
A Novel 2-Hit Zebrafish Model to Study Early Pathogenesis of Non-Alcoholic Fatty Liver Disease

 ,
,  and
and 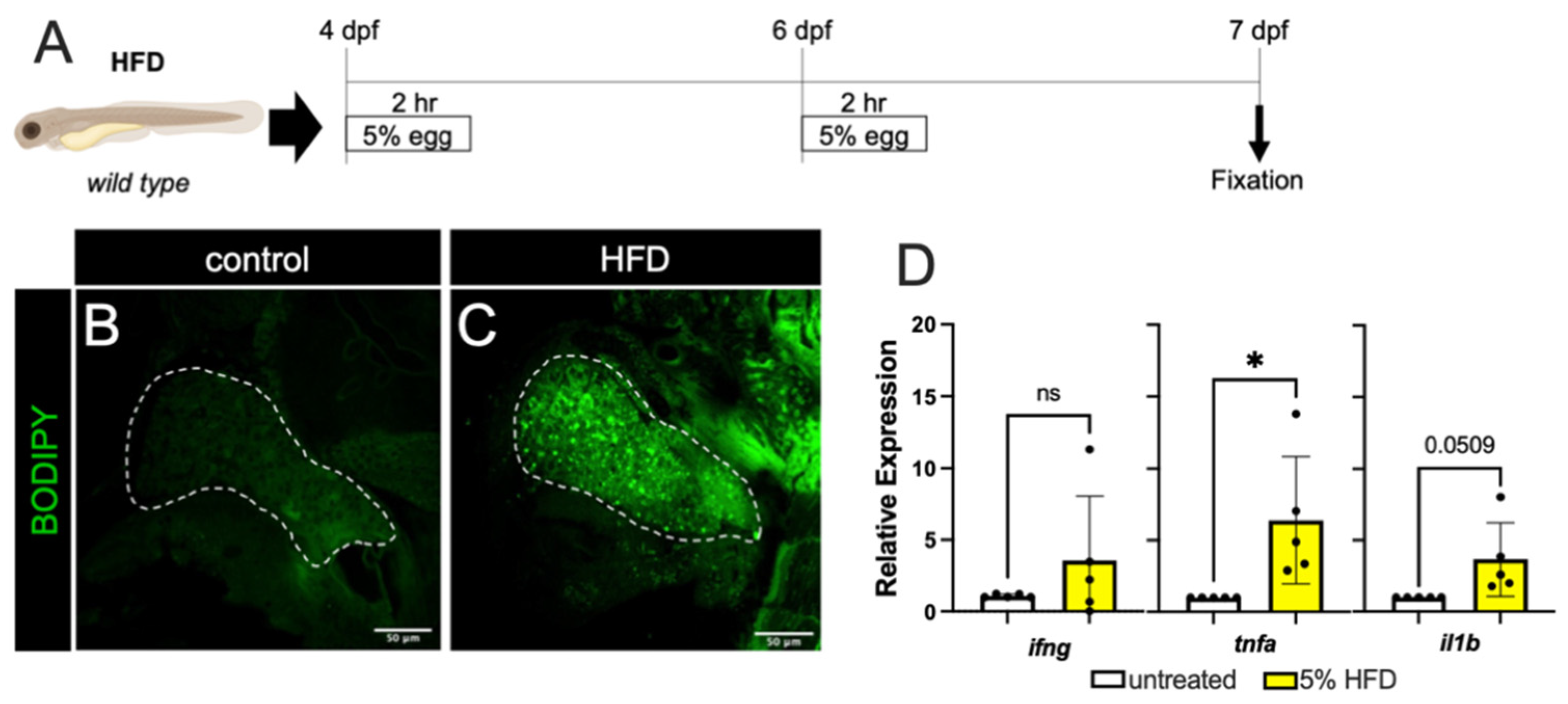{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Experiments
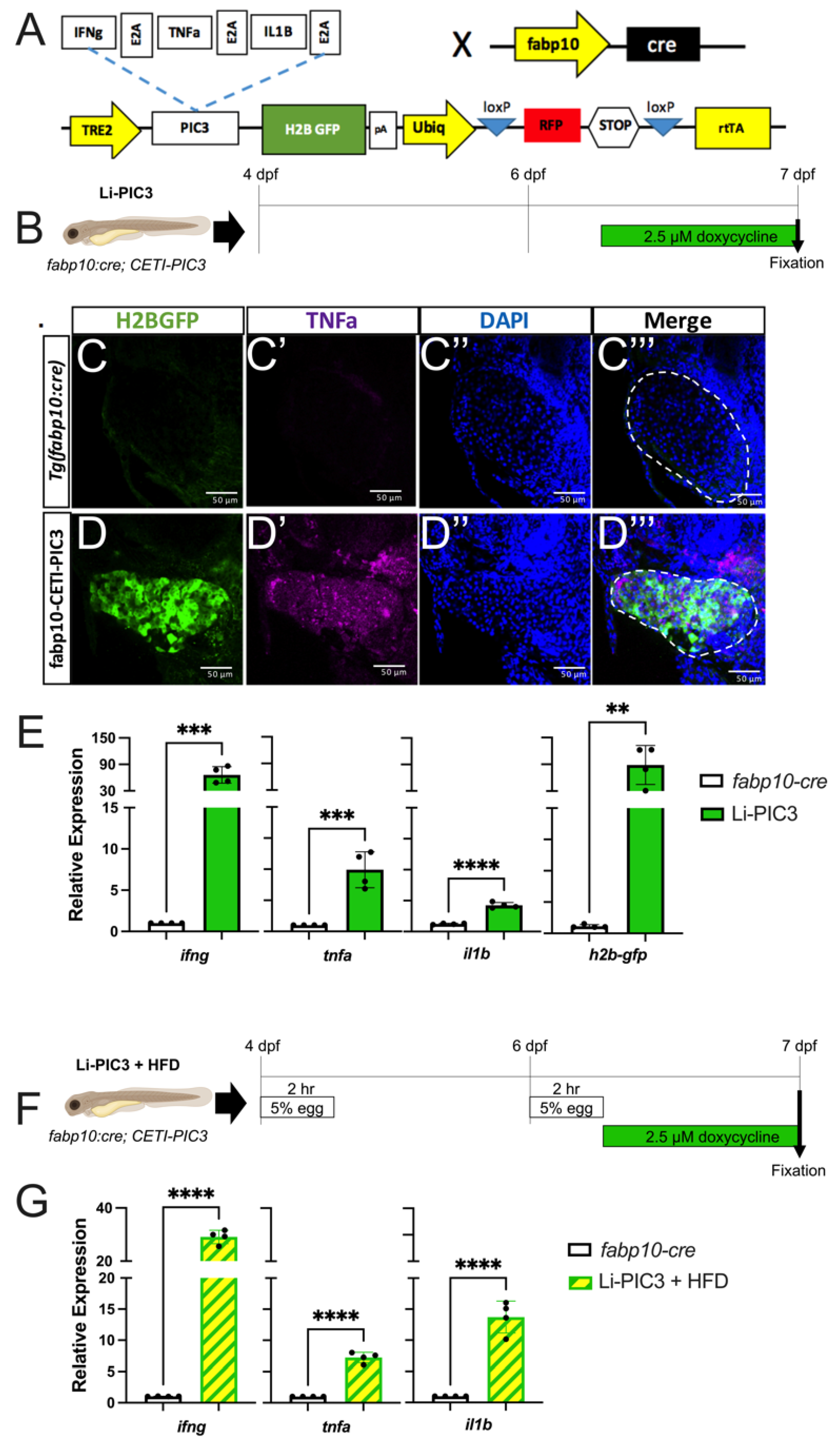
2.2. Inflammatory Liver Model
2.3. High-fat Diet Feeding and Lipid Staining
2.4. Pioglitazone Treatment
2.5. Free Glucose Assay
2.6. Combination Inflammatory Liver and High-Fat Diet Feeding Model
2.7. Detection of Reactive Oxidative Species
2.8. Immunofluorescence Staining
2.9. Quantitative Real-Time PCR
2.10. Statistical Analysis
3. Results
3.1. Models Exhibit Cytokine Expression and Lipid Accumulation in the Hepatocytes
3.2. NAFLD Induction Promotes Infiltration of Macrophages
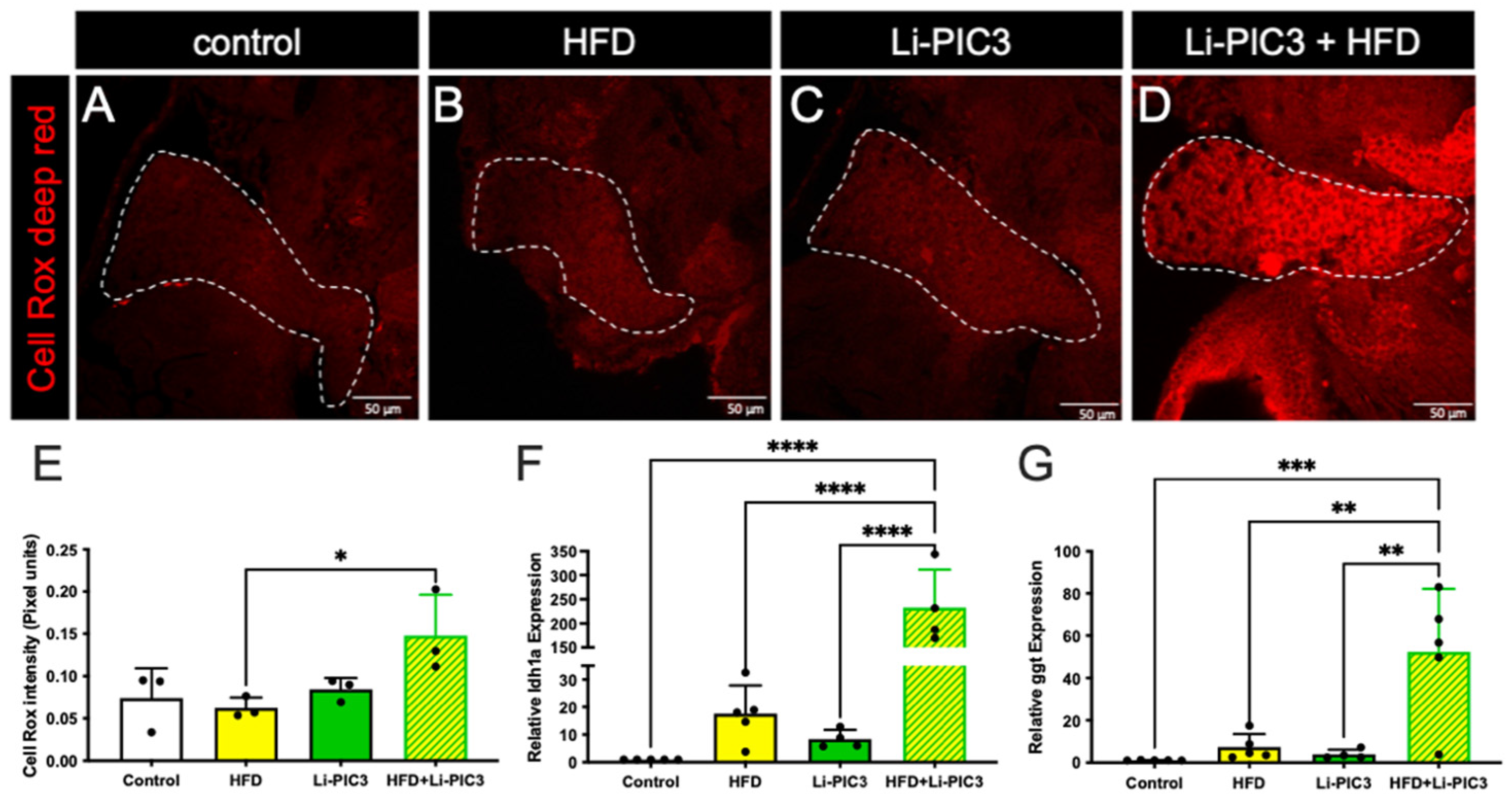
3.3. NAFLD Induction Leads to ROS Accumulation and Liver Damage
3.4. Association of NAFLD with Insulin Resistance
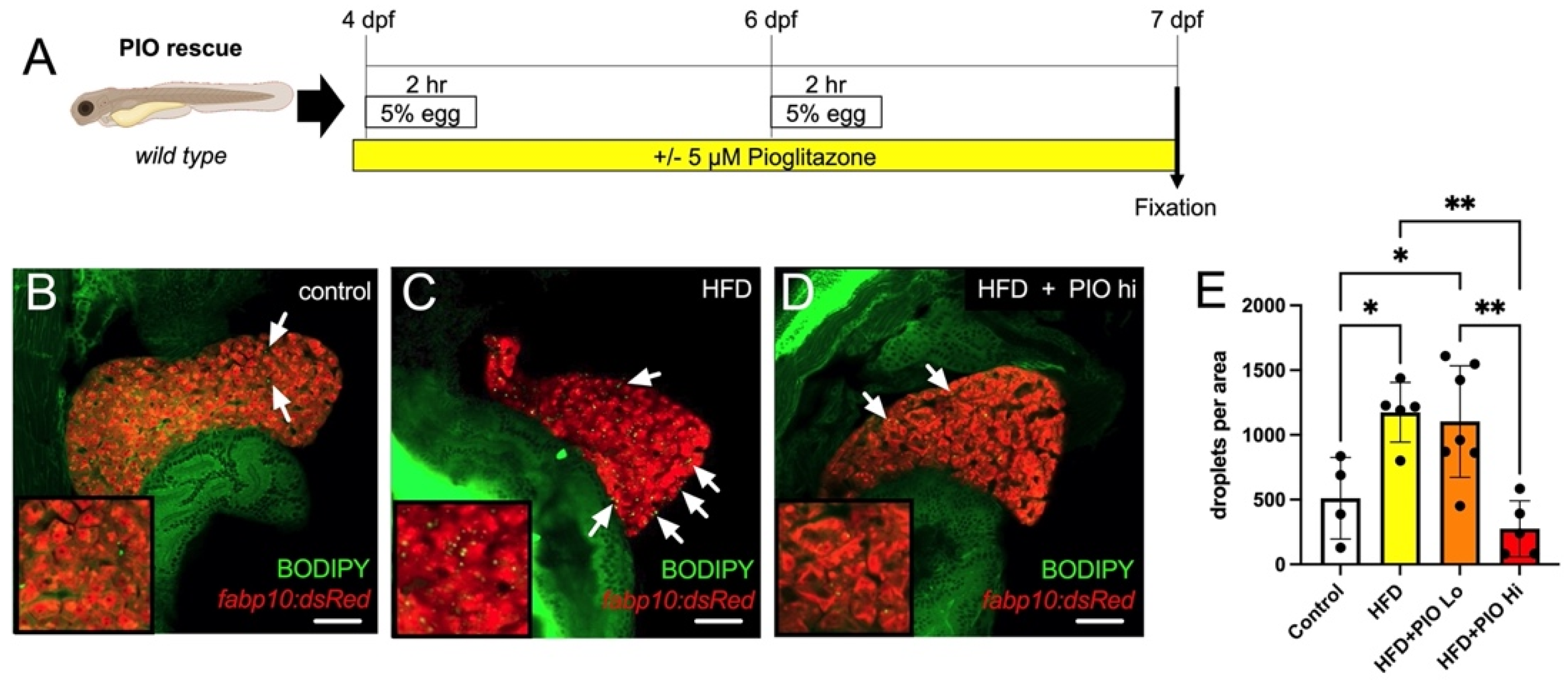
3.5. Pioglitazone Treatment Abates Steatosis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the Epidemic of Nonalcoholic Fatty Liver Disease Demonstrates an Exponential Increase in Burden of Disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Eslamparast, T.; Eghtesad, S.; Hekmatdoost, A.; Poustchi, H. Probiotics and Nonalcoholic Fatty Liver Disease. Middle East J. Dig. Dis. 2013, 5, 129–136. [Google Scholar] [PubMed]
- Dhamija, E.; Paul, S.B.; Kedia, S. Non-Alcoholic Fatty Liver Disease Associated with Hepatocellular Carcinoma: An Increasing Concern. Indian J. Med. Res. 2019, 149, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Paschos, P.; Paletas, K. Non Alcoholic Fatty Liver Disease Two-Hit Process: Multifactorial Character of the Second Hit. Hippokratia 2009, 13, 128. [Google Scholar]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, Z.; Caviglia, J.M.; Corey, K.E.; Herfel, T.M.; Masia, R.; Chung, R.; Lefkowitch, J.H.; Schwabe, R.F.; Tabas, I. Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2016, 24, 848–862. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Hoffmann, C.; Djerir, N.E.H.; Danckaert, A.; Fernandes, J.; Roux, P.; Charrueau, C.; Lachagès, A.-M.; Charlotte, F.; Brocheriou, I.; Clément, K.; et al. Hepatic Stellate Cell Hypertrophy Is Associated with Metabolic Liver Fibrosis. Sci. Rep. 2020, 10, 3850. [Google Scholar] [CrossRef]
- Van Herck, M.A.; Vonghia, L.; Francque, S.M. Animal Models of Nonalcoholic Fatty Liver Disease—A Starter’s Guide. Nutrients 2017, 9, 1072. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.L.; Carten, J.D.; Farber, S.A. Zebrafish Lipid Metabolism: From Mediating Early Patterning to the Metabolism of Dietary Fat and Cholesterol. Methods Cell Biol. 2011, 101, 111–141. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Zheng, Y.-M.; Zhang, J.-P. Comparative Study of Different Diets-Induced NAFLD Models of Zebrafish. Front. Endocrinol. 2018, 9, 366. [Google Scholar] [CrossRef] [PubMed]
- Goessling, W.; Sadler, K.C. Zebrafish: An Important Tool for Liver Disease Research. Gastroenterology 2015, 149, 1361–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otis, J.P.; Zeituni, E.M.; Thierer, J.H.; Anderson, J.L.; Brown, A.C.; Boehm, E.D.; Cerchione, D.M.; Ceasrine, A.M.; Avraham-Davidi, I.; Tempelhof, H.; et al. Zebrafish as a Model for Apolipoprotein Biology: Comprehensive Expression Analysis and a Role for ApoA-IV in Regulating Food Intake. Dis. Model Mech. 2015, 8, 295–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Harkewicz, R.; Hartvigsen, K.; Wiesner, P.; Choi, S.