
Heart Failure Severity Closely Correlates with Intestinal Dysbiosis and Subsequent Metabolomic Alterations


 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Model of Transverse Aortic Constriction
2.3. DNA Extraction and Sequencing of Bacterial 16S rDNA
2.4. Metabolite Analyses in the TAC Model
2.5. Processing of 16S rRNA Data
2.6. Bacterial Compositional Analysis
2.7. Functional Analysis
2.8. Statistics
3. Results
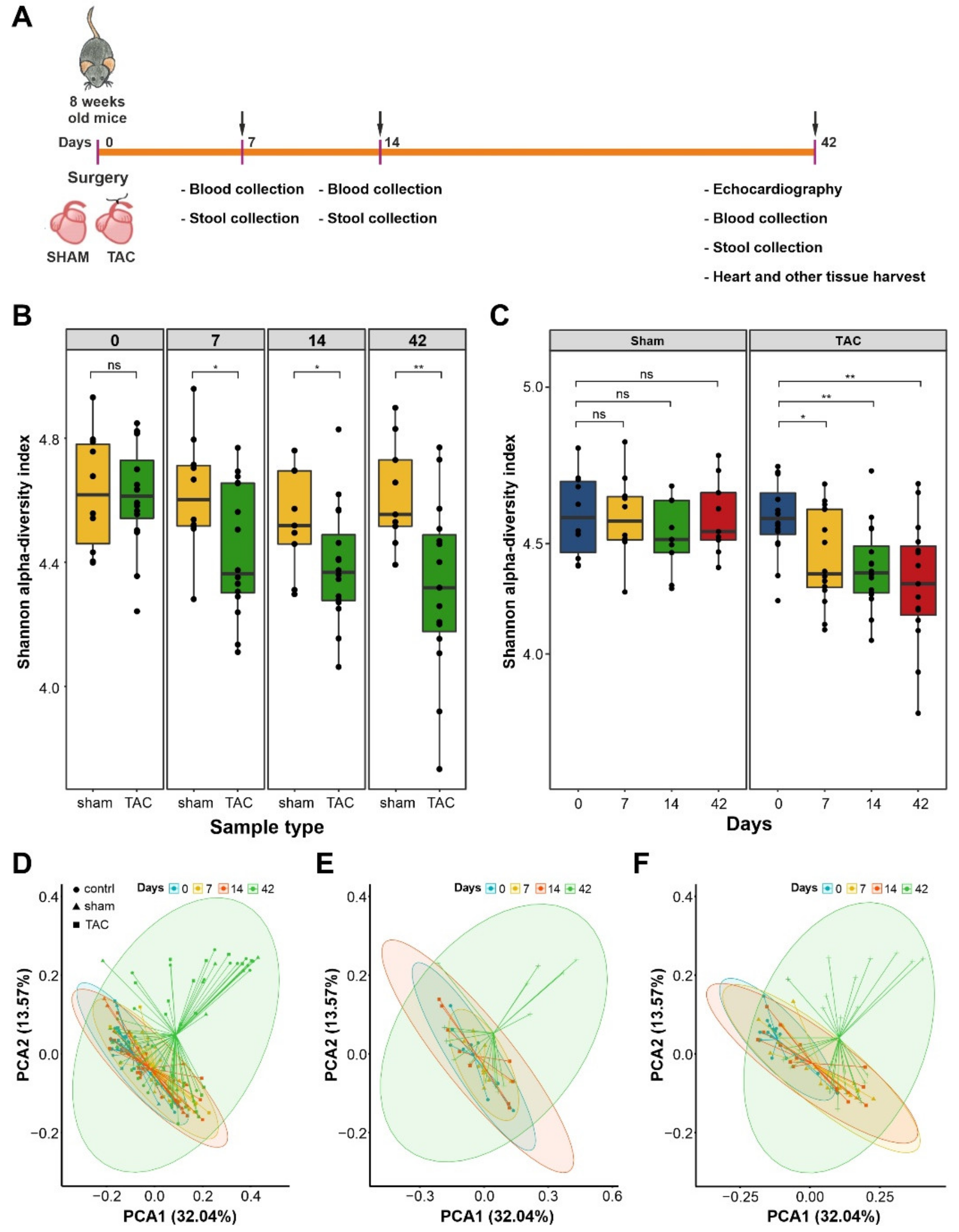
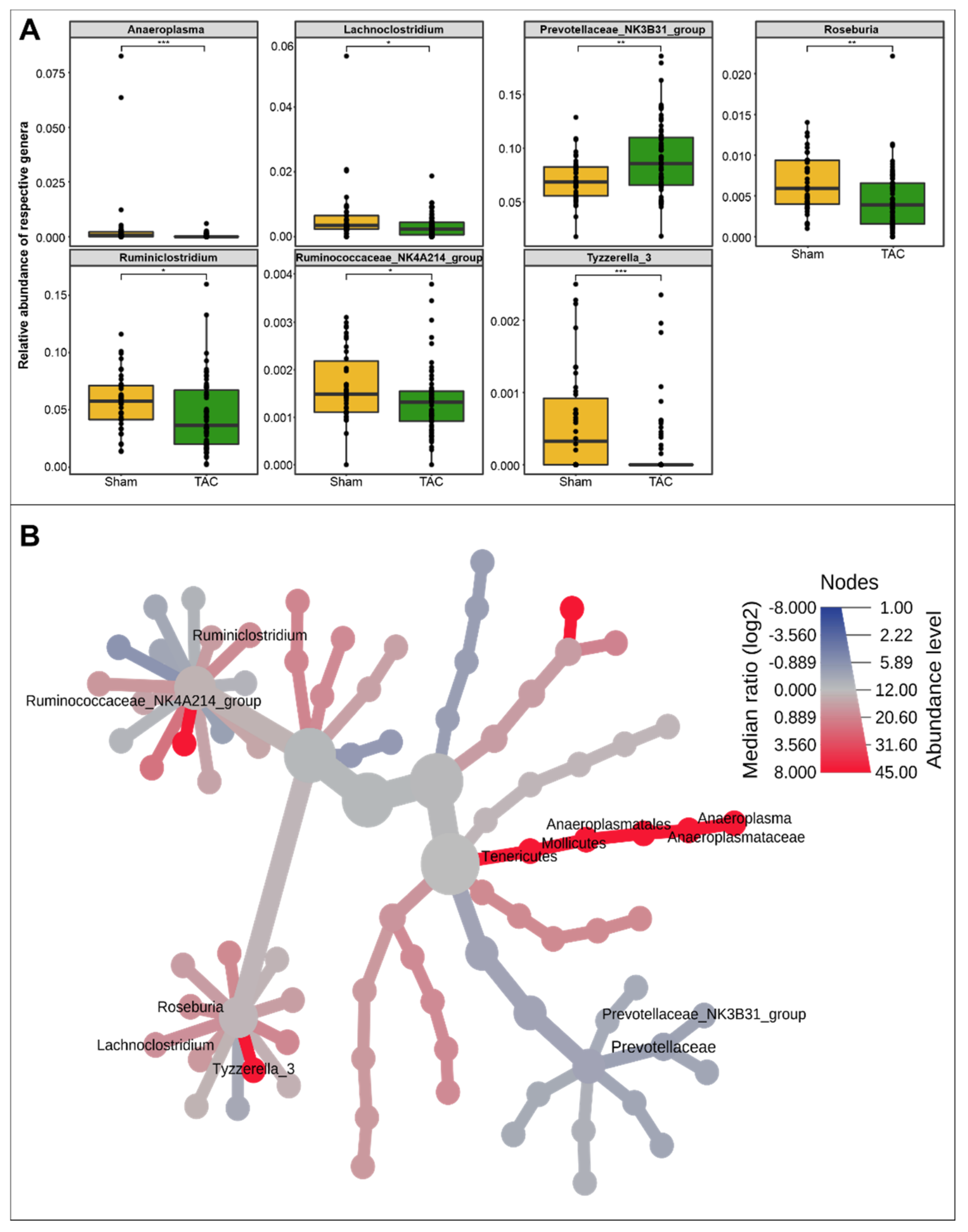
3.1. HF Induced Changes in Bacterial Composition and Loss of Diversity of Fecal Bacteria
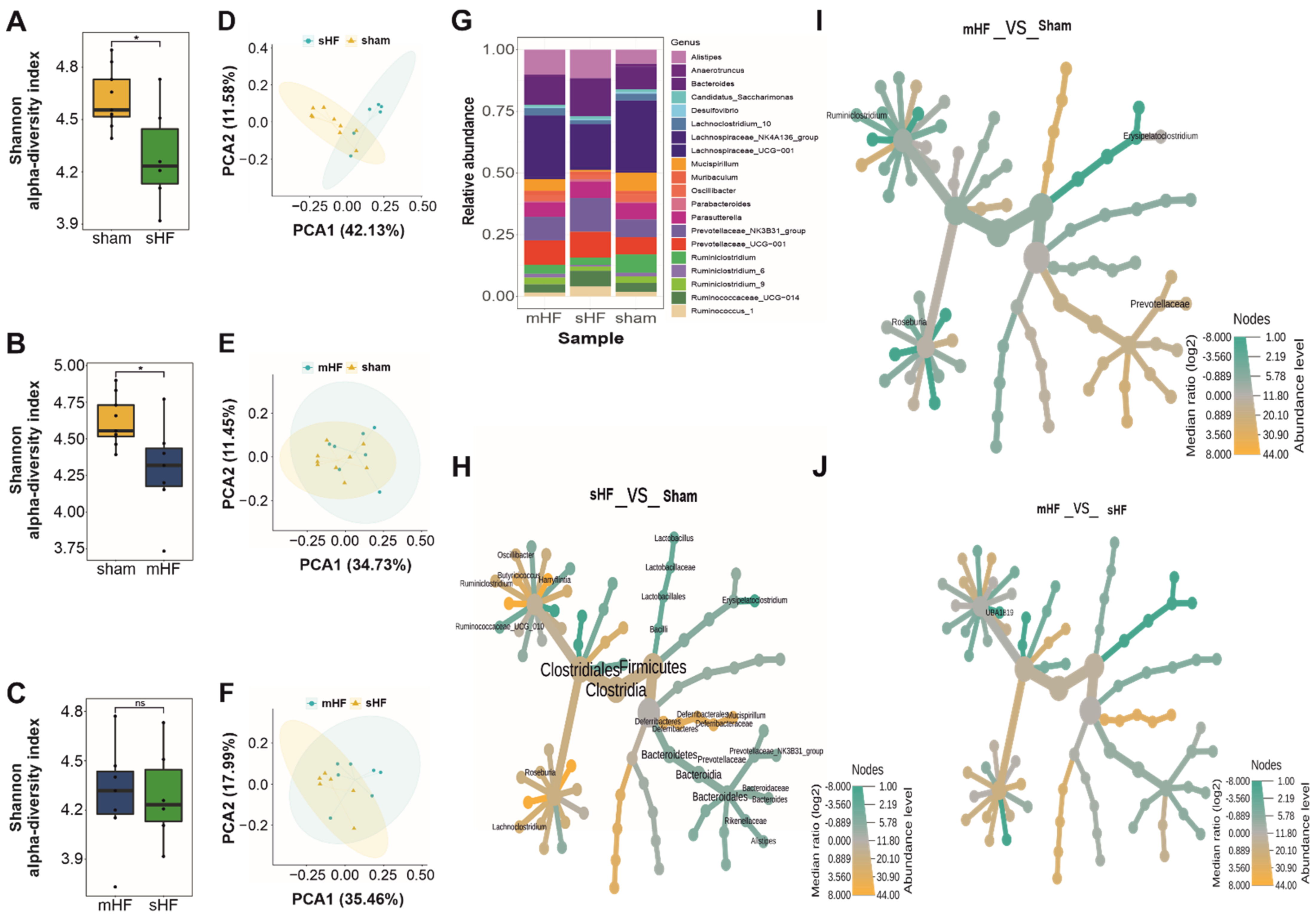
3.2. Severe HF Was Associated with Aggravated Alterations in Intestinal Microbiota Compared to Mild or Moderate HF
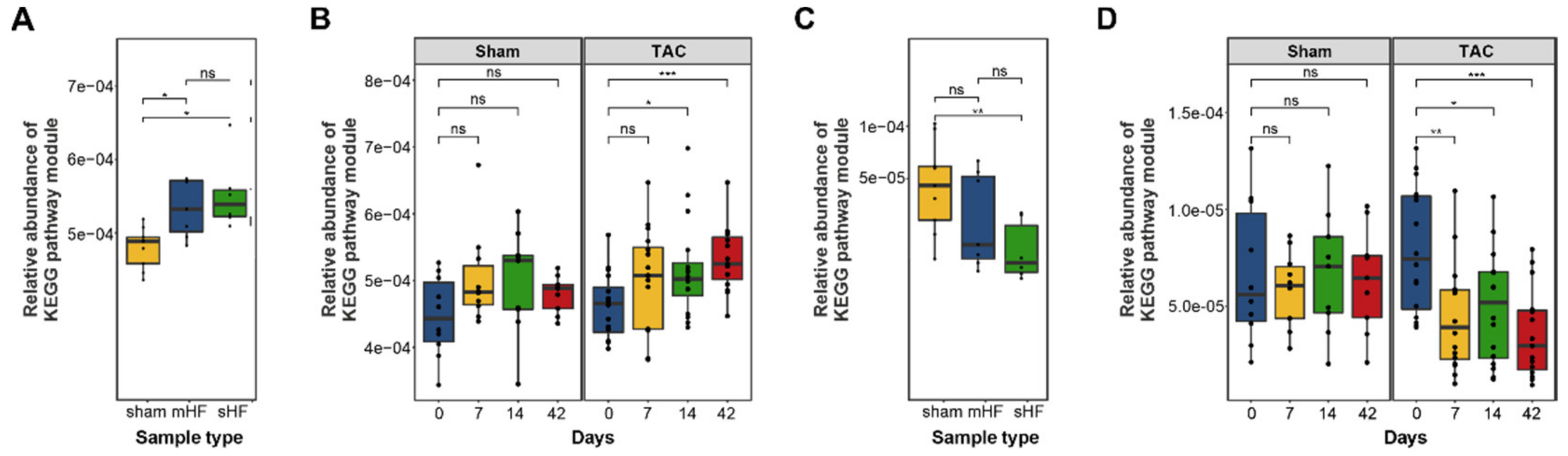
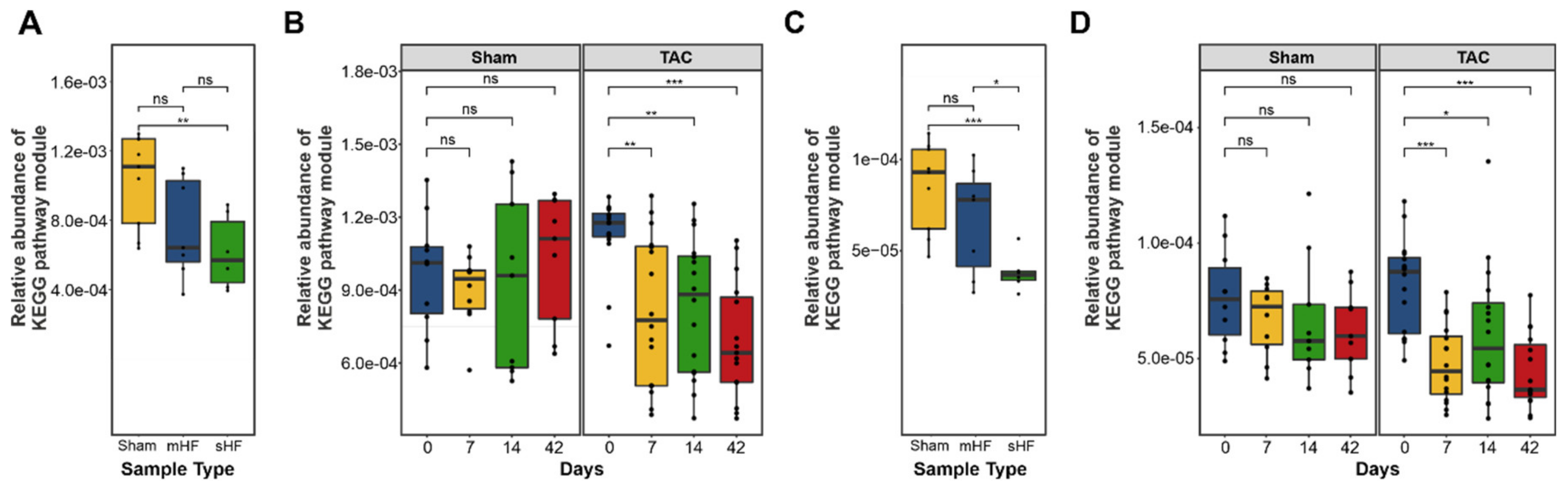
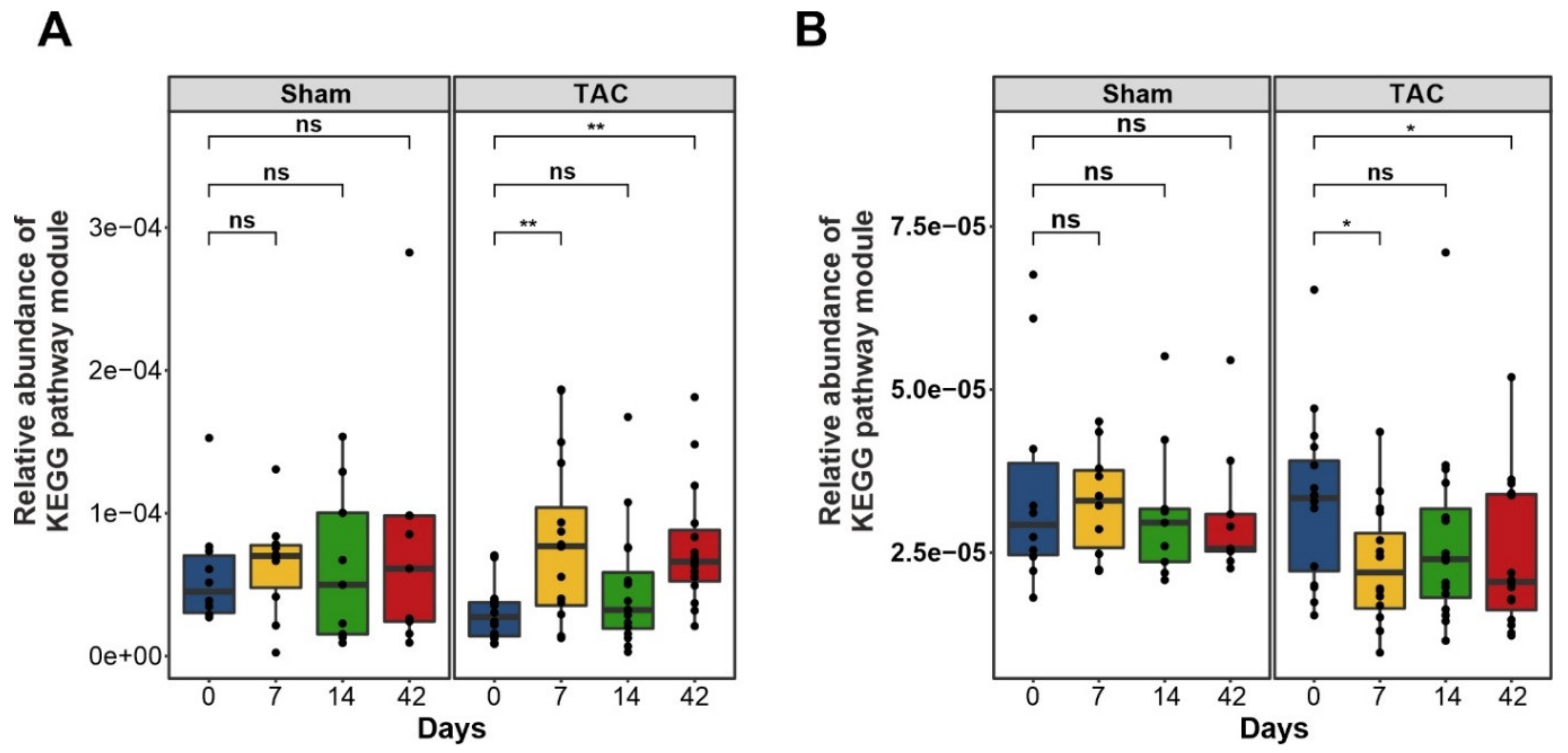
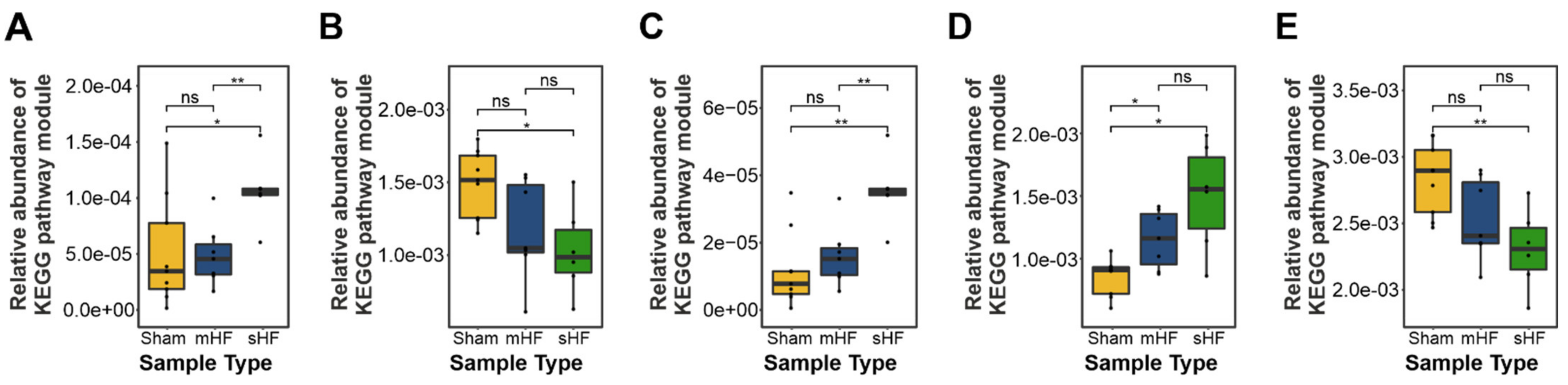
3.3. Functional Metabolic Analysis of the Gut Microbiome Revealed Altered Carbohydrate, Lipid, and Amino Acid Metabolism in Failing Hearts
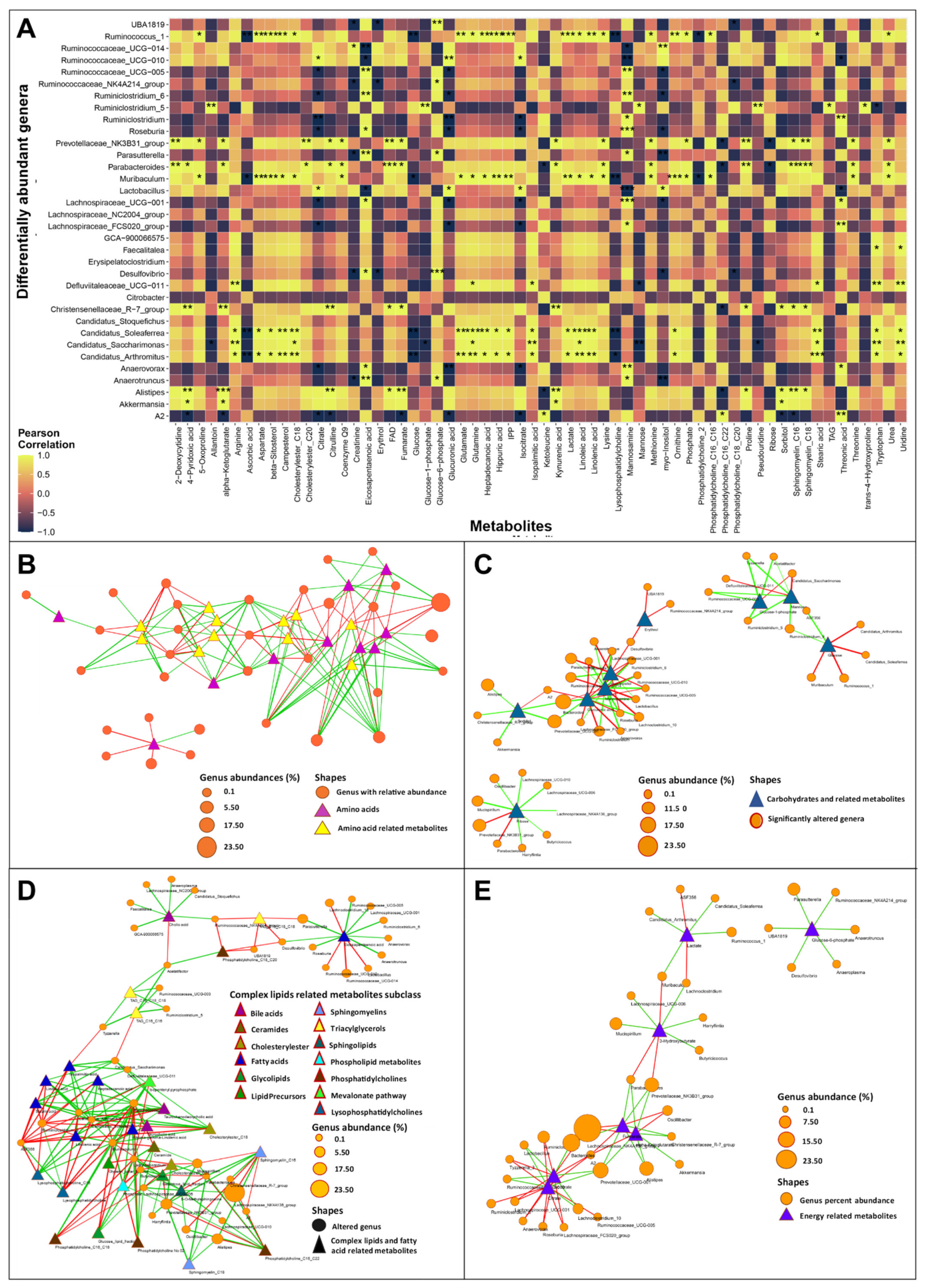
3.3.1. Bile Acid Metabolism
3.3.2. Short-Chain Fatty Acids Metabolism
3.3.3. TMA and TMAO Pathways
3.3.4. Amino Acid Metabolism
3.4. Gut Dysbiosis Caused Alterations in Circulating Metabolites
