
Role of Oxidative Stress in Cardiac Dysfunction and Subcellular Defects Due to Ischemia-Reperfusion Injury


Abstract
:1. Introduction
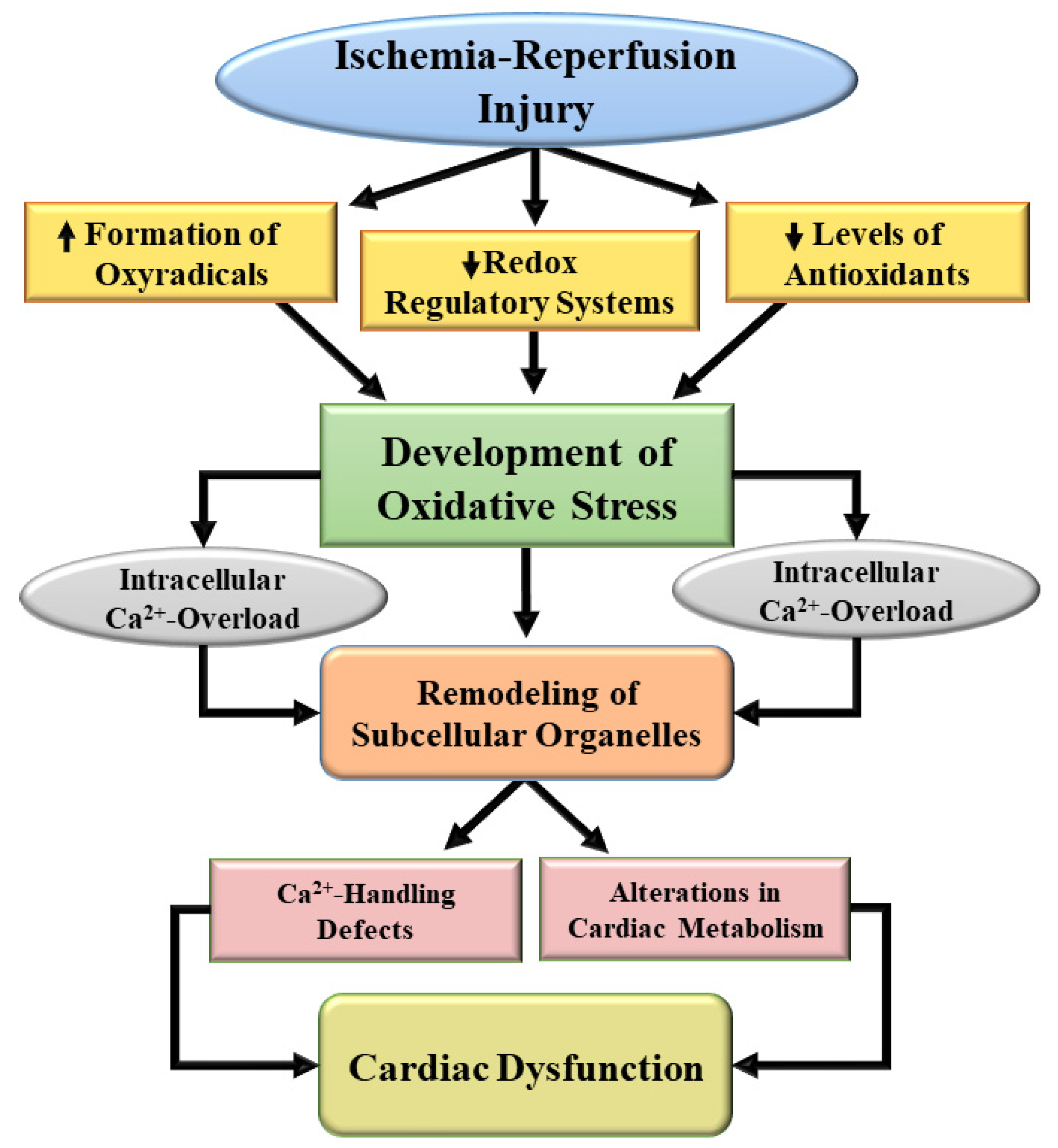
2. I/R-Induced Generation of Oxidative Stress and Its Implication in Heart Disease
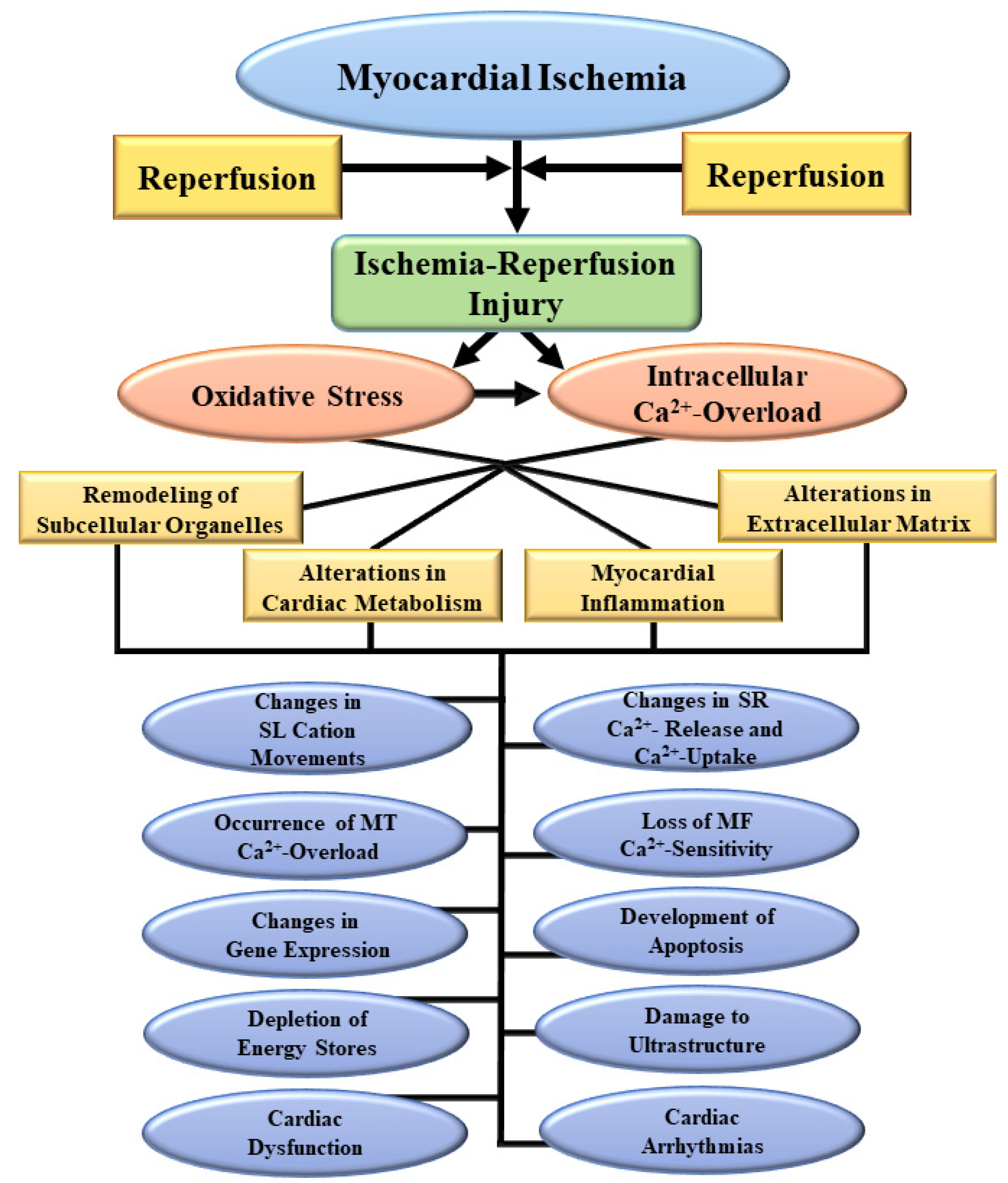
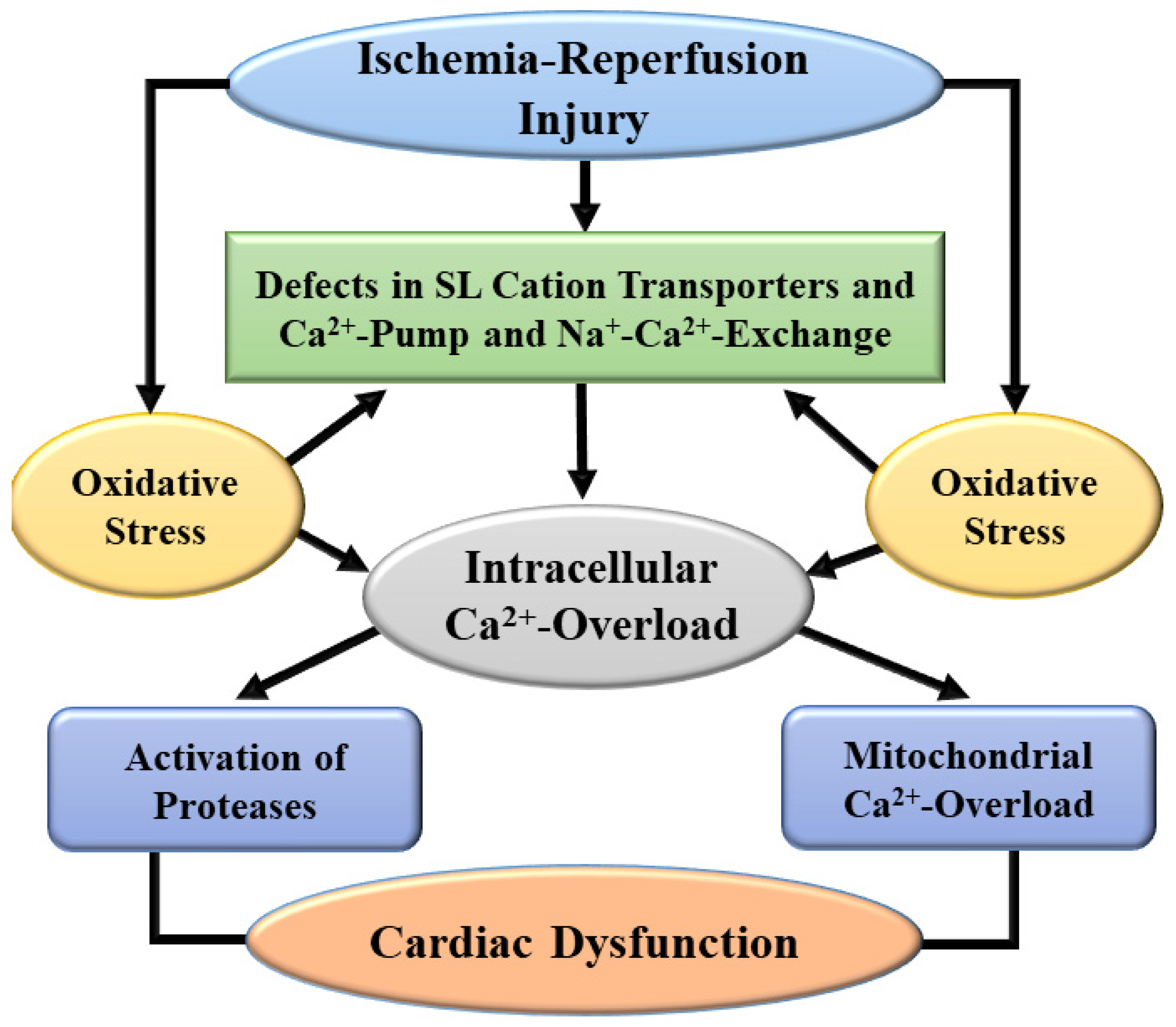
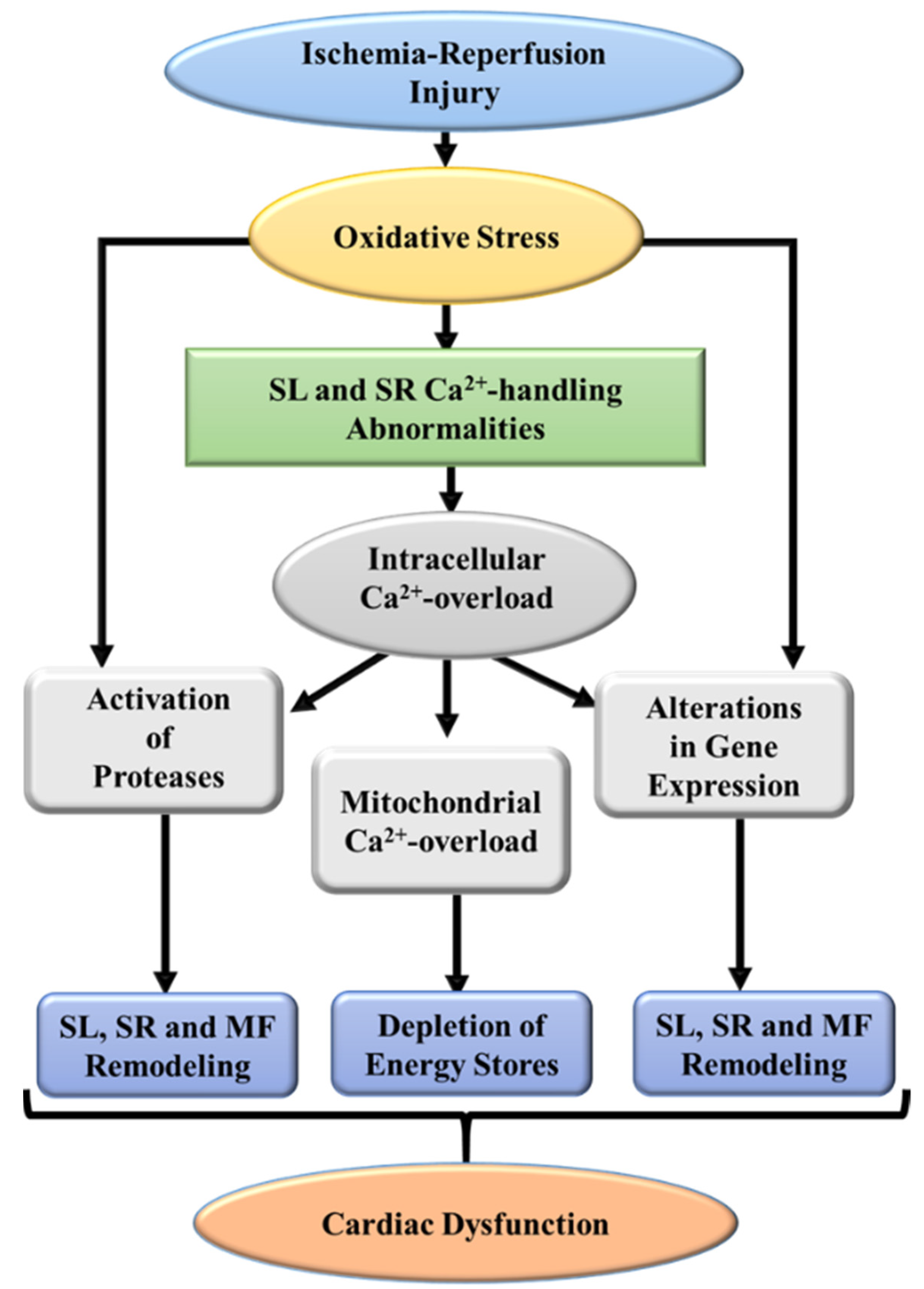
3. Pathophysiological Aspects of I/R-Induced Injury
4. Cardioprotection in Hearts Subjected to I/R Injury
5. Pharmacotherapy of I/R Injury to the Heart
6. Evidence for the Role of Oxidative Stress in I/R-Induced Cardiac Dysfunction and Subcellular Defects
7. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoffman, J.W.; Gilbert, T.B.; Poston, R.S.; Silldorff, E.P. Myocardial reperfusion injury: Etiology, mechanisms, and therapies. J. Extra Corpor. Technol. 2004, 36, 391–411. [Google Scholar] [PubMed]
- Dhalla, N.S.; Elmoselhi, A.B.; Hata, T.; Makino, N. Status of myocardial antioxidants in ischemia-reperfusion injury. Cardiovasc. Res. 2000, 47, 446–456. [Google Scholar] [CrossRef]
- Neuzil, J.; Rayner, B.S.; Lowe, H.C.; Witting, P.K. Oxidative stress in myocardial ischemia reperfusion injury: A renewed focus on a long-standing area of heart research. Redox Rep. 2005, 10, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Milei, J.; Grana, D.R.; Forcada, P.; Ambrosio, G. Mitochondrial oxidative and structural damage in ischemia-reperfusion in human myocardium. Current knowledge and future directions. Front. Biosci. 2007, 12, 1124–1130. [Google Scholar] [CrossRef] [Green Version]
- Dhalla, N.S.; Saini, H.K.; Tappia, P.S.; Sethi, R.; Mengi, S.A.; Gupta, S.K. Potential role and mechanisms of subcellular remodeling in cardiac dysfunction due to ischemic heart disease. J. Cardiovasc. Med. 2007, 8, 238–250. [Google Scholar] [CrossRef]
- Rodrigo, R.; Libuy, M.; Feliu, F.; Hasson, D. Oxidative stress-related biomarkers in essential hypertension and ischemia-reperfusion myocardial damage. Dis. Markers 2013, 35, 773–790. [Google Scholar] [CrossRef] [Green Version]
- Neri, M.; Riezzo, I.; Pascale, N.; Pomara, C.; Turillazzi, E. Ischemia/reperfusion injury following acute myocardial infarction: A critical issue for clinicians and forensic pathologists. Mediators Inflamm. 2017, 2017, 7018393. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Chuang, C.C.; Zuo, L. Molecular characterization of reactive oxygen species in myocardial ischemia-reperfusion injury. Biomed. Res. Int. 2015, 2015, 864946. [Google Scholar] [CrossRef] [Green Version]
- Kurian, G.A.; Rajagopal, R.; Vedantham, S.; Rajesh, M. The role of oxidative stress in myocardial ischemia and reperfusion injury and remodeling: Revisited. Oxidative Med. Cell. Longev. 2016, 2016, 165450. [Google Scholar] [CrossRef] [Green Version]
- Xiang, M.; Lu, Y.; Xin, L.; Gao, J.; Shang, C.; Jiang, Z.; Lin, H.; Fang, X.; Qu, Y.; Wang, Y.; et al. Role of oxidative stress in reperfusion following myocardial ischemia and its treatments. Oxidative Med. Cell. Longev. 2021, 2021, 6614009. [Google Scholar] [CrossRef]
- Bugger, H.; Pfeil, K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165768. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.L.; Theruvath, T.P.; Zhong, Z.; Nieminen, A.L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta 2009, 1787, 1395–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, H.K.; Machackova, J.; Dhalla, N.S. Role of reactive oxygen species in ischemic preconditioning of subcellular organelles in the heart. Antioxid. Redox Signal. 2004, 6, 393–404. [Google Scholar] [CrossRef]
- Steffens, S.; Montecucco, F.; Mach, F. The inflammatory response as a target to reduce myocardial ischemia and reperfusion injury. Thromb. Haemost. 2009, 102, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Monassier, J.P. Reperfusion injury in acute myocardial infarction. From bench to cath lab. Part I: Basic considerations. Arch. Cardiovasc. Dis. 2008, 101, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.K.; Bhullar, S.K.; Elimban, V.; Dhalla, N.S. Oxidative stress as a mechanism for functional alterations in cadiac hypertrophy and heart failure. Antioxidants 2021, 10, 931. [Google Scholar] [CrossRef]
- Bhullar, S.K.; Shah, A.K.; Dhalla, N.S. Role of angiotensin II in the development of subcellular remodeling in heart failure. Explor. Med. 2021, 2, 352–371. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Bhullar, S.K.; Shah, A.K. Future scope and challenges for congestive heart failure: Moving towards development of pharmacotherapy. Can. J. Physiol. Pharmacol. 2022. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Elimban, V.; Shah, A.K.; Nusier, M. Mechanisms of cardiac dysfunction in heart failure due to myocardial infarction. J. Integr. Cardiol. Open Access 2019, 2, 3–7. [Google Scholar] [CrossRef] [Green Version]
