
Macrophage C/EBPδ Drives Gemcitabine, but Not 5-FU or Paclitaxel, Resistance of Pancreatic Cancer Cells in a Deoxycytidine-Dependent Manner


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Conditioned Medium Collection and PDAC Cytotoxicity Assays
2.3. Size Separation of Proteins in Conditioned Media
2.4. RT-qPCR and RNA-Seq
2.5. Statistical Analysis
3. Results
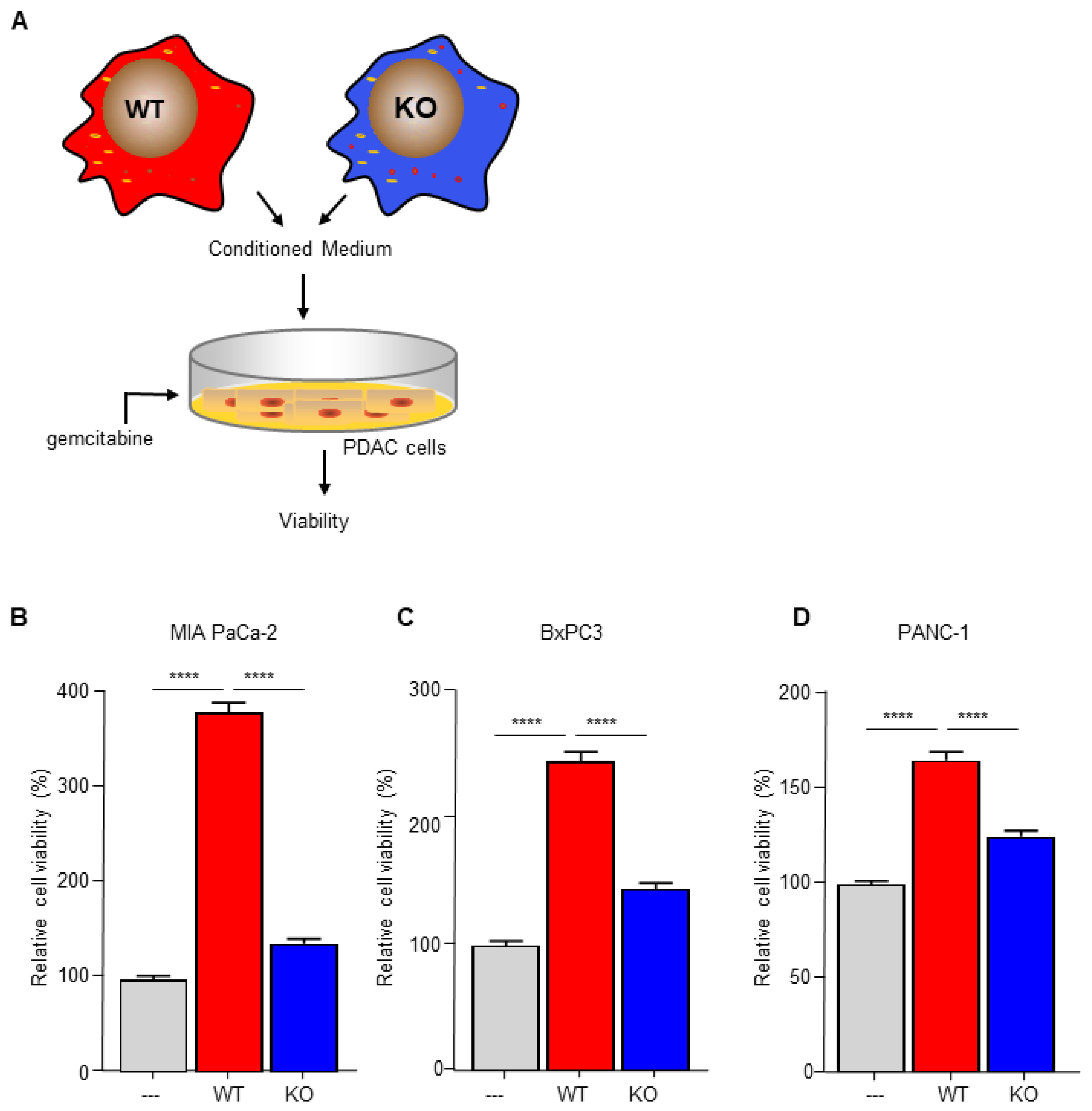