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peterson, J.; Garges, S.; Giovanni, M.; McInnes, P.; Wang, L.; Schloss, J.A.; Bonazzi, V.; McEwen, J.E.; Wetterstrand, K.A.; Deal, C.; et al. The NIH Human Microbiome Project. Genome Res. 2009, 19, 2317–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belbin, L.; Faith, D.P.; Milligan, G.W. A Comparison of Two Approaches to Beta-Flexible Clustering. Multivar. Behav. Res. 1992, 27, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.W.; Kitai, T.; Hazen, S.L. Gut Microbiota in Cardiovascular Health and Disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandek, A.; Bjarnason, I.; Volk, H.-D.; Crane, R.; Meddings, J.B.; Niebauer, J.; Kalra, P.R.; Buhner, S.; Herrmann, R.; Springer, J.; et al. Studies on bacterial endotoxin and intestinal absorption function in patients with chronic heart failure. Int. J. Cardiol. 2012, 157, 80–85. [Google Scholar] [CrossRef]
- Dick, S.A.; Epelman, S. Chronic Heart Failure and Inflammation. Circ. Res. 2016, 119, 159–176. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kalman, J.; Mayer, L.; Fillit, H.M.; Packer, M. Elevated Circulating Levels of Tumor Necrosis Factor in Severe Chronic Heart Failure. N. Engl. J. Med. 1990, 323, 236–241. [Google Scholar] [CrossRef]