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000, 18, 655–673. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Elimban, V.; Bartekova, M.; Adameova, A. Involvement of oxidative stress in the development of subcellular defects and heart disease. Biomedicines 2022, 10, 393. [Google Scholar] [CrossRef] [PubMed]
- Saini, H.K.; Xu, Y.J.; Zhang, M.; Liu, P.P.; Kirshenbaum, L.A.; Dhalla, N.S. Role of tumor necrosis factor-alpha and other cytokines in ischemia-reperfusion-induced injury in the heart. Exp. Clin. Cardiol. 2005, 10, 213–222. [Google Scholar] [PubMed]
- Ambrosio, G.; Zweier, J.L.; Flaherty, J.T. The relationship between oxygen radical generation and impairment of myocardial energy metabolism following post-ischemic reperfusion. J. Mol. Cell. Cardiol. 1991, 23, 1359–1374. [Google Scholar] [CrossRef]
- Sinning, C.; Westermann, D.; Clemmensen, P. Oxidative stress in ischemia and reperfusion: Current concepts, novel ideas and future perspectives. Biomark. Med. 2017, 11, 11031–11040. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Matsui, Y.; Sadoshima, J. The role of autophagy in mediating cell survival and death during ischemia and reperfusion in the heart. Antioxid. Redox Signal. 2007, 9, 1373–1381. [Google Scholar] [CrossRef]
- Muller, A.L.; Hryshko, L.V.; Dhalla, N.S. Extracellular and intracellular proteases in cardiac dysfunction due to ischemia-reperfusion injury. Int. J. Cardiol. 2013, 164, 39–47. [Google Scholar] [CrossRef]
- Marin, W.; Marin, D.; Ao, X.; Liu, Y. Mitochondria as a therapeutic target for cardiac ischemia-reperfusion injury (review). Int. J. Mol. Med. 2021, 47, 485–499. [Google Scholar] [CrossRef]
- Ostadal, P.; Elmoselhi, A.B.; Zdobnicka, I.; Lukas, A.; Elimban, V.; Dhalla, N.S. Role of oxidative stress in ischemia-reperfusion-induced changes in Na+, K+-ATPase isoform expression in rat heart. Antioxid. Redox Signal. 2004, 6, 914–923. [Google Scholar] [CrossRef]
- Netticadan, T.; Temsah, R.; Osada, M.; Dhalla, N.S. Status of Ca2+/calmodulin protein kinase phosphorylation of cardiac SR proteins in ischemia-reperfusion. Am. J. Physiol. 1999, 277, C384–C391. [Google Scholar] [CrossRef]
- Marczin, N.; El-Habashi, N.; Hoare, G.S.; Bundy, R.E.; Yacoub, M. Antioxidants in myocardial ischemia-reperfusion injury: Therapeutic potential and basic mechanisms. Arch. Biochem. Biophys. 2003, 420, 222–236. [Google Scholar] [CrossRef]
- Bartekova, M.; Barancik, M.; Ferenczyova, K.; Dhalla, N.S. Beneficial effects of N-acetylcysteine and N–N-mercaptopropionylglycine on Ischemia reperfusion injury in the heart. Curr. Med. Chem. 2018, 25, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Bartekova, M.; Adameova, A.; Gorbe, A.; Ferenczyova, K.; Pechanova, O.; Lazou, A.; Dhalla, N.S.; Ferdinandy, P.; Giricz, Z. Natural and synthetic antioxidants targeting cardiac oxidative stress and redox signaling in cardiometabolic diseases. Free Radic. Biol. Med. 2021, 169, 446–477. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative stress in cardiovascular diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef] [PubMed]
- Kleikers, P.W.M.; Wingler, K.; Hermans, J.J.R.; Diebold, I.; Altenhofer, S.; Radermacher, K.A.; Janssen, B.; Gorlach, A.; Schmidt, H.H.H.W. NADPH oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. J. Mol. Med. 2012, 90, 1391–1406. [Google Scholar] [CrossRef]
- Ferrari, R.; Ceconi, C.; Curello, S.; Guarnieri, C.; Caldarera, C.M.; Albertini, A.; Visioli, O. Oxygen-mediated myocardial damage during ischemia and reperfusion: Role of the cellular defenses against oxygen toxicity. J. Mol. Cell. Cardiol. 1985, 17, 937–945. [Google Scholar] [CrossRef]
- Arduini, A.; Mezzetti, A.; Porreca, E.; Lapenna, D.; DeJulia, J.; Marzio, L.; Polidoro, G.; Cuccurullo, F. Effect of ischemia and reperfusion on antioxidant enzymes and mitochondrial inner membrane proteins in perfused rat heart. Biochim. Biophys. Acta 1988, 970, 113–121. [Google Scholar] [CrossRef]
- Padmavathi, G.; Ramkumar, K.M. MicroRNA mediated regulation of the major redox homeostasis switch, Nrf2, and its impact on oxidative stress-induced ischemic/reperfusion injury. Arch. Biochem. Biophys. 2021, 698, 108725. [Google Scholar] [CrossRef]