3.1. Macrophage C/EBPδ Drives Gemcitabine Resistance of Pancreatic Cancer Cells
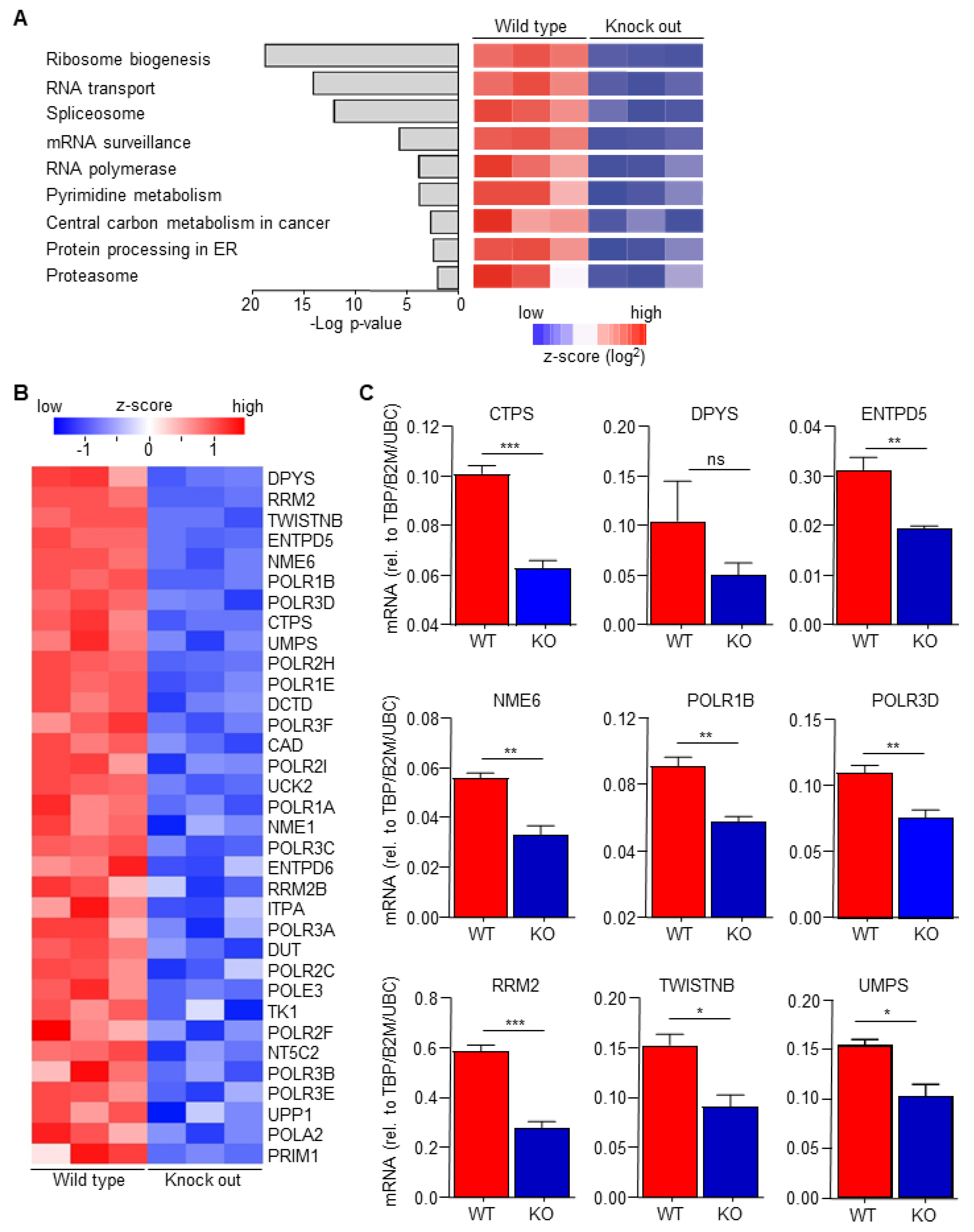
3.2. C/EBPδ Deficiency Inhibits Pyrimidine Syntheses in Macrophages
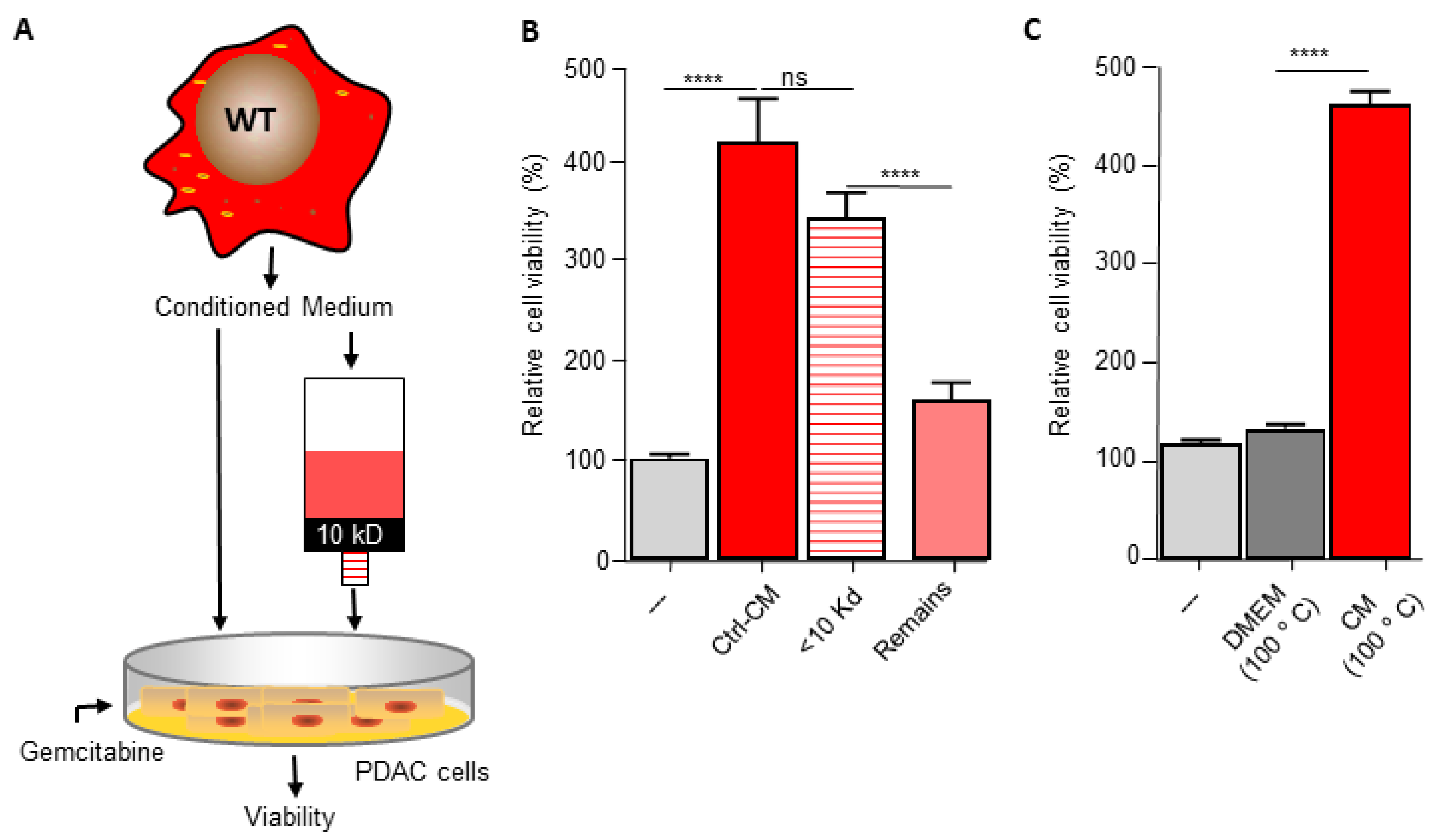
