
Targeting Src-Hic-5 Signal Cascade for Preventing Migration of Cholangiocarcinoma Cell HuCCT1

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of CCA Tissues
2.2. Cell Culture
2.3. Antibodies and Chemicals
2.4. RNA Interference
2.5. Proliferation Assay
2.6. Wound Healing Migration Assay
2.7. Transwell Migration Assay
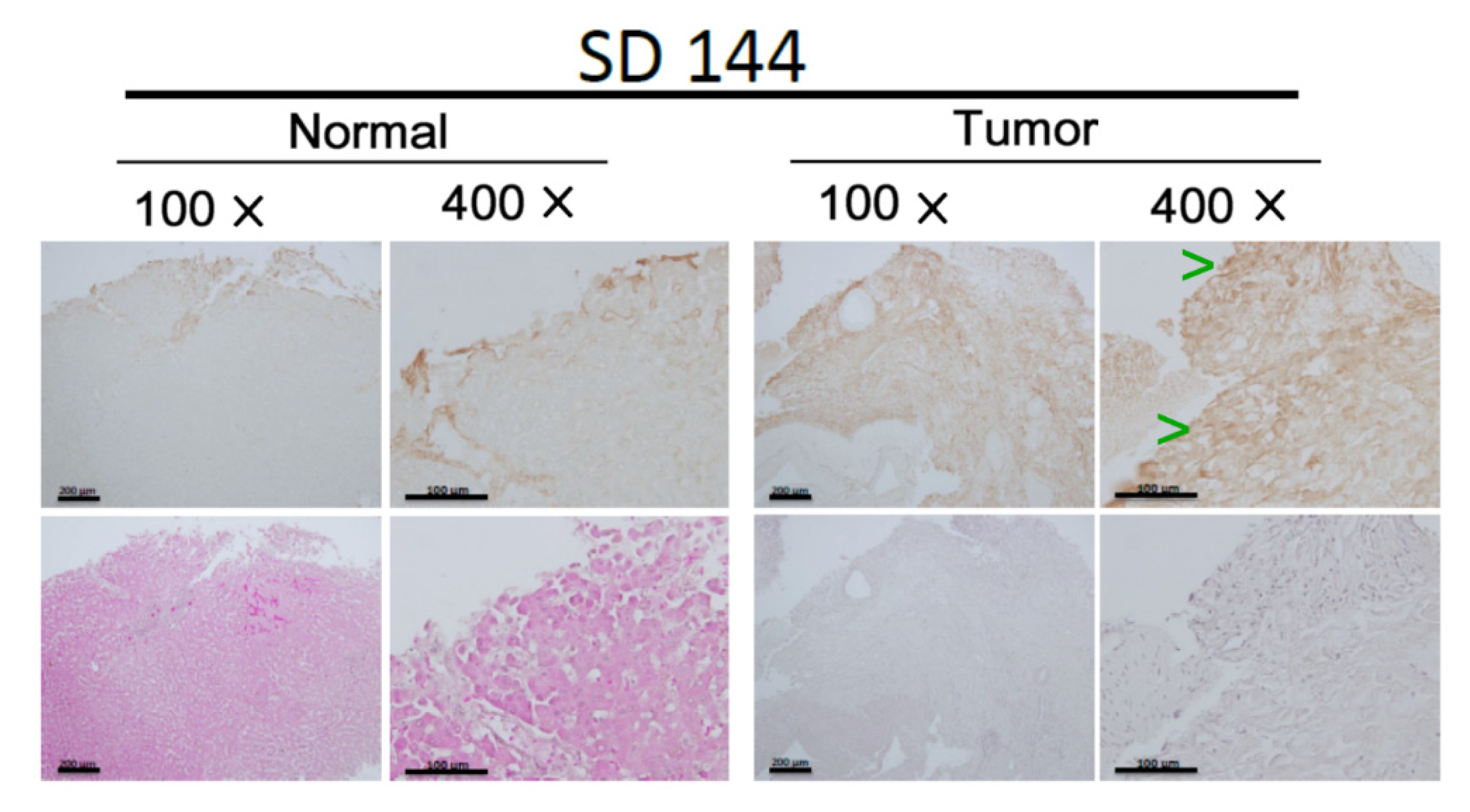
2.8. Immunohistochemistry (IHC)
2.9. Statistical Analysis
3. Results
3.1. Screening the Enhanced Signal Molecules Associated with CCA Metastasis
3.2. Cross Talk between Hic-5 and Src for Activating AKT in HuccT1
3.3. Depleting Hic-5 Expression and Inhibiting Src Activity Suppress HuccT1 Migration in a Concerted Manner
4. Discussion
4.1. The Role of Focal Adhesion Signaling Molecules Responsible for Cell Migration in Tumor Metastasis
4.2. Targeting Hic-5 and Src Is Promising in Preventing CCA Progression
4.3. Combined Therapy Is More Effective in Blocking Metastatic Signaling
5. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bergquist, A.; von Seth, E. Epidemiology of cholangiocarcinoma. Best Pract. Res. Clin. Gastroenterol. 2015, 29, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Zhu, A.X.; Fuchs, C.S.; Brooks, G.A. Forty-Year Trends in Cholangiocarcinoma Incidence in the U.S.: Intrahepatic Disease on the Rise. Oncologist 2016, 21, 594–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma-evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Simile, M.M.; Bagella, P.; Vidili, G.; Spanu, A.; Manetti, R.; Seddaiu, M.A.; Babudieri, S.; Madeddu, G.; Serra, P.A.; Altana, M.; et al. Targeted Therapies in Cholangiocarcinoma: Emerging Evidence from Clinical Trials. Medicina 2019, 55, 42. [Google Scholar] [CrossRef] [Green Version]
- Sia, D.; Tovar, V.; Moeini, A.; Llovet, J.M. Intrahepatic cholangiocarcinoma: Pathogenesis and rationale for molecular therapies. Oncogene 2013, 32, 4861–4870. [Google Scholar] [CrossRef] [Green Version]
- Sirica, A.E. Role of ErbB family receptor tyrosine kinases in intrahepatic cholangiocarcinoma. World J. Gastroenterol. 2008, 14, 7033–7058. [Google Scholar] [CrossRef] [Green Version]
- Sia, D.; Losic, B.; Moeini, A.; Cabellos, L.; Hao, K.; Revill, K.; Bonal, D.; Miltiadous, O.; Zhang, Z.; Hoshida, Y.; et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat. Commun. 2015, 6, 6087. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanian, B.; Venkatraman, S.; Janvilisri, T.; Suthiphongchai, T.; Jitkaew, S.; Sripa, J.; Tohtong, R. RTK25: A Comprehensive Molecular Profiling Strategy in Cholangiocarcinoma Using an Integrated Bioinformatics Approach. Pharmaceuticals 2021, 14, 898. [Google Scholar] [CrossRef]