- Haramaki, N.; Stewart, D.B.; Aggarwal, S.; Ikeda, H.; Reznick, A.Z.; Packer, L. Networking antioxidants in the isolated rat heart are selectively depleted by ischemia-reperfusion. Free. Radic. Biol. Med. 1998, 25, 329–339. [Google Scholar] [CrossRef]
- Chen, Y.R.; Zweier, J.L. Cardiac mitochondria reactive oxygen species generation. Circ. Res. 2014, 114, 524–537. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. Understanding and preventing mitochondrial oxidative damage. Biochem. Soc. Trans. 2016, 44, 1219–1226. [Google Scholar] [CrossRef]
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Dambrova, M.; Zuurbier, C.J.; Borutaite, V.; Liepinsh, E.; Makrecka-Kuka, M. Energy substrate metabolism and mitochondrial oxidative stress in cardiac ischemia/reperfusion injury. Free Radic. Biol. Med. 2021, 165, 24–37. [Google Scholar] [CrossRef]
- Liu, P.; Hock, C.E.; Nagele, R.; Wong, P.Y.K. Formation of nitric oxide, superoxide, and peroxynitrite in myocardial ischemia-reperfusion injury in rats. Am. J. Physiol. 1997, 272, H2327–H2336. [Google Scholar] [CrossRef] [PubMed]
- Anatoliotakis, N.; Defteros, S.; Bouras, G.; Giannopoulos, G.; Tsounis, D.; Angelidis, C.; Kaoukis, A.; Stefanadis, C. Myeloperoxidase: Expressing inflammation and oxidative stress in cardiovascular disease. Curr. Top. Med. Chem. 2013, 13, 115–138. [Google Scholar] [CrossRef] [PubMed]
- Kaluderic, N.; Carpi, A.; Menabo, R.; Di Lisa, F.; Paolocci, N. Monoamine oxidase (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim. Biophys. Acta 2011, 1813, 1323–1332. [Google Scholar] [CrossRef] [Green Version]
- Mialet-Perez, J.; Santin, Y.; Parini, A. Monoamine oxidase-A, serotonin and norepinephrine: Synergistic players in cardiac physiology and pathology. J. Neural Transm. 2018, 125, 1627–1634. [Google Scholar] [CrossRef]
- Harrison, D.G.; Cai, H.; Landmesser, U.; Griendling, K.K. Interactions of angiotensin II with NADPH oxidase, oxidant stress and cardiovascular stress. J. Renin Angiotensin Aldosterone Syst. 2003, 4, 51–61. [Google Scholar] [CrossRef]
- Kaminski, K.A.; Bonda, T.A.; Korecki, J.; Musial, W.J. Oxidative stress and neutrophil activation—The two keystones of ischemia/reperfusion injury. Int. J. Cardiol. 2002, 86, 41–59. [Google Scholar] [CrossRef]
- Li, B.; Tian, J.; Sun, Y.; Xu, T.R.; Chi, R.F.; Zhang, X.L.; Hu, X.L.; Zhang, Y.A.; Qin, F.Z.; Zhang, W.F. Activation of NADPH oxidase mediates increased endoplasmic reticulum stress and left ventricular remodeling after myocardial infarction. Biochim. Biophys. Acta 2015, 1852, 805–815. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Xuan, Y.T.; Gu, Y.; Prabhu, S.D. Prolonged oxidative stress inverts the cardiac force-frequency relation: Role of altered calcium handling and myofilament calcium responsiveness. J. Mol. Cell. Cardiol. 2006, 40, 64–75. [Google Scholar] [CrossRef]
- Sharma, G.P.; Varley, K.G.; Kim, S.W.; Barwinsky, J.; Cohen, M.; Dhalla, N.S. Alterations in energy metabolism and ultrastructure upon reperfusion of the ischemic myocardium after coronary occlusion. Am. J. Cardiol. 1975, 36, 234–243. [Google Scholar] [CrossRef]
- Nayler, W.G.; Panagiotopoulos, S.; Elz, J.S.; Daly, M.J. Calcium-mediated damage during post-ischemic reperfusion. J. Mol. Cell. Cardiol. 1988, 20 Suppl 2, 41–54. [Google Scholar] [CrossRef]
- Murphy, J.G.; Smith, T.W.; Marsh, J.D. Mechanisms of reoxygenation-induced calcium overload in cultured chick embryo heart cells. Am. J. Physiol. 1988, 254, H1133–H1141. [Google Scholar] [CrossRef] [PubMed]
- Jennings, R.B.; Reimer, K.A. The cell biology of acute myocardial ischemia. Annu. Rev. Med. 1991, 42, 225–246. [Google Scholar] [CrossRef] [PubMed]
- Bolli, R.; Marban, E. Molecular and cellular mechanisms of myocardial stunning. Physiol. Rev. 1999, 79, 609–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piper, M.H.; Meuter, K.; Schafer, C. Cellular mechanisms of ischemia-reperfusion injury. Ann. Thorac. Surg. 2003, 75, S644–S648. [Google Scholar] [CrossRef]
- Bhosale, G.; Sharpe, J.A.; Sundier, S.; Duchen, M. Calcium signaling as mediator of cell energy demand and a trigger to cell death. Ann. N. Y. Acad. Sci. 2015, 1350, 107–116. [Google Scholar] [CrossRef]
- Badalzadeh, R.; Mokhtari, B.; Yavari, R. Contribution of apoptosis in myocardial reperfusion injury and loss of cardioprotection in diabetes mellitus. J. Physiol. Sci. 2015, 65, 201–215. [Google Scholar] [CrossRef]
- Ferrari, R.; Guardigli, G.; Mele, D.; Percoco, G.F.; Ceconi, C.; Curello, S. Oxidative stress during myocardial ischemia and heart failure. Curr. Pharm. Des. 2004, 10, 1699–1711. [Google Scholar] [CrossRef]