3.3. Macrophage C/EBPδ Drives Gemcitabine Resistance in a Deoxycytidine-Dependent Manner
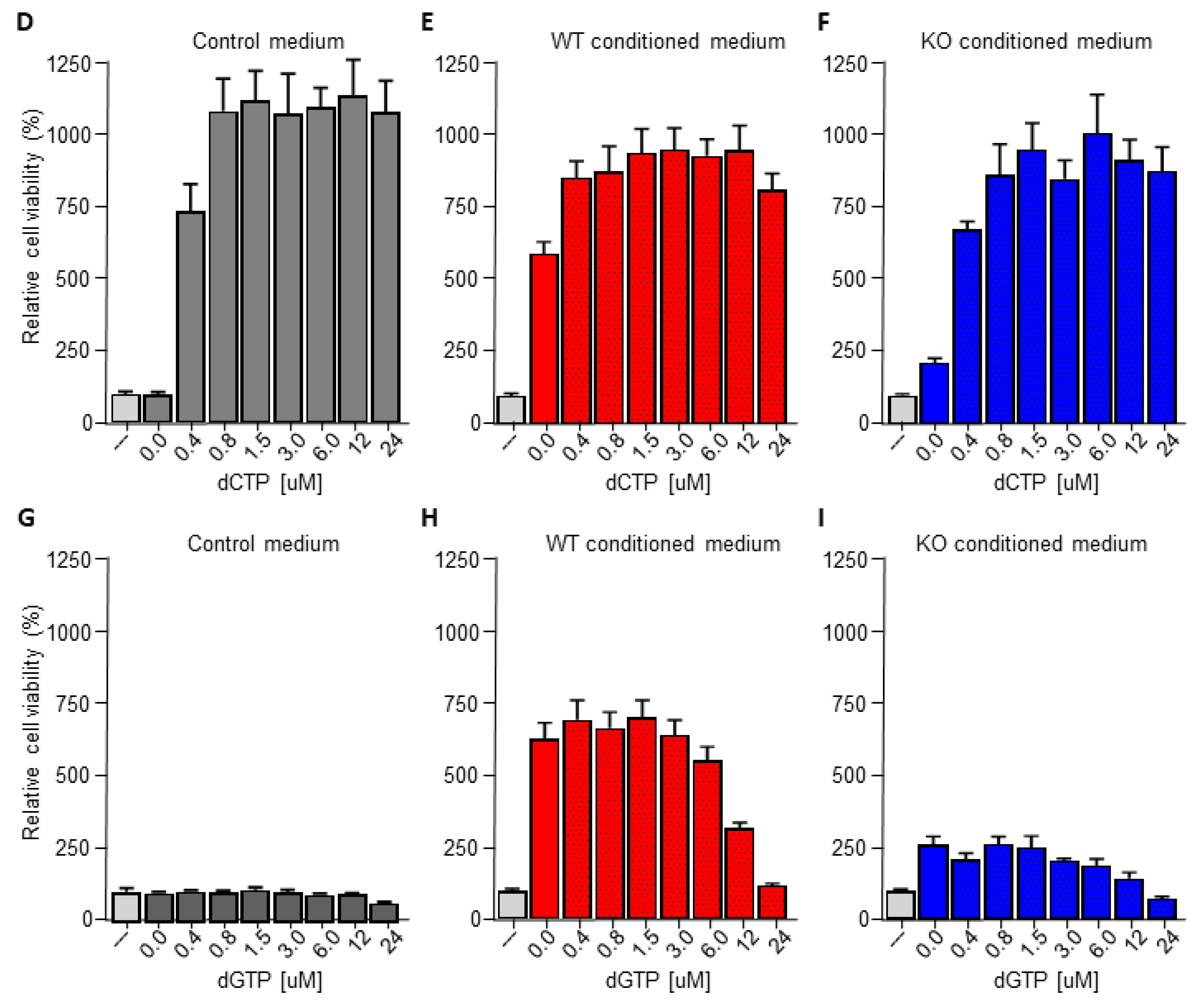
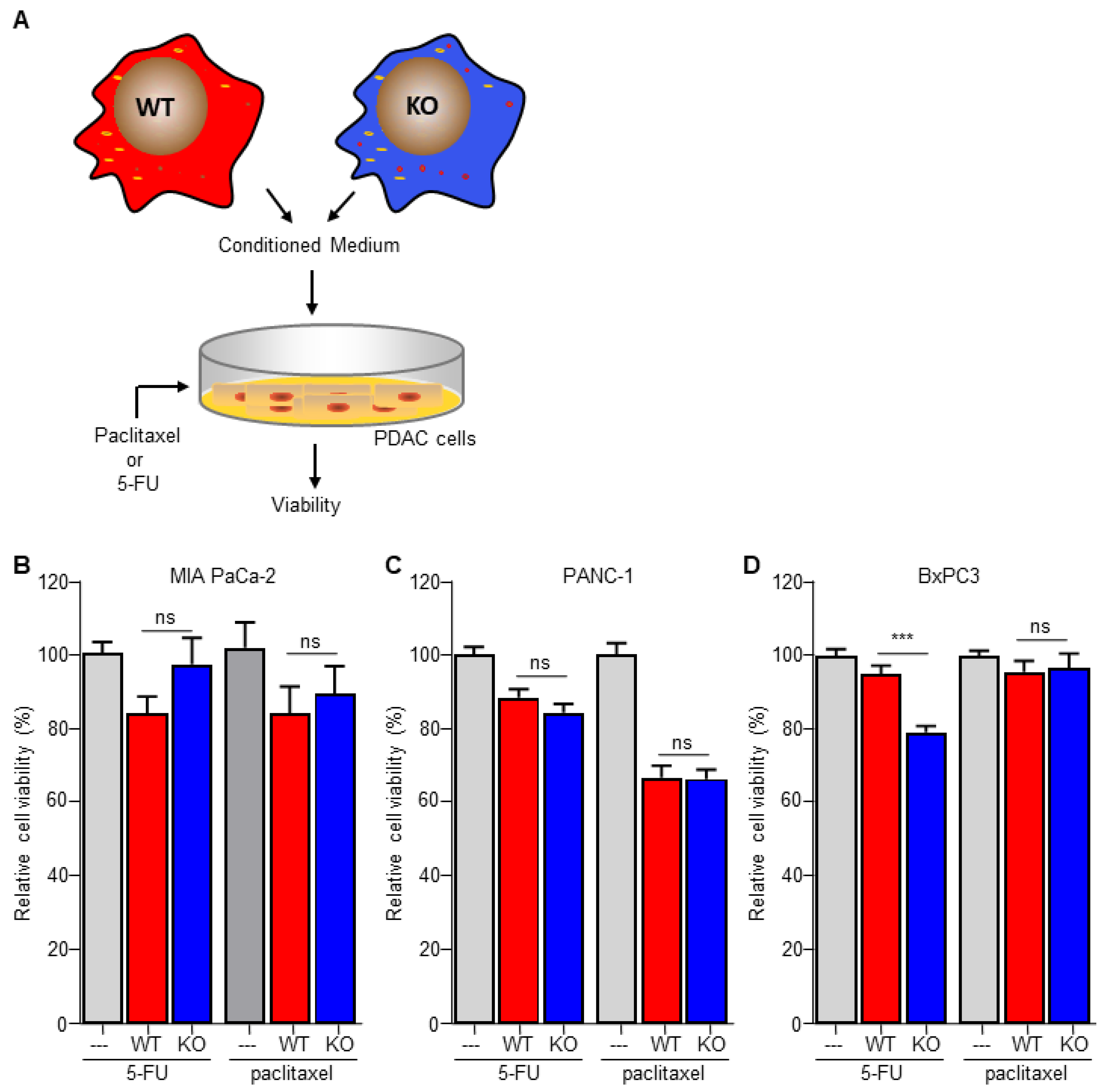
3.4. Macrophage C/EBPδ-Dependent Resistance of Pancreatic Cancer Cells Is Specific for Deoxycytidine Analogs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today. Lyon: International Agency for Research on Cancer; 2020. Available online: https://gco.iarc.fr/today (accessed on 11 January 2022).