- Jin, W. ErBb Family Proteins in Cholangiocarcinoma and Clinical Implications. J. Clin. Med. 2020, 9, 2255. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Peiris, M.N.; Donoghue, D.J. Functions of FGFR2 corrupted by translocations in intrahepatic cholangiocarcinoma. Cytokine Growth Factor Rev. 2020, 52, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Lu, D.; Chen, R.; Lin, Y.; Zhu, H.; Zhang, Z.; Cai, S.; Cui, P.; Song, G.; Rao, D.; et al. Proteogenomic characterization identifies clinically relevant subgroups of intrahepatic cholangiocarcinoma. Cancer Cell 2022, 40, 70–87 e15. [Google Scholar] [CrossRef] [PubMed]
- Smoot, R.L.; Werneburg, N.W.; Sugihara, T.; Hernandez, M.C.; Yang, L.; Mehner, C.; Graham, R.P.; Bronk, S.F.; Truty, M.J.; Gores, G.J. Platelet-derived growth factor regulates YAP transcriptional activity via Src family kinase dependent tyrosine phosphorylation. J. Cell Biochem. 2018, 119, 824–836. [Google Scholar] [CrossRef] [PubMed]
- Correnti, M.; Cappon, A.; Pastore, M.; Piombanti, B.; Lori, G.; Oliveira, D.; Munoz-Garrido, P.; Lewinska, M.; Andersen, J.B.; Coulouarn, C.; et al. The protease-inhibitor SerpinB3 as a critical modulator of the stem-like subset in human cholangiocarcinoma. Liver Int. 2022, 42, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Lepore, A.; Choy, P.M.; Lee, N.C.W.; Carella, M.A.; Favicchio, R.; Briones-Orta, M.A.; Glaser, S.S.; Alpini, G.; D’Santos, C.; Tooze, R.M.; et al. Phosphorylation and Stabilization of PIN1 by JNK Promote Intrahepatic Cholangiocarcinoma Growth. Hepatology 2021, 74, 2561–2579. [Google Scholar] [CrossRef]
- Jaidee, R.; Kukongviriyapan, V.; Senggunprai, L.; Prawan, A.; Jusakul, A.; Laphanuwat, P.; Kongpetch, S. Inhibition of FGFR2 enhances chemosensitivity to gemcitabine in cholangiocarcinoma through the AKT/mTOR and EMT signaling pathways. Life Sci. 2022, 296, 120427. [Google Scholar] [CrossRef]
- Deng, S.; Zhang, L.; Li, J.; Jin, Y.; Wang, J. Activation of the PI3K-AKT signaling pathway by SPARC contributes to the malignant phenotype of cholangiocarcinoma cells. Tissue Cell 2022, 76, 101756. [Google Scholar] [CrossRef]
- Zhou, S.; Qu, K.L.; Li, J.A.; Chen, S.L.; Zhang, Y.G.; Zhu, C.; Jin, H.; Wang, Y.; Pang, Q.; Liu, H.C. YY1 activates EMI2 and promotes the progression of cholangiocarcinoma through the PI3K/Akt signaling axis. Cancer Cell Int. 2021, 21, 699. [Google Scholar] [CrossRef]
- Liang, S.; Guo, H.; Ma, K.; Li, X.; Wu, D.; Wang, Y.; Wang, W.; Zhang, S.; Cui, Y.; Liu, Y.; et al. A PLCB1-PI3K-AKT Signaling Axis Activates EMT to Promote Cholangiocarcinoma Progression. Cancer Res. 2021, 81, 5889–5903. [Google Scholar] [CrossRef]
- Wu, J.R.; Hu, C.T.; You, R.I.; Pan, S.M.; Cheng, C.C.; Lee, M.C.; Wu, C.C.; Chang, Y.J.; Lin, S.C.; Chen, C.S.; et al. Hydrogen peroxide inducible clone-5 mediates reactive oxygen species signaling for hepatocellular carcinoma progression. Oncotarget 2015, 6, 32526–32544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.R.; You, R.I.; Hu, C.T.; Cheng, C.C.; Rudy, R.; Wu, W.S. Hydrogen peroxide inducible clone-5 sustains NADPH oxidase-dependent reactive oxygen species-c-jun