- Luedde, M.; Winkler, T.; Heinsen, F.-A.; Rühlemann, M.C.; Spehlmann, M.E.; Bajrovic, A.; Lieb, W.; Franke, A.; Ott, S.J.; Frey, N. Heart failure is associated with depletion of core intestinal microbiota. ESC Heart Fail. 2017, 4, 282–290. [Google Scholar] [CrossRef]
- Cui, X.; Ye, L.; Li, J.; Jin, L.; Wang, W.; Li, S.; Bao, M.; Wu, S.; Li, L.; Geng, B.; et al. Metagenomic and metabolomic analyses unveil dysbiosis of gut microbiota in chronic heart failure patients. Sci. Rep. 2018, 8, 635. [Google Scholar] [CrossRef]
- Kamo, T.; Akazawa, H.; Suda, W.; Saga-Kamo, A.; Shimizu, Y.; Yagi, H.; Liu, Q.; Nomura, S.; Naito, A.T.; Takeda, N.; et al. Dysbiosis and compositional alterations with aging in the gut microbiota of patients with heart failure. PLoS ONE 2017, 12, e0174099. [Google Scholar] [CrossRef]
- Yuzefpolskaya, M.; Bohn, B.; Nasiri, M.; Zuver, A.M.; Onat, D.D.; Royzman, E.A.; Nwokocha, J.; Mabasa, M.; Pinsino, A.; Brunjes, D.; et al. Gut microbiota, endotoxemia, inflammation, and oxidative stress in patients with heart failure, left ventricular assist device, and transplant. J. Heart Lung Transplant. 2020, 39, 880–890. [Google Scholar] [CrossRef]
- Pasini, E.; Aquilani, R.; Testa, C.; Baiardi, P.; Angioletti, S.; Boschi, F.; Verri, M.; Dioguardi, F.S. Pathogenic Gut Flora in Patients With Chronic Heart Failure. JACC Heart Fail. 2016, 4, 220–227. [Google Scholar] [CrossRef]