-H.; Almazan, F.; Pattison, J.; Deer, E.; Sayaphupha, T.; Dennis, E.A.; et al. Oxidized Cholesteryl Esters and Phospholipids in Zebrafish Larvae Fed a High Cholesterol Diet: Macrophage Binding and Activation. J. Biol. Chem. 2010, 285, 32343–32351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Gaudet, D.; Miller, Y.I. Deficient Cholesterol Esterification in Plasma of Apoc2 Knockout Zebrafish and Familial Chylomicronemia Patients. PLoS ONE 2017, 12, e0169939. [Google Scholar] [CrossRef]
- Ka, J.; Jin, S.-W. Zebrafish as an Emerging Model for Dyslipidemia and Associated Diseases. J. Lipid Atheroscler. 2021, 10, 42–56. [Google Scholar] [CrossRef]
- Schlegel, A. Studying Non-Alcoholic Fatty Liver Disease with Zebrafish: A Confluence of Optics, Genetics, and Physiology. Cell Mol. Life Sci. 2012, 69, 3953–3961. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Wang, K.; Zheng, X.; Chen, X.; Zhang, W.; Zhang, Y.; Hou, J.; Liu, L. High Fat plus High Cholesterol Diet Lead to Hepatic Steatosis in Zebrafish Larvae: A Novel Model for Screening Anti-Hepatic Steatosis Drugs. Nutr. Metab. 2015, 12, 42. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, S.; Harris-Kawano, A.; Haider, I.; Mirmira, R.G.; Sims, E.K.; Anderson, R.M. A Novel Cre-Enabled Tetracycline-Inducible Transgenic System for Tissue-Specific Cytokine Expression in the Zebrafish: CETI-PIC3. Dis. Model Mech. 2020, 13, dmm042556. [Google Scholar] [CrossRef]
- Ni, T.T.; Lu, J.; Zhu, M.; Maddison, L.A.; Boyd, K.L.; Huskey, L.; Ju, B.; Hesselson, D.; Zhong, T.P.; Page-McCaw, P.S.; et al. Conditional Control of Gene Function by an Invertible Gene Trap in Zebrafish. Proc. Natl. Acad. Sci. USA 2012, 109, 15389–15394. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.A.; Conteh, A.M.; Sorrell, C.A.; Mirmira, A.; Tersey, S.A.; Mirmira, R.G.; Linnemann, A.K.; Anderson, R.M. An In Vivo Zebrafish Model for Interrogating ROS-Mediated Pancreatic β-Cell Injury, Response, and Prevention. Oxid. Med. Cell Longev. 