N-terminal kinase signaling in hepatocellular carcinoma. Oncogenesis 2019, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.S. The role of hydrogen peroxide-inducible clone-5 in tumor progression. Ci Ji Yi Xue Za Zhi 2020, 32, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, J.; Tumbarello, D.A.; Schmidt, R.P.; Turner, C.E. Hic-5 promotes invadopodia formation and invasion during TGF-beta-induced epithelial-mesenchymal transition. J. Cell Biol. 2012, 197, 421–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Li, X.; Song, N.; Li, A.; Hou, K.; Qu, X.; Che, X.; Liu, Y. Src promotes EGF-induced epithelial-to-mesenchymal transition and migration in gastric cancer cells by upregulating ZEB1 and ZEB2 through AKT. Cell Biol. Int. 2018, 42, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Montell, D.J. Border-cell migration: The race is on. Nat. Rev. Mol. Cell Biol. 2003, 4, 13–24. [Google Scholar] [CrossRef]
- Guo, W.; Giancotti, F.G. Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 2004, 5, 816–826. [Google Scholar] [CrossRef]
- Paul, C.D.; Mistriotis, P.; Konstantopoulos, K. Cancer cell motility: Lessons from migration in confined spaces. Nat. Rev. Cancer 2017, 17, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Majidpoor, J.; Mortezaee, K. Steps in metastasis: An updated review. Med. Oncol. 2021, 38, 3. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Isaji, T.; Zhang, G.; Qi, F.; Duan, C.; Fukuda, T.; Gu, J. EpCAM associates with integrin and regulates cell adhesion in cancer cells. Biochem. Biophys. Res. Commun. 2020, 522, 903–909. [Google Scholar] [CrossRef] [PubMed]
- de Boer, C.J.; van Krieken, J.H.; Janssen-van Rhijn, C.M.; Litvinov, S.V. Expression of Ep-CAM in normal, regenerating, metaplastic, and neoplastic liver. J. Pathol. 1999, 188, 201–206. [Google Scholar] [CrossRef]
- Pongchairerk, U.; Guan, J.L.; Leardkamolkarn, V. Focal adhesion kinase and Src phosphorylations in HGF-induced proliferation and invasion of human cholangiocarcinoma cell line, HuCCA-1. World J. Gastroenterol. 2005, 11, 5845–5852. [Google Scholar] [CrossRef] [PubMed]
- Mon, N.N.; Hasegawa, H.; Thant, A.A.; Huang, P.; Tanimura, Y.; Senga, T.; Hamaguchi, M. A role for focal adhesion kinase signaling in tumor necrosis factor-alpha-dependent matrix metalloproteinase-9 production in a cholangiocarcinoma cell line, CCKS1. Cancer Res. 2006, 66, 6778–6784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Xu, H.; Wang, P.; Wang, J.; Affo, S.; Wang, H.; Xu, M.; Liang, B.; Che, L.; Qiu, W.; et al. Focal adhesion kinase (FAK) promotes cholangiocarcinoma development and progression via YAP activation. J. Hepatol. 2021, 75, 888–899. [Google Scholar] [CrossRef]
- Nishiya, N.; Tachibana, K.; Shibanuma, M.; Mashimo, J.I.; Nose, K. Hic-5-reduced cell spreading on fibronectin: Competitive effects between paxillin and Hic-5 through interaction with focal adhesion kinase. Mol. Cell Biol. 2001, 21, 5332–5345. [Google Scholar] [CrossRef] [Green Version]
