infection |
|---|---|---|---|
| A | 12, 18, 31, 61 | CAR | Cryptic (GI tract, respiratory tract, urinary tract) |
| B | 3, 7, 11, 14, 16, 21, 34, 35, 50, 55, 66, 68, 76, 77, 78, 79 | CD46, CD68, CD80, DSG2 | Respiratory tract, eye, urinary tract, GI tract |
| C | 1, 2, 5, 6, 57, 89 | CAR, HSPG, MHC1-a2, SR, VCAM-1 | Respiratory tract, eye, lymph, liver, urinary tract, GI tract |
| D | 8, 9, 10, 13, 15, 17, 19, 20, 22-30, 32, 33, 36, 37, 38, 39, 42–49, 51, 53, 54, 56, 58, 59, 60, 62-65, 67, 69–75, 80–88, 90–103 | CAR, CD46, SA | Eye, GI tract |
| E | 4 | CAR | Respiratory tract, eye |
| F | 40, 41 | CAR | GI tract |
| G | 52 | CAR, SA | GI tract |
| Modifications Strategies | Effects | Name | Modification Details | Results |
|---|---|---|---|---|
| Modifications at the transduction level | Enhance the infection efficiency of HAdV | Ad5-D24RGD | Link Arg-Gly-Asp (RGD) peptide chains in the fibre portion of HAdV and mutation of the Rb binding site of E1A | Can specifically bind to the integrin αvβ3 or αvβ5 on the cell surface and can specifically replicate in Rb-mutated cells |
| AxdAdB3/F-RGD | Link Arg-Gly-Asp (RGD) peptide chains in the fibre portion of HAdV and mutation of E1A and E1B | Can specifically bind to the integrin αvβ3 or αvβ5 on the cell surface and can replicate in Rb-mutated cells and p53-mutated cells | ||
| Ad.5/3-CTV | Replace Ad5’s fibre knob with Ad3’s fibre knob | Can specifically target the Ad3 receptor CD46 | ||
| Modifications at the cell cycle-dependent replication selectivity | Enhance the replication selectivity of HAdV | ONYX-015 (dl1520) | Deletion of E1B55KD | Can specifically replicate in p53-mutated cells |
| dl922-947 (AxE1AdB) and Δ24 | Mutation of Rb binding site of E1A | Can specifically replicate in Rb-mutated cells | ||
| AxdAdB-3 | Mutation of E1A and E1B | Can replicate in Rb-mutated and p53-mutated cells | ||
| AdΔΔ | Mutation of Rb binding site in E1A region and deletion of E1B19kD and retains E3 region | Can specifically replicate in Rb-mutated cells and can sensitize apoptosis of normal cells and can suppress host immune response | ||
| Modifications at the tissue-specific promoter-regulated replication selectivity | Enhance the replication selectivity of HAdV | CN706 (CV706, Ad-PSEE1a) | Insert PSA promoter/enhancer in E1A region | Can selectively replicate in PSA-producing cells |
| CV764 | Insert PSA enhancer (PSE) in E1A region and insert hK2 enhancer/promoter in E1B region | Can selectively replicate in PSA-producing cells | ||
| CV787 | Insert probasin promoter in E1A region and insert PSE in E1B region and retains E3 region | Can selectively replicate in PSA- and probasin-producing cells and can suppress host immune response | ||
| Ad5PB-RSV-NIS | Insert probasin promoter in E1A region and replace E3 with NIS gene | Can selectively replicate in probasin-producing cells and can express NIS | ||
| Ad.PSMApro-hNIS | Insert PSMA promoter and Insert NIS gene | Can selectively replicate in PSMA-producing cells and can express NIS | ||
| Ad-PSMA(E-P)-CD | Insert PSMA enhancers and promoters | Can selectively replicate in PSMA-producing cells | ||
| Ad-PSES-luc | Insert PSE and PSMA enhancer | Can selectively replicate in PSA- and PSMA-producing cells | ||
| Ad[I/PPT-Luc] and Ad[I/PPT-E1A] | Insert PSE, PSMA enhancer and TARP promoter | Can selectively replicate in PSA-, PSMA-, and TARP-producing cells | ||