- Beale, A.L.; O’Donnell, J.A.; Nakai, M.E.; Nanayakkara, S.; Vizi, D.; Carter, K.; Dean, E.; Ribeiro, R.V.; Yiallourou, S.; Carrington, M.J.; et al. The Gut Microbiome of Heart Failure With Preserved Ejection Fraction. J. Am. Heart Assoc. 2021, 10, e020654. [Google Scholar] [CrossRef]
- Huang, Z.; Mei, X.; Jiang, Y.; Chen, T.; Zhou, Y. Gut Microbiota in Heart Failure Patients with Preserved Ejection Fraction (GUMPTION Study). Front. Cardiovasc. Med. 2022, 8, 13. [Google Scholar] [CrossRef]
- Sun, W.; Du, D.; Fu, T.; Han, Y.; Li, P.; Ju, H. Alterations of the Gut Microbiota in Patients with Severe Chronic Heart Failure. Front. Microbiol. 2022, 12, 14. [Google Scholar] [CrossRef]
- Hayashi, T.; Yamashita, T.; Takahashi, T.; Tabata, T.; Watanabe, H.; Gotoh, Y.; Shinohara, M.; Kami, K.; Tanaka, H.; Matsumoto, K.; et al. Uncovering the Role of Gut Microbiota in Amino Acid Metabolic Disturbances in Heart Failure Through Metagenomic Analysis. Front. Cardiovasc. Med. 2021, 8, 789325. [Google Scholar] [CrossRef]
- Clavel, T.; Lagkouvardos, I.; Blaut, M.; Stecher, B. The mouse gut microbiome revisited: From complex diversity to model ecosystems. Int. J. Med. Microbiol. 2016, 306, 316–327. [Google Scholar] [CrossRef]