- Colon-Bolea, P.; Garcia-Gomez, R.; Casar, B. RAC1 Activation as a Potential Therapeutic Option in Metastatic Cutaneous Melanoma. Biomolecules 2021, 11, 1554. [Google Scholar] [CrossRef]
- Xiong, J.; Yan, L.; Zou, C.; Wang, K.; Chen, M.; Xu, B.; Zhou, Z.; Zhang, D. Integrins regulate stemness in solid tumor: An emerging therapeutic target. J. Hematol. Oncol. 2021, 14, 177. [Google Scholar] [CrossRef]
- Rosenzweig, S.A. Acquired Resistance to Drugs Targeting Tyrosine Kinases. Adv. Cancer Res. 2018, 138, 71–98. [Google Scholar] [CrossRef]
- Phuchareon, J.; McCormick, F.; Eisele, D.W.; Tetsu, O. EGFR inhibition evokes innate drug resistance in lung cancer cells by preventing Akt activity and thus inactivating Ets-1 function. Proc. Natl. Acad. Sci. USA 2015, 112, E3855–E3863. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, S.; Colombero, C.; Panelo, L.; Dakarapu, R.; Falck, J.R.; Costas, M.A.; Nowicki, S. GPR75 receptor mediates 20-HETE-signaling and metastatic features of androgen-insensitive prostate cancer cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158573. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.T.; Wu, J.R.; Cheng, C.C.; Wu, W.S. The Therapeutic Targeting of HGF/c-Met Signaling in Hepatocellular Carcinoma: Alternative Approaches. Cancers 2017, 9, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omoto, T.; Kim-Kaneyama, J.R.; Lei, X.F.; Orimo, A.; Ohnishi, K.; Yoshihara, K.; Miyauchi, A.; Li, S.; Gao, L.; Umemoto, T.; et al. The impact of stromal Hic-5 on the tumorigenesis of colorectal cancer through lysyl oxidase induction and stromal remodeling. Oncogene 2018, 37, 1205–1219. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, S.; Lyu, H.; Riker, A.I.; Zhang, Y.; Liu, B. Development of Effective Therapeutics Targeting HER3 for Cancer Treatment. Biol. Proced. Online 2019, 21, 5. [Google Scholar] [CrossRef] [Green Version]
- Girotti, M.R.; Lopes, F.; Preece, N.; Niculescu-Duvaz, D.; Zambon, A.; Davies, L.; Whittaker, S.; Saturno, G.; Viros, A.; Pedersen, M.; et al. Paradox-breaking RAF inhibitors that also target SRC are effective in drug-resistant BRAF mutant melanoma. Cancer Cell 2015, 27, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Qiu, Y.; Zhu, L.; Zhao, Y.; Lin, Y.; Ma, D.; Qin, Z.; Sun, C.; Shen, X.; Li, T.; et al. NOD1 inhibits proliferation and enhances response to chemotherapy via suppressing SRC-MAPK pathway in hepatocellular carcinoma. J. Mol. Med. 2020, 98, 221–232. [Google Scholar] [CrossRef]
- Sugihara, T.; Werneburg, N.W.; Hernandez, M.C.; Yang, L.; Kabashima, A.; Hirsova, P.; Yohanathan, L.; Sosa, C.; Truty, M.J.; Vasmatzis, G.; et al. YAP Tyrosine Phosphorylation and Nuclear Localization in Cholangiocarcinoma Cells Are Regulated by LCK and Independent of LATS Activity. Mol. Cancer Res. 2018, 16, 1556–1567. [Google Scholar] [CrossRef] [Green Version]