2018, 2018, 1324739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Robertson, M.A.; Hesselson, D.; Stainier, D.Y.R.; Anderson, R.M. Glucagon Is Essential for Alpha Cell Transdifferentiation and Beta Cell Neogenesis. Development 2015, 142, 1407–1417. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Perez, M.; Kulkarni, A.; Samala, N.; Sorrell, C.; El, K.; Haider, I.; Mukhtar Aleem, A.; Holman, T.R.; Rai, G.; Tersey, S.A.; et al. A 12-Lipoxygenase-Gpr31 Signaling Axis Is Required for Pancreatic Organogenesis in the Zebrafish. FASEB J. 2020, 34, 14850–14862. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Agrawal, I.; Gong, Z. Reversion of Tumor Hepatocytes to Normal Hepatocytes during Liver Tumor Regression in an Oncogene-Expressing Transgenic Zebrafish Model. Dis. Model Mech. 2019, 12, dmm039578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oates, J.R.; McKell, M.C.; Moreno-Fernandez, M.E.; Damen, M.S.M.A.; Deepe, G.S.; Qualls, J.E.; Divanovic, S. Macrophage Function in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: The Mac Attack. Front. Immunol. 2019, 10, 2893. [Google Scholar] [CrossRef] [Green Version]
- Koenig, G.; Seneff, S. Gamma-Glutamyltransferase: A Predictive Biomarker of Cellular Antioxidant Inadequacy and Disease Risk. Dis. Markers 2015, 2015, 818570. [Google Scholar] [CrossRef] [Green Version]
- Kotoh, K.; Kato, M.; Kohjima, M.; Tanaka, M.; Miyazaki, M.; Nakamura, K.; Enjoji, M.; Nakamuta, M.; Takayanagi, R. Lactate Dehydrogenase Production in Hepatocytes Is Increased at an Early Stage of Acute Liver Failure. Exp. Med. 2011, 2, 195–199. [Google Scholar] [CrossRef] [Green Version]
- Dharmalingam, M.; Yamasandhi, P.G. Nonalcoholic Fatty Liver Disease and Type 2 Diabetes Mellitus. Indian J. Endocrinol. Metab. 2018, 22, 421–428. [Google Scholar] [CrossRef]