- Neri, M.; Fineschi, V.; Di Paolo, M.; Pomara, C.; Riezzo, I.; Turillazzi, E.; Cerretani, D. Cardiac oxidative stress and inflammation cytokines response after myocardial infarction. Curr. Vasc. Pharmacol. 2015, 13, 26–36. [Google Scholar] [CrossRef]
- Li, X.; Zhang, F.; Zhou, H.; Hu, Y.; Guo, D.; Fang, X.; Chen, Y. Interplay of TNF-α, soluble TNF receptors and oxidative stress in coronary chronic total occlusion of the oldest patients with coronary heart disease. Cytokine 2020, 125, 154836. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Qu, Y.; Chen, H.; Qian, J. Insight into long noncoding RNA-miRNA-mRNA axes in myocardial ischemia-reperfusion injury: The implications for mechanism and therapy. Epigenomics 2019, 11, 1733–1748. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Sun, W.; Guo, Z.; Liu, B.; Yu, H.; Zhang, J. Long noncoding RNAs in myocardial ischemia-reperfusion inury. Oxidative Med. Cell. Longev. 2021, 2021, 8889123. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, Z.; Fan, Z.; Yang, Y.; Lu, C. Involvement of non-coding RNAs in the pathogenesis of myocardial ischemia/reperfusion injury (review). Intl. J. Mol. Med. 2021, 47, 42. [Google Scholar] [CrossRef]
- Wang, B.F.; Yoshioka, J. The emerging role of thioredoxin-interacting protein in myocardial ischemia-reperfusion injury. J. Cardiovasc. Pharmacol. Ther. 2017, 22, 219–229. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; He, M.; Ni, L.; He, K.; Su, K.; Deng, Y.; Li, Y.; Xia, H. The role of arachidonic acid metabolism in myocardial ischemia-reperfusion injury. Cell Biochem. Biophys. 2020, 78, 255–265. [Google Scholar] [CrossRef]
- Laude, K.; Richard, V.; Thuillez, C. Coronary endothelial cells: A target of ischemia reperfusion and its treatment? Arch. Mal. Coeur. Vaiss. 2004, 97, 250–254. [Google Scholar]
- Rohrbach, S.; Troidl, C.; Hamm, C.; Schulz, R. Ischemia and reperfusion related myocardial inflammation: A network of cells and mediators targeting the cardiomyocyte. IUBMB Life 2015, 67, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Gunata, M.; Parlakpinar, H. A review of myocardial ischemia/reperfusion injury: Pathophysiology, experimental models, biomarkers, genetics and pharmacological treatment. Cell. Biochem. Func. 2021, 39, 190–217. [Google Scholar] [CrossRef]
- Schanze, N.; Bode, C.; Duerschmied, D. Platelet contributions to myocardial ischemia/reperfusion injury. Front. Immunol. 2019, 10, 1260. [Google Scholar] [CrossRef] [Green Version]
- Sadek, H.A.; Nulton-Persson, A.C.; Szweda, P.A.; Szweda, L.I. Cardiac ischemia/reperfusion, aging, and redox-dependent alterations in mitochondrial function. Arch. Biochem. Biophys. 2003, 420, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Vercesi, A.E.; Kowaltowski, A.J.; Oliveira, H.C.F.; Castilho, R.F. Mitchondrial Ca2+ transport, permeability transition and oxidative stress in cell death: Implications in cardiotoxicity, neurodegeneration and dyslipidemias. Front. Biosci. 2006, 11, 2554–2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halestrap, A.P. Mitochondria and reperfusion injury of the heart—A holey death but not beyond salvation. J. Bioenerg, Biomembr. 2009, 41, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Shi, D.; Guo, M. The roles of PKC-δ and PKC-ε in myocardial ischemia/reperfusion injury. Pharmacol. Res. 2021, 170, 105716. [Google Scholar] [CrossRef]
- Ruan, Y.; Zeng, J.; Jin, Q.; Chu, M.; Ji, K.; Wang, Z.; Li, L. Endoplasmic reticulum stress serves an important role in cardiac ischemia/reperfusion injury (review). Exp. Ther. Med. 2020, 20, 268. [Google Scholar] [CrossRef]
- Zima, A.V.; Mazurek, S.R. Functional impact of ryanodine receptor oxidation on intracellular calcium regulation in the heart. Rev. Physiol. Biochem. Pharmacol. 2016, 171, 39–62. [Google Scholar] [CrossRef]
- Frohlich, G.M.; Meier, P.; White, S.K.; Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury: Looking beyond primary PCI. Eur. Heart J. 2013, 34, 1714–1722. [Google Scholar] [CrossRef] [Green Version]
- Pagliaro, P.; Penna, C. Redox signalling and cardioprotection: Translatability and mechanism. Br. J. Pharmacol. 2015, 172, 1974–1995. [Google Scholar] [CrossRef] [Green Version]
- Daiber, A.; Andreadou, I.; Oelze, M.; Davidson, S.M.; Hausenloy, D.J. Discovery of new therapeutic redox targets for cardioprotection against ischemia/reperfusion injury and heart failure. Free Radic. Biol. Med. 2021, 163, 325–343. [Google Scholar] [CrossRef]
- Tappia, P.S.; Shah, A.K.; Ramjiawan, B.; Dhalla, N.S. Modification of ischemia/reperfusion-induced alterations in subcellular organelles by ischemic preconditioning. Int. J. Mol. Sci. 2022, 23, 3425. [Google Scholar] [CrossRef]