| Ad.DD3-E1A-IL-24 and Ad-DD3p-E1A | Insert DD3PC3 promoter | Can selectively replicate in DD3PC3-producing cells | ||
| Ad-OC-E1a | Insert OC promoter in E1A region | Can selectively replicate in OC-producing cells | ||
| OBP-301 | Insert hTERT promoter in E1A region | Can selectively replicate in hTERT-producing cells | ||
| Ad-BSP-Ela | Insert BSP promoter in E1A region | Can selectively replicate in BSP-producing cells | ||
| Modifications with multiple targeting strategies | Enhance the infection efficiency and replication selectivity of HAdV | Ad5/35PSES.mRFP/ttk | Replace Ad5’s fibre knob with Ad35’s fibre knob and insert PSE and PSMA enhancer and insert mRFP/ttk fusion protein | Can specifically target the CD46 and can selectively replicate in PSA- and PSMA-producing cells and can express mRFP/ttk |
| ORCA-010 (Ad5-D24RGDT1) | Link RGD peptide chains in the fibre portion and Mutation of Rb binding site of E1A and mutation of E3/19K gene | Can specifically bind to the integrin αvβ3 or αvβ5 and can specifically replicate in Rb-mutated cells and can promote host cells lysis and viruses release | ||
| Oncolytic adenovirus as vector-mediated gene therapy | Make HAdV have the dual anti-tumour effect of oncolysis and gene therapy | Ad.sTβRFc | Insert sTGFbRIIFc | Can express sTGFbRIIFc and inhibit the TGF-β pathway |
| Ad5-Δ24-sOPG-Fc-RGD | Link RGD peptide chains in the fibre portion and mutation of Rb binding site of E1A and insert sOPG and Fc | Can specifically bind to the integrin αvβ3 or αvβ5 and can specifically replicate in Rb-mutated cells and can express sOPG and Fc | ||
| Ad5-PSE/PBN-E1A-ARC685Y | Insert PSE and insert mutated androgen receptor (AR) cDNA in E1A region | Can selectively replicate in PSA-producing cells and can replicate in cells with both high and low androgen levels | ||
| Ad-PL-PPT-E1A | Insert PSE, PSMA enhancer and TARP promoter and insert PSA-IZ-CD40L fusion gene | Can selectively replicate in PSA-, PSMA-, and TARP-producing cells and can activate the body’s anti-tumour immune response | ||
| Fusion of genes from different oncolytic adenoviruses | By combining the gene fragments of different HAdV to improve the therapeutic effect and reduce the side effects | Ad657 | Use the HVR region of Ad57 to replace this region of Ad6 by homologous recombination | Compared with Ad5 and Ad6, Ad657 has similar in vitro oncolytic activity. Ad657 is associated with the lowest hepatotoxicity. |
| RCAd11pADP | ADP gene, located from 29,491 to 29,772 nt in the Ad5 E3 region, was cloned into the Ad11pe1 shuttle vector; RCAd11p vector carrying ADP was constructed by inserting the gene into the Ad11p E1 shuttle vector at 451 nt upstream of the E1A region | Can significantly promote tumour apoptosis and effectively prolong the period of mice survival |
| Study Title | Official Title | Intervention | Study Description | Phase | Status | Completion Date |
|---|---|---|---|---|---|---|
| Use of Recombinant Adenovirus Therapy to Treat Localized Prostate Cancer (NCT01931046) | A Phase 1/2a Study of In-situ REIC/Dkk-3 Therapy in Patients With Localized Prostate Cancer (MTG-REIC-PC003) | Drug: Ad5-SGE-REIC/Dkk3 | The purpose of this study is to evaluate the safety and effectiveness of AD5-SGE-REIC/Dkk-3 in patients with localized prostate cancer. | Phase 1 Phase 2 | Completed, no results | March 2020 |