- Webb, C.R.; Koboziev, I.; Furr, K.L.; GriSham, M.B. Protective and pro-inflammatory roles of intestinal bacteria. Pathophysiology 2016, 23, 67–80. [Google Scholar] [CrossRef] [Green Version]
- Barin, J.G.; Talor, M.V.; Diny, N.L.; Ong, S.; Schaub, J.A.; Gebremariam, E.; Bedja, D.; Chen, G.; Choi, H.S.; Hou, X.; et al. Regulation of autoimmune myocarditis by host responses to the microbiome. Exp. Mol. Pathol. 2017, 103, 141–152. [Google Scholar] [CrossRef]
- Hu, X.-F.; Zhang, W.-Y.; Wen, Q.; Chen, W.-J.; Wang, Z.-M.; Chen, J.; Zhu, F.; Liu, K.; Cheng, L.-X.; Yang, J.; et al. Fecal microbiota transplantation alleviates myocardial damage in myocarditis by restoring the microbiota composition. Pharmacol. Res. 2019, 139, 412–421. [Google Scholar] [CrossRef]
- Kuhn, C.; Frank, D.; Dierck, F.; Oehl, U.; Krebs, J.; Will, R.; Lehmann, L.H.; Backs, J.; Katus, H.A.; Frey, N. Cardiac remodeling is not modulated by overexpression of muscle LIM protein (MLP). Basic Res. Cardiol. 2012, 107, 262. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Müller, O.J.; Heckmann, M.B.; Ding, L.; Rapti, K.; Rangrez, A.Y.; Gerken, T.; Christiansen, N.; Rennefahrt, U.E.E.; Witt, H.; Maldonado, S.G.; et al. Comprehensive plasma and tissue profiling reveals systemic metabolic alterations in cardiac hypertrophy and failure. Cardiovasc. Res. 2019, 115, 1296–1305. [Google Scholar] [CrossRef]