- Sanada, S.; Komuro, I.; Kitakaze, M. Pathophysiology of myocardial reperfusion injury: Preconditioning, postconditioning, and translational aspects of protective measures. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1723–H1741. [Google Scholar] [CrossRef] [Green Version]
- Ferdinandy, P.; Hausenloy, D.J.; Heusch, G.; Baxter, G.F.; Schulz, R. Interaction of risk factors, comorbidties, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol. Rev. 2014, 66, 1142–1174. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, N.; Oka, S.; Sadoshima, J. Modulation of signaling mechanisms in the heart by thioredoxin 1. Free Radic. Biol. Med. 2017, 109, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.L.; Ho, Y.C.; Yet, S.F. A central role of heme oxygenase-1 in cardiovascular protection. Antioxid. Redox Signal. 2011, 15, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.B.; Chen, X.; Zhou, X.Y.; Wang, X.B. The role of peroxisome proliferator-activated receptor γ in mediating cardioprotection against ischemia/reperfusion injury. J. Cardiovasc. Pharmacol. Ther. 2018, 23, 46–56. [Google Scholar] [CrossRef]
- Wang, W.L.; Ge, T.Y.; Chen, X.; Mao, Y.; Zhu, Y.Z. Advances in the protective mechanism of NO, H2S, and H2 in myocardial ischemic injury. Front. Cardiovasc. Med. 2020, 7, 588206. [Google Scholar] [CrossRef] [PubMed]
- Dongo, E.; Hornyak, I.; Benko, Z.S.; Kiss, L. The cardioprotective potential of hydrogen sulfide in myocardial ischemia/reperfusion injury (review). Acta Physiol. Hung 2011, 98, 369–381. [Google Scholar] [CrossRef]
- Chohan, P.K.; Singh, R.B.; Dhalla, N.S.; Netticadan, T. L-arginine administration recovers sarcoplasmic reticulum function in ischemic reperfused hearts by preventing calpain activation. Cardiovasc. Res. 2006, 69, 152–163. [Google Scholar] [CrossRef] [Green Version]
- Jensen, R.V.; Andreadou, I.; Hausenloy, D.J.; Botker, H.E. The role of O-GlcNAcylation for protection against ischemia-reperfusion injury. Int. J. Mol. Sci. 2019, 20, 404. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Chen, P.; Zhong, J.; Cheng, Y.; Chen, H.; He, Y.; Chen, C. HIF-1α in myocardial ischemia-reperfusion injury (review). Mol. Med. Rep. 2021, 23, 352. [Google Scholar] [CrossRef]
- Ding, J.; Yang, Z.; Ma, H.; Zhang, H. Mitochondrial aldehyde dehydrogenase in myocardial ischemic and ischemia-reperfusion injury. Adv. Exp. Med. Biol. 2019, 1193, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Zhang, H.; Shengshou, H. Mitochondrial aldehyde dehydrogenase 2 activation and cardioprotection. J. Mol. Cell. Cardiol. 2013, 55, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, T.; Gomes, A.V. MicroRNAs in the regulation of cellular redox status and its implications in myocardial ischemia-reperfusion injury. Redox Biol. 2020, 36, 101607. [Google Scholar] [CrossRef]
- Shen, Y.; Liu, X.; Shi, J.; Wu, X. Involvement of Nrf2 in myocardial ischemia and reperfusion injury. Int. J. Biol. Macromol. 2019, 125, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Sun, W.; Zhang, Z.; Zheng, Y. The role of Nrf2-mediated pathway in cardiac remodeling and heart failure. Oxidative Med. Cell. Longev. 2014, 2014, 260429. [Google Scholar] [CrossRef] [Green Version]
- Bocci, V.; Valachi, G. Nrf2 activation as target to implement therapeutic treatments. Front. Chem. 2015, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Barzegar, M.; Kaur, G.; Wang, Y.; Boyer, C.J.; Alexander, J.S. Potential therapeutic roles of stem cells in ischemia-reperfusion injury. Stem Cells Res. 2019, 37, 101421. [Google Scholar] [CrossRef]
- Jeroudi, M.O.; Hartley, C.J.; Bolli, R. Myocardial reperfusion injury: Role of oxygen radicals and potential therapy with antioxidants. Am. J. Cardiol 1994, 73, 2B–7B. [Google Scholar] [CrossRef]
- Garcia-Dorado, D.; Rodriguez-Sinovas, A.; Ruiz-Meena, M.; Inserte, J. Protection against myocardial ischemia-reperfusion injury in clinical practice. Rev. Esp. Cardiol. 2014, 67, 394–404. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, Y.J.; Saini, H.K.; Turan, B.; Liu, P.P.; Dhalla, N.S. Pentoxifylline attenuates cardiac dysfunction and reduces TNF-α level in ischemic-reperfused heart. Am. J. Physiol. Heart. Circ. Physiol. 2005, 289, H832–H839. [Google Scholar] [CrossRef]