| A Phase I/II, Safety Clinical Trial of DCVAC/PCa and ONCOS-102 in Men With Metastatic Castration-resistant Prostate Cancer (NCT03514836) | A Phase I/II, Clinical Trial to Evaluate the Safety and Immune Activation of the Combination of DCVAC/PCa, and ONCOS-102, in Men With Advanced Metastatic Castration-resistant Prostate Cancer. | Biological: DCVac/PCa (ONCOS-102) Drug: Cyclophosphamide | This open label, dose escalating study is a phase I/IIa first in man study designed to evaluate the safety and tolerability of intratumoral administration of a novel oncolytic adenovirus (ORCA-010) in treating diagnosed treatment naïve Patients with localized prostate cancer. | Phase 1 Phase 2 | Terminated (Insufficient Accrual) | 25 January 2021 |
| A Clinical Trial of AdNRGM Plus CB1954 in Prostate Cancer (AdUP) (NCT04374240) | Phase I Trial of Replicative Defective Type 5 Adenovirus Vector Expressing Nitroreductase & GMCSF Given Via Trans-perineal Template-guided Intra-prostatic Injection Followed by iv CB1954 in Locally Relapsed Prostate Cancer Patients | Genetic: AdNRGM | This is an open label, non-randomised, phase I, sequential group trial which will explore the safety and tolerability of ascending doses of AdNRGM, in combination with CB1954. | Phase 1 | Completed, no results | November 2021 |
| Phase 1 Trial of Interleukin 12 Gene Therapy for Locally Recurrent Prostate Cancer (NCT02555397) | Phase 1 Trial of Oncolytic Adenovirus-Mediated Cytotoxic and Interleukin 12 Gene Therapy for Locally Recurrent Prostate Cancer After Definitive Radiotherapy. | Biological: Ad5-yCD/mutTKSR39rep-hIL12 | The primary purpose of this phase 1 study is to determine the dose-dependent toxicity and maximum tolerated dose (MTD) of oncolytic adenovirus-mediated cytotoxic and IL-12 gene therapy in men with locally recurrent prostate cancer after definitive radiotherapy. | Phase 1 | Active, not recruiting | 14 February 2023 |
| First in Man Clinical Study to Evaluate Safety and Tolerability of an Oncolytic Adenovirus in Prostate Cancer Patients (NCT04097002) | A Phase I/IIa Study Evaluating the Safety and Tolerability of Intratumoral Administration of ORCA-010 in Treatment-Naïve Patients With Localized Prostate Cancer. | Biological: ORCA-010 | This open label, dose escalating study is a phase I/IIa first in man study designed to evaluate the safety and tolerability of intratumoral administration of a novel oncolytic adenovirus (ORCA-010) in treating diagnosed treatment naïve Patients with localized prostate cancer. | Phase 1 Phase 2 | Recruiting | December 2023 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, K.; Feng, S.; Luo, Z. Oncolytic Adenovirus, a New Treatment Strategy for Prostate Cancer. Biomedicines 2022, 10, 3262. https://doi.org/10.3390/biomedicines10123262
Yang K, Feng S, Luo Z. Oncolytic Adenovirus, a New Treatment Strategy for Prostate Cancer. Biomedicines. 2022; 10(12):3262. https://doi.org/10.3390/biomedicines10123262
Chicago/Turabian StyleYang, Kaiyi, Shenghui Feng, and Zhijun Luo. 2022. "Oncolytic Adenovirus, a New Treatment Strategy for Prostate Cancer" Biomedicines 10, no. 12: 3262. https://doi.org/10.3390/biomedicines10123262
APA StyleYang, K., Feng, S., & Luo, Z. (2022). Oncolytic Adenovirus, a New Treatment Strategy for Prostate Cancer. Biomedicines, 10(12), 3262. https://doi.org/10.3390/biomedicines10123262