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Principe, D.R.; Underwood, P.W.; Korc, M.; Trevino, J.G.; Munshi, H.G.; Rana, A. The Current Treatment Paradigm for Pancreatic Ductal Adenocarcinoma and Barriers to Therapeutic Efficacy. Front. Oncol. 2021, 11, 688377. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with Nab-Paclitaxel Plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Porta, C.; Riboldi, E.; Totaro, M.G.; Strauss, L.; Sica, A.; Mantovani, A. Macrophages in cancer and infectious diseases: The ‘good’ and the ‘bad’. Immunotherapy 2011, 3, 1185–1202. [Google Scholar] [CrossRef]
- Jadus, M.R.; Irwin, M.C.; Irwin, M.R.; Horansky, R.D.; Sekhon, S.; Pepper, K.A.; Kohn, D.B.; Wepsic, H.T. Macrophages can recognize and kill tumor cells bearing the membrane isoform of macrophage colony-stimulating factor. Blood 1996, 87, 5232–5241. [Google Scholar] [CrossRef] [Green Version]
- Goswami, K.K.; Ghosh, T.; Ghosh, S.; Sarkar, M.; Bose, A.; Baral, R. Tumor promoting role of anti-tumor macrophages in tumor microenvironment. Cell Immunol. 2017, 316, 1–10. [Google Scholar] [CrossRef]
- Jung, K.Y.; Cho, S.W.; Kim, Y.A.; Kim, D.; Oh, B.C.; Park, D.J.; Park, Y.J. Cancers with Higher Density of Tumor-Associated Macrophages Were Associated with Poor Survival Rates. J. Pathol. Transl. Med. 2015, 49, 318–324. [Google Scholar] [CrossRef] [Green Version]
- Lindsten, T.; Hedbrant, A.; Ramberg, A.; Wijkander, J.; Solterbeck, A.; Eriksson, M.; Delbro, D.; Erlandsson, A. Effect of macrophages on breast cancer cell proliferation.; and on expression of hormone receptors.; uPAR and HER-2. Int. J. Oncol. 2017, 51, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.M.; Jing, Y.Y.; Yu, G.F.; Kou, X.R.; Ye, F.; Gao, L.; Li, R.; Zhao, Q.D.; Yang, Y.; Lu, Z.H.; et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014, 352, 160–168. [Google Scholar] [CrossRef]
- Liu, C.Y.; Xu, J.Y.; Shi, X.Y.; Huang, W.; Ruan, T.Y.; Xie, P.; Ding, J.L. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells; partially through TLR4/IL-10 signaling pathway. Lab. Investig. 2013, 93, 844–854. [Google Scholar] [CrossRef] [Green Version]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells.; relieves immunosuppression.; and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [Green Version]
- Weizman, N.; Krelin, Y.; Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Wong, R.J.; Gil, Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 2014, 33, 3812–3819. [Google Scholar] [CrossRef] [Green Version]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Buchholz, S.M.; Goetze, R.G.; Singh, S.K.; Ammer-Herrmenau, C.; Richards, F.M.; Jodrell, D.I.; Buchholz, M.; Michl, P.; Ellenrieder, V.; Hessmann, E.; et al. Depletion of Macrophages Improves Therapeutic Response to Gemcitabine in Murine Pancreas Cancer. Cancers 2020, 12, 1978. [Google Scholar] [CrossRef] [PubMed]
- Lekstrom-Himes, J.; Xanthopoulos, K.G. Biological role of the CCAAT/enhancer-binding protein family of transcription factors. J. Biol. Chem. 1998, 273, 28545–28548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gery, S.; Tanosaki, S.; Hofmann, W.K.; Koppel, A.; Koeffler, H.P. C/EBPdelta expression in a BCR-ABL-positive cell line induces growth arrest and myeloid differentiation. Oncogene 2005, 24, 1589–1597. [Google Scholar] [CrossRef] [Green Version]
- Stein, T.; Salomonis, N.; Nuyten, D.S.; van de Vijver, M.J.; Gusterson, B.A. A mouse mammary gland involution mRNA signature identifies biological pathways potentially associated with breast cancer metastasis. J. Mammary Gland Biol. Neoplasia 2009, 14, 99–116. [Google Scholar] [CrossRef]