- Zhou, X.; Sheng, X.; Chen, M.; Wang, Z.; Yu, L.; Jiang, H. Tumor necrosis factor-α inhibitor: A promising therapeutic approach for attenuating myocardial ischemia-reperfusion by antioxidants stress. Int. J. Cardiol. 2015, 190, 282–283. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.B.; Dhalla, N.S. Ischemia-reperfusion-induced changes in sarcolemmal Na+-K+-ATPase are due to the activation of calpain in the heart. Can. J. Physiol. Pharmacol. 2010, 88, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.L.; Freed, D.; Dhalla, N.S. Activation of proteases and changes in Na+-K+-ATPase subunits in hearts subjected to ischemia-reperfusion. J. Appl. Physiol. 2013, 114, 351–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragasevic, N.; Jakovljevic, V.; Zivkovic, V.; Draginic, N.; Andjic, M.; Bolevich, S.; Jovic, S. The role of aldosterone inhibitors in cardiac ischemia-reperfusion injury. Can. J. Physiol. Pharmacol. 2021, 99, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Bell, R.M.; Botker, H.E.; Zuurbier, C.J. SGLT2 inhibitors reduce infarct size in reperfused ischemic heart and improve cardiac function during ischemic episodes in preclinical models. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165770. [Google Scholar] [CrossRef]
- Rout, A.; Tantry, U.S.; Novakovic, M.; Sukhi, A.; Gurbel, P.A. Targeted pharmacotherapy for ischemia reperfusion injury in acute myocardial infarction. Expert. Opin. Pharmacother. 2020, 21, 1851–1865. [Google Scholar] [CrossRef]
- Correa, F.; Martinez-Abundis, E.; Hernandez-Resendiz, S.; Garcia, N.; Buelna-Chontal, M.; Arreguin, F.; Zazueta, C. Pharmacological strategies to contend against myocardial reperfusion damage: Diverse chemicals for multiple targets. Curr. Med. Chem. 2010, 17, 2261–2273. [Google Scholar] [CrossRef]
- Kryzwonos-Zawadzka, A.; Franczak, A.; Sawicki, G.; Wozniak, M.; Bil-Lula, I. Multidrug prevention or therapy of ischemia-reperfusion injury of the heart—Mini-review. Environ. Toxicol. Pharmacol. 2017, 55, 55–59. [Google Scholar] [CrossRef]
- Wang, W.; Kang, P.M. Oxidative stress and antioxidant treatments in cardiovascular diseases. Antioxidants 2020, 9, 1292. [Google Scholar] [CrossRef]
- Hamilton, K.L. Antioxidants and cardioprotection. Med. Sci. Sports Exerc. 2007, 39, 1544–1553. [Google Scholar] [CrossRef]
- Braunersreuther, V.; Jaquet, V. Reactive oxygen species in myocardial reperfusion injury: From physiopathology to therapeutic approaches. Curr. Pharm. Biotechnol. 2012, 13, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, R.; Libuy, M.; Feliu, F.; Hasson, D. Molecular basis of cardioprotective effect of antioxidant vitamins in myocardial infarction. Biomed. Res. Int. 2013, 2013, 437613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.Y.; Xu, X.; Li, X.C. Cardiovascular diseases: Oxidative damage and antioxidant protection. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 3091–3096. [Google Scholar]
- Leopold, J.A. Antioxidants and coronary artery disease: From pathophysiology to preventive therapy. Coron. Artery. Dis. 2015, 26, 176–183. [Google Scholar] [CrossRef] [Green Version]
- Lai, L.N.; Zhang, X.J.; Zhang, X.Y.; Song, L.H.; Guo, C.H.; Lei, J.W.; Song, X.L. Lazaroid U83836E protects the heart against ischemia reperfusion injury via inhibition of oxidative stress and activation of PKC. Mol. Med. Rep. 2016, 13, 3993–4000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moya-Lillo, J.; Rojas-Sole, C.; Munoz-Salamanca, D.; Panieri, E.; Saso, L.; Rodrigo, R. Targeting ferrptosis against ischemia-reperfusion cardiac injury. Antioxidants 2021, 10, 667. [Google Scholar] [CrossRef] [PubMed]
- Boucher, F.R.; Jouan, M.G.; Moro, C.; Rakotavo, A.N.; Tanguy, S.; de Leiris, J. Does selenium exert cardioprotective effects against oxidative stress in myocardial ischemia? Acta Physiol. Hung. 2008, 95, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Venardos, K.M.; Perkins, A.; Headrick, J.; Kaye, D.M. Myocardial ischemia-reperfusion injury, antioxidant enzymes systems, and selenium: A review. Curr. Med. Chem. 2007, 14, 1539–1549. [Google Scholar] [CrossRef]
- Camara, A.K.S.; Bienengraeber, M.; Stowe, D.F. Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury. Front. Physiol. 2011, 2, 13. [Google Scholar] [CrossRef] [Green Version]
- Ramachandra, C.J.A.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Lin, Y.H.; Hausenloy, D.J. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine 2020, 57, 102884. [Google Scholar] [CrossRef]
- Lotz, C.; Herrmann, J.; Notz, Q.; Meybohm, P.; Kehl, F. Mitochondria and pharmacologic cardiac conditioning at the heart of ischemic injury. Int. J. Mol. Sci. 2021, 22, 3224. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Mitochondrial bioenergetics and cardiolipin alterations in myocardial ischemia-reperfusion injury: Implications for pharmacological cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1341–H1352. [Google Scholar] [CrossRef] [PubMed]