- Regan-Fendt, K.; Li, D.; Reyes, R.; Yu, L.; Wani, N.A.; Hu, P.; Jacob, S.T.; Ghoshal, K.; Payne, P.R.O.; Motiwala, T. Transcriptomics-Based Drug Repurposing Approach Identifies Novel Drugs against Sorafenib-Resistant Hepatocellular Carcinoma. Cancers 2020, 12, 2730. [Google Scholar] [CrossRef]
- Qian, B.; Wei, L.; Yang, Z.; He, Q.; Chen, H.; Wang, A.; Yang, D.; Li, Q.; Li, J.; Zheng, S.; et al. Hic-5 in pancreatic stellate cells affects proliferation, apoptosis, migration, invasion of pancreatic cancer cells and postoperative survival time of pancreatic cancer. Biomed. Pharmacother. 2020, 121, 109355. [Google Scholar] [CrossRef]
- Sun, C.; Jing, W.; Xiong, G.; Ma, D.; Lin, Y.; Lv, X.; Zhao, Y.; Ma, X.; Zhu, L.; Shen, X.; et al. Inhibiting Src-mediated PARP1 tyrosine phosphorylation confers synthetic lethality to PARP1 inhibition in HCC. Cancer Lett 2022, 526, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Hoque Apu, E.; Akram, S.U.; Rissanen, J.; Wan, H.; Salo, T. Desmoglein 3-Influence on oral carcinoma cell migration and invasion. Exp. Cell Res. 2018, 370, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Salo, T.; Sutinen, M.; Hoque Apu, E.; Sundquist, E.; Cervigne, N.K.; de Oliveira, C.E.; Akram, S.U.; Ohlmeier, S.; Suomi, F.; Eklund, L.; et al. A novel human leiomyoma tissue derived matrix for cell culture studies. BMC Cancer 2015, 15, 981. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metastatic Potential | ||||
|---|---|---|---|---|
| Metastatic Index | +++ a | ++ | + | 0 |
| Lympho/vascular invasion | ∨ b | ∨ | ∨ | - |
| Perineural invasion | ∨ | - | - | - |
| Regional lymph nodes involved | M c > 10 | 5 < M < 10 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, W.-S.; Ling, C.-H.; Lee, M.-C.; Cheng, C.-C.; Chen, R.-F.; Lin, C.-F.; You, R.-I.; Chen, Y.-C. Targeting Src-Hic-5 Signal Cascade for Preventing Migration of Cholangiocarcinoma Cell HuCCT1. Biomedicines 2022, 10, 1022. https://doi.org/10.3390/biomedicines10051022
Wu W-S, Ling C-H, Lee M-C, Cheng C-C, Chen R-F, Lin C-F, You R-I, Chen Y-C. Targeting Src-Hic-5 Signal Cascade for Preventing Migration of Cholangiocarcinoma Cell HuCCT1. Biomedicines. 2022; 10(5):1022. https://doi.org/10.3390/biomedicines10051022
Chicago/Turabian StyleWu, Wen-Sheng, Chin-Hsien Ling, Ming-Che Lee, Chuan-Chu Cheng, Rui-Fang Chen, Chen-Fang Lin, Ren-In You, and Yen-Cheng Chen. 2022. "Targeting Src-Hic-5 Signal Cascade for Preventing Migration of Cholangiocarcinoma Cell HuCCT1" Biomedicines 10, no. 5: 1022. https://doi.org/10.3390/biomedicines10051022
APA StyleWu, W. -S., Ling, C. -H., Lee, M. -C., Cheng, C. -C., Chen, R. -F., Lin, C. -F., You, R. -I., & Chen, Y. -C. (2022). Targeting Src-Hic-5 Signal Cascade for Preventing Migration of Cholangiocarcinoma Cell HuCCT1. Biomedicines, 10(5), 1022. https://doi.org/10.3390/biomedicines10051022