- Helmering, J.; Juan, T.; Li, C.M.; Chhoa, M.; Baron, W.; Gyuris, T.; Richards, W.G.; Turk, J.R.; Lawrence, J.; Cosgrove, P.A.; et al. A mutation in Ampd2 is associated with nephrotic syndrome and hypercholesterolemia in mice. Lipids Health Dis. 2014, 13, 167. [Google Scholar] [CrossRef] [Green Version]
- Van Ravenzwaay, B.; Cunha, G.C.-P.; Leibold, E.; Looser, R.; Mellert, W.; Prokoudine, A.; Walk, T.; Wiemer, J. The use of metabolomics for the discovery of new biomarkers of effect. Toxicol. Lett. 2007, 172, 21–28. [Google Scholar] [CrossRef]
- Bordag, N.; Klie, S.; Jürchott, K.; Vierheller, J.; Schiewe, H.; Albrecht, V.; Tonn, J.-C.; Schwartz, C.; Schichor, C.; Selbig, J. Glucocorticoid (dexamethasone)-induced metabolome changes in healthy males suggest prediction of response and side effects. Sci. Rep. 2015, 5, 15954. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.; Wang, Q.; Cole, J.R.; Rosen, G.L. Using the RDP Classifier to Predict Taxonomic Novelty and Reduce the Search Space for Finding Novel Organisms. PLoS ONE 2012, 7, e32491. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Vegan: Community Ecology Package. R Package Version 2.2-0. 2014. Available online: http://CRAN.Rproject.org/package=vegan (accessed on 10 July 2021).