- Eshraghian, A. Current and Emerging Pharmacological Therapy for Non-Alcoholic Fatty Liver Disease. World J. Gastroenterol. 2017, 23, 7495–7504. [Google Scholar] [CrossRef]
- Lian, J.; Fu, J. Pioglitazone for NAFLD Patients With Prediabetes or Type 2 Diabetes Mellitus: A Meta-Analysis. Front. Endocrinol. 2021, 12, 615409. [Google Scholar] [CrossRef]
- Yu, J.; Marsh, S.; Hu, J.; Feng, W.; Wu, C. The Pathogenesis of Nonalcoholic Fatty Liver Disease: Interplay between Diet, Gut Microbiota, and Genetic Background. Gastroenterol. Res. Pr. 2016, 2016, 2862173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caballero, F.; Fernández, A.; Matías, N.; Martínez, L.; Fucho, R.; Elena, M.; Caballeria, J.; Morales, A.; Fernández-Checa, J.C.; García-Ruiz, C. Specific Contribution of Methionine and Choline in Nutritional Nonalcoholic Steatohepatitis: Impact on Mitochondrial S-Adenosyl-L-Methionine and Glutathione. J. Biol. Chem. 2010, 285, 18528–18536. [Google Scholar] [CrossRef] [Green Version]
- Rinella, M.E.; Green, R.M. The Methionine-Choline Deficient Dietary Model of Steatohepatitis Does Not Exhibit Insulin Resistance. J. Hepatol. 2004, 40, 47–51. [Google Scholar] [CrossRef]
- Soltis, A.R.; Kennedy, N.J.; Xin, X.; Zhou, F.; Ficarro, S.B.; Yap, Y.S.; Matthews, B.J.; Lauffenburger, D.A.; White, F.M.; Marto, J.A.; et al. Hepatic Dysfunction Caused by Consumption of a High-Fat Diet. Cell Rep. 2017, 21, 3317–3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, B.; Tsukamoto, H. Inflammation in Alcoholic and Nonalcoholic Fatty Liver Disease: Friend or Foe? Gastroenterology 2016, 150, 1704–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson-Baucum, E.; Piñeros, A.R.; Kulkarni, A.; Webb-Robertson, B.-J.; Maier, B.; Anderson, R.M.; Wu, W.; Tersey, S.A.; Mastracci, T.L.; Casimiro, I.; et al. Deoxyhypusine Synthase Promotes a Pro-Inflammatory Macrophage Phenotype. Cell. Metab. 