- Ikezoe, T.; Gery, S.; Yin, D.; O’Kelly, J.; Binderup, L.; Lemp, N.; Taguchi, H.; Koeffler, H.P. CCAAT/enhancer-binding protein delta: A molecular target of 1.;25-dihydroxyvitamin D3 in androgen-responsive prostate cancer LNCaP cells. Cancer Res. 2005, 65, 4762–4768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivko, G.S.; Sanford, D.C.; Dearth, L.D.; Tang, D.; DeWille, J.W. CCAAT/Enhancer binding protein delta (c/EBPdelta) regulation and expression in human mammary epithelial cells: II. Analysis of activating signal transduction pathways; transcriptional; post-transcriptional; and post-translational control. J. Cell Biochem. 2004, 93, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Hartl, L.; Duitman, J.; Aberson, H.L.; Chen, K.; Dijk, F.; Roelofs, J.J.T.H.; Dings, M.P.G.; Hooijer, G.K.J.; Hernanda, P.Y.; Pan, Q.; et al. CCAAT/Enhancer-Binding Protein Delta (C/EBPδ): A Previously Unrecognized Tumor Suppressor that Limits the Oncogenic Potential of Pancreatic Ductal Adenocarcinoma Cells. Cancers 2020, 12, 2546. [Google Scholar] [CrossRef] [PubMed]
- Duitman, J.; Hartl, L.; Roelofs, J.J.T.H.; Bijlsma, M.F.; Spek, C.A. Non-Tumor CCAAT/Enhancer-Binding Protein Delta Potentiates Tumor Cell Extravasation and Pancreatic Cancer Metastasis Formation. Biomolecules 2021, 11, 1079. [Google Scholar] [CrossRef]
- Chang, L.H.; Huang, H.S.; Wu, P.T.; Jou, I.M.; Pan, M.H.; Chang, W.C.; Wang, D.D.; Wang, J.M. Role of macrophage CCAAT/enhancer binding protein delta in the pathogenesis of rheumatoid arthritis in collagen-induced arthritic mice. PLoS ONE 2012, 7, e45378. [Google Scholar] [CrossRef]
- Banerjee, S.; Xie, N.; Cui, H.; Tan, Z.; Yang, S.; Icyuz, M.; Abraham, E.; Liu, G. MicroRNA let-7c regulates macrophage polarization. J. Immunol. 2013, 190, 6542–6549. [Google Scholar] [CrossRef]
- Jones, V.S.; Huang, R.Y.; Chen, L.P.; Chen, Z.S.; Fu, L.; Huang, R.P. Cytokines in cancer drug resistance: Cues to new therapeutic strategies. Biochim. Biophys. Acta 2016, 1865, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Spek, C.A.; Aberson, H.L.; Butler, J.M.; de Vos, A.F.; Duitman, J. CEBPD Potentiates the Macrophage Inflammatory Response but CEBPD Knock-Out Macrophages Fail to Identify CEBPD-Dependent Pro-Inflammatory Transcriptional Programs. Cells 2021, 10, 2233. [Google Scholar] [CrossRef]
- Slapak, E.J.; Kong, L.; El Mandili, M.; Nieuwland, R.; Kros, A.; Bijlsma, M.F.; Spek, C.A. ADAM9-Responsive Mesoporous Silica Nanoparticles for Targeted Drug Delivery in Pancreatic Cancer. Cancers 2021, 13, 3321. [Google Scholar] [CrossRef] [PubMed]
- Halbrook, C.J.; Pontious, C.; Kovalenko, I.; Lapienyte, L.; Dreyer, S.; Lee, H.J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P.; et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. 2019, 29, 1390–1399.e6. [Google Scholar] [CrossRef]
- Shukla, S.K.; Purohit, V.; Mehla, K.; Gunda, V.; Chaika, N.V.; Vernucci, E.; King, R.J.; Abrego, J.; Goode, G.D.; Dasgupta, A.; et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 2017, 32, 71–87.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar]
- Cunningham, D.; Chau, I.; Stocken, D.D.; Valle, J.W.; Smith, D.; Steward, W.; Harper, P.G.; Dunn, J.; Tudur-Smith, C.; West, J.; et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J. Clin. Oncol. 2009, 27, 5513–5518. [Google Scholar] [CrossRef]
- Heinemann, V.; Quietzsch, D.; Gieseler, F.; Gonnermann, M.; Schönekäs, H.; Rost, A.; Neuhaus, H.; Haag, C.; Clemens, M.; Heinrich, B.; et al. Randomized phase III trial of gemcitabine plus cisplatin compared with gemcitabine alone in advanced pancreatic cancer. J. Clin. Oncol. 2006, 24, 3946–3952. [Google Scholar] [CrossRef]