- Joseph, L.C.; Barca, E.; Subramanyam, P.; Komrowaski, M.; Pajvani, U.; Colecraft, H.M.; Hirano, M.; Morrow, J.P. Inhibition of NAPDH oxidase 2 (NOX2) prevents oxidative stress and mitochondrial abnormalities caused by saturated fat in cardiomyocytes. PLoS ONE 2016, 11, 145750. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Guan, Q.; Guo, L.; Zhang, H.; Pang, X.; Cheng, Y.; Zhang, X.; Sun, Y. Gypenosides alleviate myocardial ischemia-reperfusion injury via attenuation of oxidative stress and preservation of mitochondrial function in rat heart. Cell Stress Chaperones 2016, 21, 429–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penna, C.; Perrelli, M.G.; Pagliaro, P. Mitochondrial pathways, permeability transition pore, and redox signaling in cardioprotection: Therapeutic implications. Antioxid. Redox Signal. 2013, 18, 556–599. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Jang, S.; Parodi-Rullan, R.; Khuchua, Z.; Kuznetsov, A.V. Mitochondrial permeability transition in cardiac ischemia-reperfusion: Wether cyclophilin D is a viable target for cardioprotection? Cell. Mol. Life Sci. 2017, 74, 2795–2813. [Google Scholar] [CrossRef]
- Halladin, N.L. Oxidaitve and inflammatory biomarkers of ischemia and reperfusion injuries. Dan. Med. J. 2015, 62, B5054. [Google Scholar]
- Giacomo, C.G.; Antonio, M. Melatonin in cardiac ischemia/reperfusion-induced mitochondrial adaptive changes. Cardiovasc. Hematol. Disord. Drug Targets 2007, 7, 163–169. [Google Scholar] [CrossRef]
- Jiang, X.; Wu, D.; Jiang, Z.; Ling, W.; Qian, G. Protective effect of nicorandil on cardiac microvascular injury: Role of mitochondrial integrity. Oxidative Med. Cell. Longev. 2021, 2021, 4665632. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Golfman, L.; Takeda, S.; Takeda, N.; Nagano, M. Evidence for the role of oxidative stress in acute ischemic heart disease: A brief review. Can. J. Cardiol. 1999, 15, 587–593. [Google Scholar]
- Singh, R.B.; Hryshko, L.; Freed, D.; Dhalla, N.S. Activation of proteolytic enzymes and depression of the sarcolemma Na+-K+-ATPase in ischemia-reperfused heart may be mediated through oxidative stress. Can. J. Physiol. Pharmacol. 2012, 90, 249–260. [Google Scholar] [CrossRef]
- Dixon, I.M.C.; Kaneko, M.; Hata, T.; Panagia, V.; Dhalla, N.S. Alterations in cardiac membrane Ca2+ transport during oxidative stress. Mol. Cell. Biochem. 1990, 99, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Dhalla, N.S. Effect of oxygen free radicals on cardiac contractile activity and sarcolemmal Na+-Ca2+ exchange. J. Cardiovasc. Pharmacol. Ther. 1996, 1, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Dhalla, N.S. Relationship between mechanical dysfunction and depression of sarcolemmal Ca2+-pump activity in hearts perfused with oxygen free radicals. Mol. Cell. Biochem. 1996, 160/161, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Temsah, R.M.; Netticadan, T.; Chapman, D.; Takeda, S.; Mochizuki, S.; Dhalla, N.S. Alterations in sarcoplasmic reticulum function and gene expression in ischemic-reperfused rat heart. Am. J. Physiol. 1999, 277, H584–H594. [Google Scholar] [CrossRef]
- Makazan, Z.; Saini, H.K.; Dhalla, N.S. Role of oxidative stress in alterations of mitochondrial function in ischemic-reperfused hearts. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1986–H1994. [Google Scholar] [CrossRef]
- Maddika, S.; Elimban, V.; Chapman, D.; Dhalla, N.S. Role of oxidative stress in ischemia-reperfusion-induced alterations in myofibrillar ATPase activities and gene expression in the heart. Can. J. Physiol. Pharmacol. 2009, 87, 120–129. [Google Scholar] [CrossRef]
- Suzuki, S.; Kaneko, M.; Chapman, D.C.; Dhalla, N.S. Alterations in cardiac contractile proteins due to oxygen free radicals. Biochim. Biophys. Acta. 1991, 1074, 95–100. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Control | I/R | I/R + SOD Plus CAT |
|---|---|---|---|
| A. Cardiac function: | |||
| LV DP (mmHg) | 98 ± 3.6 | 40 ± 2.9 * | 72 ± 4.2 † |
| LV EDP (mmHg) | 6.2 ± 0.4 | 64 ± 4.1 * | 36 ± 3.1 † |
| LV + dP/dt (%) | 100 ± 4.2 | 44 ± 3.1 * | 80 ± 3.6 † |
| LV – dP/dt (%) | 100 ± 3.6 | 35 ± 2.4 * | 74 ± 3.0 † |
| B. Oxidative stress markers: | |||
| H2O2 content (nmol/g wet wt) | 8.4 ± 1.2 | 38.6 ± 3.9 * | 12.3 ± 1.5 † |
| MDA content (nmol/mg tissue lipids) | 3.8 ± 0.6 | 17.5 ± 3.1 * | 5.6 ± 0.8 † |