2021, 33, 1883–1893.e7. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Pineros, A.R.; Walsh, M.A.; Casimiro, I.; Ibrahim, S.; Hernandez-Perez, M.; Orr, K.S.; Glenn, L.; Nadler, J.L.; Morris, M.A.; et al. 12-Lipoxygenase Governs the Innate Immune Pathogenesis of Islet Inflammation and Autoimmune Diabetes. JCI Insight 2021, 6, 14. [Google Scholar] [CrossRef]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef]
- Kulkarni, A.; Nadler, J.L.; Mirmira, R.G.; Casimiro, I. Regulation of Tissue Inflammation by 12-Lipoxygenases. Biomolecules 2021, 11, 717. [Google Scholar] [CrossRef]
- Tian, S.; Li, J.; Guo, Y.; Dong, W.; Zheng, X. Expression Status and Prognostic Significance of Gamma-Glutamyl Transpeptidase Family Genes in Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 731144. [Google Scholar] [CrossRef]
- El Hassouni, B.; Franczak, M.; Capula, M.; Vonk, C.M.; Gomez, V.M.; Smolenski, R.T.; Granchi, C.; Peters, G.J.; Minutolo, F.; Giovannetti, E. Lactate Dehydrogenase A Inhibition by Small Molecular Entities: Steps in the Right Direction. Oncoscience 2020, 7, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Ullah, R.; Rauf, N.; Nabi, G.; Ullah, H.; Shen, Y.; Zhou, Y.-D.; Fu, J. Role of Nutrition in the Pathogenesis and Prevention of Non-Alcoholic Fatty Liver Disease: Recent Updates. Int. J. Biol. Sci. 2019, 15, 265–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, L.; Jornayvaz, F.R. Endocrine Causes of Nonalcoholic Fatty Liver Disease. World J. Gastroenterol. 2015, 21, 11053–11076. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ding, Z.; Ishaq, M.; Bacha, A.S.; Khan, I.; Hanif, A.; Li, W.; Guo, X. Understanding the Effects of Gut Microbiota Dysbiosis on Nonalcoholic Fatty Liver Disease and the Possible Probiotics Role: Recent Updates. Int. J. Biol. Sci. 2021, 17, 818–833. [Google Scholar] [CrossRef]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann. Intern Med. 2016, 165, 305–315. [Google Scholar] [CrossRef]