- Lahti, L.; Shetty, S. (2012–2019) “Microbiome R Package”. Available online: https://www.bioconductor.org/ (accessed on 10 July 2021).
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PIC-RUSt2: An improved and customizable approach for metagenome inference. BioRxiv 2019, 672295. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, D.A. Biochemistry of tryptophan in health and disease. Mol. Asp. Med. 1983, 6, 101–197. [Google Scholar] [CrossRef]
- Liu, G.; Chen, S.; Zhong, J.; Teng, K.; Yin, Y. Crosstalk between Tryptophan Metabolism and Cardiovascular Disease, Mechanisms, and Therapeutic Implications. Oxidative Med. Cell. Longev. 2017, 2017, 1602074. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.; Nordrehaug, J.E.; Slettom, G.; Solvang, S.-E.H.; Pedersen, E.K.R.; Midttun, Ø.; Ulvik, A.; Ueland, P.M.; Nygård, O.; Giil, L.M. Plasma kynurenines and prognosis in patients with heart failure. PLoS ONE 2020, 15, e0227365. [Google Scholar] [CrossRef]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef] [Green Version]
- Leustean, A.M.; Ciocoiu, M.; Sava, A.; Costea, C.F.; Floria, M.; Tarniceriu, C.C.; Tanase, D.M. Implications of the Intestinal Microbiota in Diagnosing the Progression of Diabetes and the Presence of Cardiovascular Complications. J. Diabetes Res. 2018, 2018, 5205126. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.L.; Davidson, M.T.; Kurishima, C.; Vega, R.B.; Powers, J.C.; Matsuura, T.R.; Petucci, C.; Lewandowski, E.D.; Crawford, P.A.; Muoio, D.M.; et al. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. JCI Insight 2019, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Campbell, R.B.P.; Duffourc, M.M.; Schoborg, R.V.; Xu, Y.; Liu, X.; KenKnight, B.H.; Beaumont, E. Aberrant fecal flora observed in guinea pigs with pressure overload is mitigated in animals receiving vagus nerve stimulation therapy. Am. J. Physiol. Liver Physiol. 2016, 311, G754–G762. [Google Scholar] [CrossRef] [Green Version]
- Zheng, A.; Yi, H.; Li, F.; Han, L.; Yu, J.; Cheng, X.; Su, H.; Hong, K.; Li, J. Changes in Gut Microbiome Structure and Function of Rats with Isoproterenol-Induced Heart Failure. Int. Heart J. 2019, 60, 1176–1183. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Huang, X.; Tong, Y.; Gao, H. Butyrate improves cardiac function and sympathetic neural remodeling following myocardial infarction in rats. Can. J. Physiol. Pharmacol. 2020, 98, 391–399. [Google Scholar] [CrossRef]
- Zhang, L.; Deng, M.; Lu, A.; Chen, Y.; Chen, Y.; Wu, C.; Tan, Z.; Boini, K.M.; Yang, T.; Zhu, Q.; et al. Sodium butyrate attenuates angiotensin II-induced cardiac hypertrophy by inhibiting COX2/PGE2 pathway via a HDAC5/HDAC6-dependent mechanism. J. Cell. Mol. Med. 2019, 23, 8139–8150. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Liu, F.; Lu, J.; Shi, J.; Guan, J.; Yan, F.; Li, B.; Huo, G. Probiotic Mixture of Lactobacillus plantarum Strains Improves Lipid Metabolism and Gut Microbiota Structure in High Fat Diet-Fed Mice. Front. Microbiol. 2020, 11, 512. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wu, T.; Li, N.; Wang, X.; Chen, G.; Lyu, X. Bilberry anthocyanin extract promotes intestinal barrier function and inhibits digestive enzyme activity by regulating the gut microbiota in aging rats. Food Funct. 2019, 10, 333–343. [Google Scholar] [CrossRef]
- Liu, Y.-W.; Liong, M.-T.; Tsai, Y.-C. New perspectives of Lactobacillus plantarum as a probiotic: The gut-heart-brain axis. J. Microbiol. 2018, 56, 601–613. [Google Scholar] [CrossRef]
- Koppinger, M.P.; Lopez-Pier, M.A.; Skaria, R.; Harris, P.R.; Konhilas, J.P. Lactobacillus reuteri attenuates cardiac injury without lowering cholesterol in low-density lipoprotein receptor-deficient mice fed standard chow. Am. J. Physiol. Circ. Physiol. 2020, 319, H32–H41. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, Z.; Zhang, H.; Summah, B.S.; Liu, H.; An, D.; Zhan, Q.; Lai, W.; Zeng, Q.; Ren, H.; et al. Ferulic acid increases intestinal Lactobacillus and improves cardiac function in TAC mice. Biomed. Pharmacother. 2019, 120, 109482. [Google Scholar] [CrossRef]
- Abu-Elsaad, N.M.; Elhameed, A.G.A.; El-Karef, A.; Ibrahim, T.M. Yogurt Containing the ProbacteriaLactobacillus acidophilusCombined with Natural Antioxidants Mitigates Doxorubicin-Induced Cardiomyopathy in Rats. J. Med. Food 2015, 18, 950–959. [Google Scholar] [CrossRef]
- Urbańska, E.M.; Luchowski, P.; Luchowska, E.; Pniewski, J.; Woźniak, R.; Chodakowska-Zebrowska, M.; Lazarewicz, J. Serum kynurenic acid positively correlates with cardiovascular disease risk factor, homocysteine: A study in stroke patients. Pharmacol. Rep. 2006, 58, 507–511. [Google Scholar]