- Louvet, C.; Labianca, R.; Hammel, P.; Lledo, G.; Zampino, M.G.; André, T.; Zaniboni, A.; Ducreux, M.; Aitini, E.; Taïeb, J.; et al. Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: Results of a GERCOR and GISCAD phase III trial. J. Clin. Oncol. 2005, 23, 3509–3516. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- de Jesus, V.H.F.; Camandaroba, M.P.G.; Donadio, M.D.S.; Cabral, A.; Muniz, T.P.; de Moura Leite, L.; Sant’Ana, L.F. Retrospective comparison of the efficacy and the toxicity of standard and modified FOLFIRINOX regimens in patients with metastatic pancreatic adenocarcinoma. J. Gastrointest. Oncol. 2018, 9, 694–707. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clappaert, E.J.; Murgaski, A.; Van Damme, H.; Kiss, M.; Laoui, D. Diamonds in the Rough: Harnessing Tumor-Associated Myeloid Cells for Cancer Therapy. Front. Immunol. 2018, 9, 2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullmann, T.; Luckhardt, S.; Wolf, M.; Parnham, M.J.; Resch, E. High-Throughput Screening for CEBPD-Modulating Compounds in THP-1-Derived Reporter Macrophages Identifies Anti-Inflammatory HDAC and BET Inhibitors. Int. J. Mol. Sci. 2021, 22, 3022. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.Y.; Hsiao, Y.W.; Li, C.F.; Lo, Y.C.; Lin, Z.Y.; Hong, J.Y.; Liu, Y.M.; Han, X.; Wang, S.M.; Chen, B.K.; et al. Targeting chemotherapy-induced PTX3 in tumor stroma to prevent the progression of drug-resistant cancers. Oncotarget 2015, 6, 23987–24001. [Google Scholar] [CrossRef] [Green Version]
- Zeng, S.; Pöttler, M.; Lan, B.; Grützmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4504. [Google Scholar] [CrossRef] [Green Version]
- Schniewind, B.; Christgen, M.; Kurdow, R.; Haye, S.; Kremer, B.; Kalthoff, H.; Ungefroren, H. Resistance of pancreatic cancer to gemcitabine treatment is dependent on mitochondria-mediated apoptosis. Int. J. Cancer 2004, 109, 182–188. [Google Scholar] [CrossRef]
- Fu, Y.; Ricciardiello, F.; Yang, G.; Qiu, J.; Huang, H.; Xiao, J.; Cao, Z.; Zhao, F.; Liu, Y.; Luo, W.; et al. The Role of Mitochondria in the Chemoresistance of Pancreatic Cancer Cells. Cells 2021, 10, 497. [Google Scholar] [CrossRef]
- Rauchwerger, D.R.; Firby, P.S.; Hedley, D.W.; Moore, M.J. Equilibrative-sensitive nucleoside transporter and its role in gemcitabine sensitivity. Cancer Res. 2000, 60, 6075–6079. [Google Scholar]
- Yang, S.; Liu, Q.; Liao, Q. Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Origin, Polarization, Function, and Reprogramming. Front. Cell Dev. Biol. 2021, 8, 607209. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spek, C.A.; Aberson, H.L.; Duitman, J. Macrophage C/EBPδ Drives Gemcitabine, but Not 5-FU or Paclitaxel, Resistance of Pancreatic Cancer Cells in a Deoxycytidine-Dependent Manner. Biomedicines 2022, 10, 219. https://doi.org/10.3390/biomedicines10020219
Spek CA, Aberson HL, Duitman J. Macrophage C/EBPδ Drives Gemcitabine, but Not 5-FU or Paclitaxel, Resistance of Pancreatic Cancer Cells in a Deoxycytidine-Dependent Manner. Biomedicines. 2022; 10(2):219. https://doi.org/10.3390/biomedicines10020219
Chicago/Turabian StyleSpek, C. Arnold, Hella L. Aberson, and JanWillem Duitman. 2022. "Macrophage C/EBPδ Drives Gemcitabine, but Not 5-FU or Paclitaxel, Resistance of Pancreatic Cancer Cells in a Deoxycytidine-Dependent Manner" Biomedicines 10, no. 2: 219. https://doi.org/10.3390/biomedicines10020219
APA StyleSpek, C. A., Aberson, H. L., & Duitman, J. (2022). Macrophage C/EBPδ Drives Gemcitabine, but Not 5-FU or Paclitaxel, Resistance of Pancreatic Cancer Cells in a Deoxycytidine-Dependent Manner. Biomedicines, 10(2), 219. https://doi.org/10.3390/biomedicines10020219