- Shadid, S.; Jensen, M.D. Effect of Pioglitazone on Biochemical Indices of Non-Alcoholic Fatty Liver Disease in Upper Body Obesity. Clin. Gastroenterol. Hepatol. 2003, 1, 384–387. [Google Scholar] [CrossRef]
- Berger, J.; Moller, D.E. The Mechanisms of Action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [Green Version]
- Hanefeld, M. Pharmacokinetics and Clinical Efficacy of Pioglitazone. Int. J. Clin. Pr. Suppl. 2001, 121, 19–25. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulkarni, A.; Ibrahim, S.; Haider, I.; Basha, A.; Montgomery, E.; Ermis, E.; Mirmira, R.G.; Anderson, R.M. A Novel 2-Hit Zebrafish Model to Study Early Pathogenesis of Non-Alcoholic Fatty Liver Disease. Biomedicines 2022, 10, 479. https://doi.org/10.3390/biomedicines10020479
Kulkarni A, Ibrahim S, Haider I, Basha A, Montgomery E, Ermis E, Mirmira RG, Anderson RM. A Novel 2-Hit Zebrafish Model to Study Early Pathogenesis of Non-Alcoholic Fatty Liver Disease. Biomedicines. 2022; 10(2):479. https://doi.org/10.3390/biomedicines10020479
Chicago/Turabian StyleKulkarni, Abhishek, Sara Ibrahim, Isra Haider, Amina Basha, Emma Montgomery, Ebru Ermis, Raghavendra G. Mirmira, and Ryan M. Anderson. 2022. "A Novel 2-Hit Zebrafish Model to Study Early Pathogenesis of Non-Alcoholic Fatty Liver Disease" Biomedicines 10, no. 2: 479. https://doi.org/10.3390/biomedicines10020479
APA StyleKulkarni, A., Ibrahim, S., Haider, I., Basha, A., Montgomery, E., Ermis, E., Mirmira, R. G., & Anderson, R. M. (2022). A Novel 2-Hit Zebrafish Model to Study Early Pathogenesis of Non-Alcoholic Fatty Liver Disease. Biomedicines, 10(2), 479. https://doi.org/10.3390/biomedicines10020479