- Moradi, H.; Sica, D.A.; Kalantar-Zadeh, K. Cardiovascular Burden Associated with Uremic Toxins in Patients with Chronic Kidney Disease. Am. J. Nephrol. 2013, 38, 136–148. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.W.; Li, D.Y.; Hazen, S.L. Dietary metabolism, the gut microbiome, and heart failure. Nat. Rev. Cardiol. 2019, 16, 137–154. [Google Scholar] [CrossRef]
- Mayerhofer, C.C.; Awoyemi, A.O.; Moscavitch, S.D.; Lappegård, K.T.; Hov, J.R.; Aukrust, P.; Hovland, A.; Lorenzo, A.; Halvorsen, S.; Seljeflot, I.; et al. Design of the GutHeart-targeting gut microbiota to treat heart failure-trial: A Phase II, randomized clinical trial. ESC Heart Fail. 2018, 5, 977–984. [Google Scholar] [CrossRef] [Green Version]
- Awoyemi, A.; Mayerhofer, C.; Felix, A.S.; Hov, J.R.; Moscavitch, S.D.; Lappegård, K.T.; Hovland, A.; Halvorsen, S.; Halvorsen, B.; Gregersen, I.; et al. Rifaximin or Saccharomyces boulardii in heart failure with reduced ejection fraction: Results from the randomized GutHeart trial. EBioMedicine 2021, 70, 103511. [Google Scholar] [CrossRef]
- Mouse Genome Sequencing Consortium. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The Effect of Diet on the Human Gut Microbiome: A Metagenomic Analysis in Humanized Gnotobiotic Mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [Green Version]
- Krych, L.; Hansen, C.H.F.; Hansen, A.K.; Berg, F.W.J.V.D.; Nielsen, D.S. Quantitatively Different, yet Qualitatively Alike: A Meta-Analysis of the Mouse Core Gut Microbiome with a View towards the Human Gut Microbiome. PLoS ONE 2013, 8, e62578. [Google Scholar] [CrossRef] [Green Version]
- Zmora, N.; Suez, J.; Elinav, E. You are what you eat: Diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 35–56. [Google Scholar] [CrossRef]
- Hugenholtz, F.; De Vos, W.M. Mouse models for human intestinal microbiota research: A critical evaluation. Cell. Mol. Life Sci. 2018, 75, 149–160. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BRITE Hierarchy Pathway Module (Level 2) | p-Value—mHF vs. sHF vs. Sham | p-Value—Sham vs. TAC |
|---|---|---|
| Environmental adaptation | ** | * |
| Protein families: metabolism | ** | ns |
| Biosynthesis of other secondary metabolites | * | ns |
| Cellular community—prokaryotes | * | * |
| Unclassified: genetic information processing | * | ns |
| Cell growth and death | * | ns |
| Digestive system | * | ns |
| Metabolism of terpenoids and polyketides | * | ns |
| Transcription | * | * |
| Carbohydrate metabolism | * | * |
| Membrane transport | * | * |
| Metabolism of other amino acids | * | ns |
| Unclassified: metabolism | * | * |
| Nucleotide metabolism | * | ns |
| Lipid metabolism | * | ns |
| Glycan biosynthesis and metabolism | * | ns |
| Signal transduction | * | ns |
| Protein families: signaling and cellular processes | * | ns |
| Cell motility | * | ns |
| Energy metabolism | * | ns |
| Protein families: genetic information processing | * | ns |
| Unclassified: signaling and cellular processes | * | ns |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spehlmann, M.E.; Rangrez, A.Y.; Dhotre, D.P.; Schmiedel, N.; Chavan, N.; Bang, C.; Müller, O.J.; Shouche, Y.S.; Franke, A.; Frank, D.; et al. Heart Failure Severity Closely Correlates with Intestinal Dysbiosis and Subsequent Metabolomic Alterations. Biomedicines 2022, 10, 809. https://doi.org/10.3390/biomedicines10040809
Spehlmann ME, Rangrez AY, Dhotre DP, Schmiedel N, Chavan N, Bang C, Müller OJ, Shouche YS, Franke A, Frank D, et al. Heart Failure Severity Closely Correlates with Intestinal Dysbiosis and Subsequent Metabolomic Alterations. Biomedicines. 2022; 10(4):809. https://doi.org/10.3390/biomedicines10040809
Chicago/Turabian StyleSpehlmann, Martina E., Ashraf Y. Rangrez, Dhiraj P. Dhotre, Nesrin Schmiedel, Nikita Chavan, Corinna Bang, Oliver J. Müller, Yogesh S. Shouche, Andre Franke, Derk Frank, and et al. 2022. "Heart Failure Severity Closely Correlates with Intestinal Dysbiosis and Subsequent Metabolomic Alterations" Biomedicines 10, no. 4: 809. https://doi.org/10.3390/biomedicines10040809
APA StyleSpehlmann, M. E., Rangrez, A. Y., Dhotre, D. P., Schmiedel, N., Chavan, N., Bang, C., Müller, O. J., Shouche, Y. S., Franke, A., Frank, D., & Frey, N. (2022). Heart Failure Severity Closely Correlates with Intestinal Dysbiosis and Subsequent Metabolomic Alterations. Biomedicines, 10(4), 809. https://doi.org/10.3390/biomedicines10040809