| C. Myocardial Ca2+: | |||
| Ca2+ content (µmol/g dry wt) | 8.4 ± 1.2 | 22.6 ± 2.9 * | 9.8 ± 1.6 † |
| Parameters | Left Ventricle Developed Pressure (mmHg) | Na+-K+ ATPase Activity ((µmol) Pi/mg/h) | Protease Activities (Relative Fluorescence Units) | |
|---|---|---|---|---|
| MMP Activity | Calpain Activity | |||
| A. I/R injury/antioxidants: | ||||
| Control | 119 ± 5.7 | 28.7 ± 3.8 | 50 ± 4.3 | 36 ± 3.1 |
| I/R | 44 ± 9.8 * | 10.9 ± 3.6 * | 525 ± 26.9 * | 592 ± 25.9 * |
| I/R + NAC | 114 ± 11.6 † | 32.5 ± 3.5 † | 163 ± 8.3 † | 215 ± 16.5 † |
| I/R + MGP | 121 ± 13.2 † | 33.2 ± 4.1 † | 152 ± 9.6 † | 240 ± 23.7 † |
| B. Oxidative stress: | ||||
| Control | 94 ± 7.9 | 26.9 ± 4.1 | 56 ± 3.9 | 40 ± 4.2 |
| X + XO | 40 ± 4.2 * | 7.6 ± 3.6 * | 608 ± 23.8 * | 600 ± 15.9 * |
| H2O2 | 55 ± 6.1 * | 6.8 ± 2.7 * | 450 ± 15.6 * | 665 ± 22.7 * |
| Parameters | Na+-Ca2+ Exchange (nmol Ca2+/mg/2 s) | ATP-Dependent Ca2+-Uptake (nmol Ca2+/mg/5 min) | Ca2+-Stimulated ATPase Activity (µmol Pi/mg/h) |
|---|---|---|---|
| A. I/R injury/oxyradical scavenger: | |||
| Control | 5.2 ± 0.31 | 23.4 ± 1.2 | 11.2 ± 0.7 |
| I/R | 3.1 ± 0.29 * | 9.7 ± 0.7 * | 4.4 ± 0.7 * |
| I/R + SOD plus CAT | 4.7 ± 0.21 † | 20.8 ± 1.1 † | 9.8 ± 0.6 † |
| B. Oxidative stress: | |||
| Control | 3.8 ± 0.15 | 24.4 ± 1.0 | 11.7 ± 1.0 |
| X + XO | 1.4 ± 0.20 * | 3.6 ± 1.2 * | 4.1 ± 0.9 * |
| X + XO + SOD plus CAT | 3.0 ± 0.33 † | 22.1 ± 1.4 † | 9.2 ± 1.2 † |
| Parameters | Left Ventricular Developed Pressure (mm Hg) | Ca2+-Uptake Activity (nmol/mg/min) | Ca2+-Stimulated ATPase Content (% of Control) | Ca2+-Release Activity (nmol/mg/15 s) | Ryanodine Binding (pmol/mg) |
|---|---|---|---|---|---|
| A. I/R injury/oxyradical scavenger: | |||||
| Control | 100 ± 5.2 | 24.7 ± 1.9 | 100 | 9.6 ± 1.5 | 2.4 ± 0.11 |
| I/R | 27 ± 2.8 * | 12.5 ± 1.3 * | 25 ± 1.9 * | 2.8 ± 0.3 * | 0.8 ± 0.02 * |
| I/R + SOD plus CAT | 86 ± 4.2 † | 22.4 ± 2.8 † | 20 ± 2.1 | 5.3 ± 0.6 † | 1.8 ± 0.09 † |
| B. Oxidative stress: | |||||
| Control | 100 ± 2.9 | 28.1 ± 0.7 | 100 | 10.1± 1.9 | 2.3 ± 0.10 |
| X + XO | 16 ± 1.8 * | 9.3 ± 0.8 * | 31 ± 1.4 * | 1.5 ± 0.1 * | 1.0 ± 0.05 * |
| H2O2 | 27 ± 0.9 * | 13.9 ± 1.4 * | 58 ± 3.8 * | 2.3 ± 0.1 * | 0.9 ± 0.04 * |
| Parameters | Left Ventricular Developed Pressure (mm Hg) | Left Ventricular end Diastolic Pressure (mm Hg) | State 3 Respiration (ng Atoms O/mg/min) | ADP to O Ratio (nmol ADP/ng atom O) |
|---|---|---|---|---|
| A. I/R injury/oxyradical scavenger: | ||||
| Control | 95 ± 7 | 8.6 ± 0.6 | 402 ± 12 | 2.94 ± 0.06 |
| I/R | 24 ± 2 * | 87 ± 5 * | 303 ± 15 * | 2.58 ± 0.05 * |
| I/R + SOD plus CAT | 60 ± 2 † | 40 ± 4 † | 403 ± 21 † | 2.80 ± 0.06 † |
| B. Oxidative stress: | ||||
| Control | 115 ± 11 | 10.5 ± 0.7 | 483 ± 11 | 2.79 ± 0.07 |
| X + XO | 14.6 ± 4.6 * | 128 ± 8 * | 264 ± 12 * | 2.48 ± 0.03 * |
| H2O2 | 28.2 ± 2.3 * | 35.7 ± 3.5 * | 403 ± 5 * | 2.50 ± 0.08 * |
| Parameters | Left Ventricular Developed Pressure (mm Hg) | Myofibrillar ATPase Activities (µmol Pi/mg/h) | |
|---|---|---|---|
| Mg2+-ATPase | Ca2+-Stimulated | ||
| A. I/R injury/oxyradical scavenger/antioxidant: | |||
| Control | 105 ± 20.3 | 3.5 ± 0.5 | 13.3 ± 0.3 |
| I/R | 36.4 ± 12.1 * | 4.0 ± 0.2 | 10.7 ± 0.4 * |
| I/R + SOD plus CAT | 71.5 ± 9.5 † | 3.1 ± 0.3 | 12.9 ± 0.2 † |
| I/R + NAC | 117 ± 14.4 † | 3.1 ± 0.1 | 13.9 ± 0.1 † |
| B. Oxidative stress: | |||
| Control | 115 ± 10.1 | 3.6 ± 0.1 | 12.7 ± 0.1 |
| X + XO | 31 ± 2.8 * | 10.7 ± 0.2 * | 6.9 ± 0.2 * |
| H2O2 | –––– | 5.5 ± 0.2 * | 10.9 ± 0.4 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhalla, N.S.; Shah, A.K.; Adameova, A.; Bartekova, M. Role of Oxidative Stress in Cardiac Dysfunction and Subcellular Defects Due to Ischemia-Reperfusion Injury. Biomedicines 2022, 10, 1473. https://doi.org/10.3390/biomedicines10071473
Dhalla NS, Shah AK, Adameova A, Bartekova M. Role of Oxidative Stress in Cardiac Dysfunction and Subcellular Defects Due to Ischemia-Reperfusion Injury. Biomedicines. 2022; 10(7):1473. https://doi.org/10.3390/biomedicines10071473
Chicago/Turabian StyleDhalla, Naranjan S., Anureet K. Shah, Adriana Adameova, and Monika Bartekova. 2022. "Role of Oxidative Stress in Cardiac Dysfunction and Subcellular Defects Due to Ischemia-Reperfusion Injury" Biomedicines 10, no. 7: 1473. https://doi.org/10.3390/biomedicines10071473
APA StyleDhalla, N. S., Shah, A. K., Adameova, A., & Bartekova, M. (2022). Role of Oxidative Stress in Cardiac Dysfunction and Subcellular Defects Due to Ischemia-Reperfusion Injury. Biomedicines, 10(7), 1473. https://doi.org/10.3390/biomedicines10071473