
Pathogenesis of Paradoxical Reactions Associated with Targeted Biologic Agents for Inflammatory Skin Diseases


Abstract
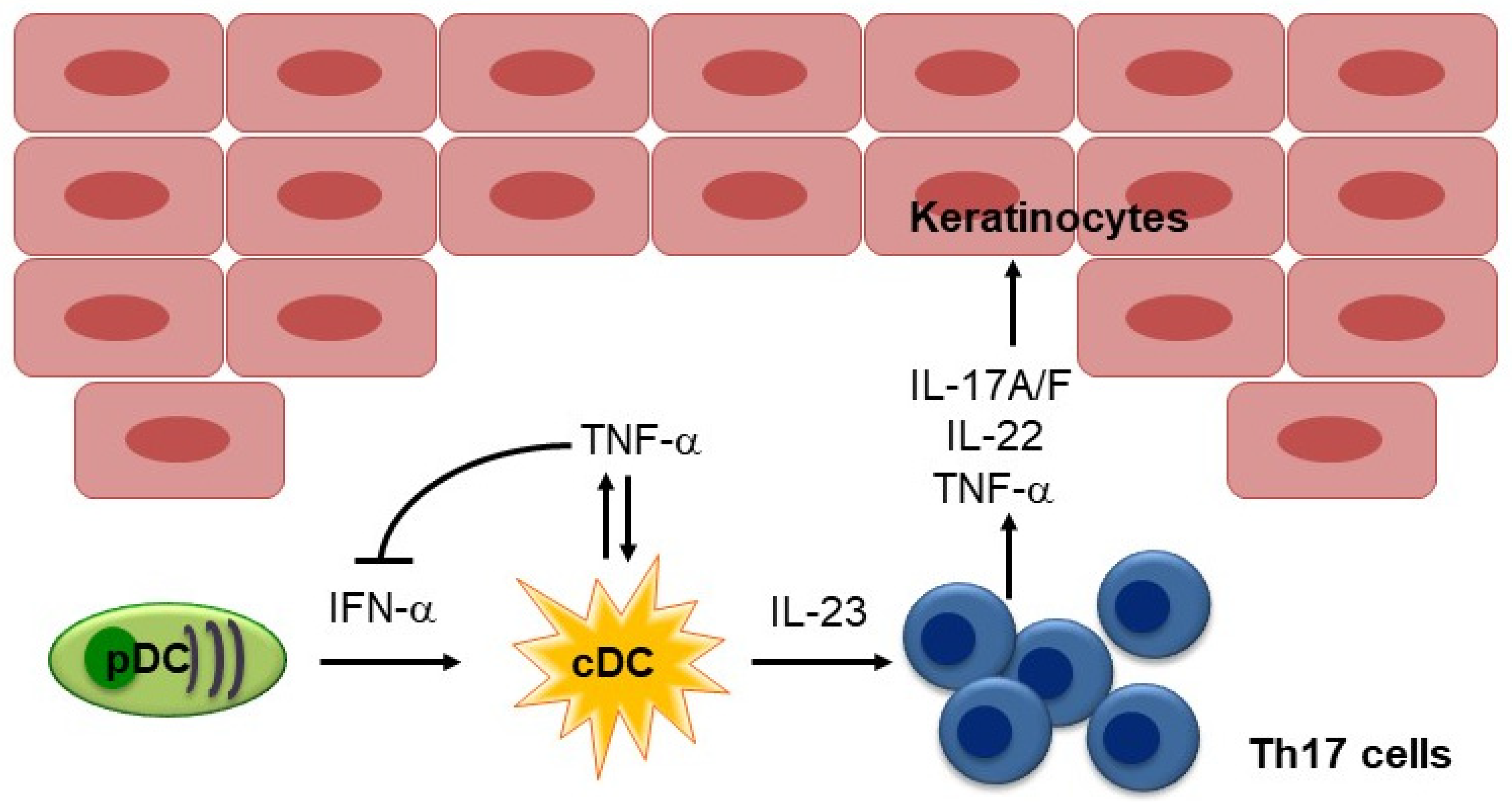
:1. Introduction
2. Signature Cytokines and Associated Molecular Pathways Involved in the Idiopathic Counterparts of PRs Caused by Biologic Agents
2.1. IL-17 and IL-23 in Psoriasis
2.2. IL-4, IL-13, and IL-5 in Atopic Dermatitis
2.3. Type I IFNs in Systemic Lupus Erythematosus
3. Paradoxical Reactions Caused by Each Type of Biologic Agent
3.1. TNF-α Inhibitors
3.1.1. Psoriasiform Reactions
Mechanism Responsible for Psoriasiform Reactions Induced by TNF-α Inhibitors
3.1.2. Eczematous Reactions
Mechanism Responsible for Eczematous Reactions Induced by TNF-α Inhibitors
3.1.3. Lupus-Like Reactions
Clinical Features of Lupus-like Reactions Induced by TNF-α Inhibitors
Mechanism Responsible for Lupus-like Reactions Induced by TNF-α Inhibitors
3.2. IL-12/23 p40 Inhibitors
3.2.1. Psoriasiform Reactions
3.2.2. Eczematous Reactions
3.2.3. Ustekinumab for Skin Reactions Associated with TNF-α Inhibitors
3.3. IL-17 Inhibitors
3.3.1. Psoriasiform Reactions
3.3.2. Eczematous Reactions
Mechanism Responsible for Eczematous Reactions Induced by IL-17 Inhibitors
3.4. IL-23p19 Inhibitors
3.4.1. Eczematous Reactions
Mechanism Responsible for Eczematous Reactions Induced by IL-23p19 Inhibitors
3.5. IL-4Rα Inhibitors
3.5.1. Psoriasiform Reactions
Mechanism Responsible for Psoriasiform Reactions Induced by IL-4Rα Inhibitors
3.5.2. Eczematous Reactions
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Murphy, M.J.; Cohen, J.M.; Vesely, M.D.; Damsky, W. Paradoxical eruptions to targeted therapies in dermatology: A systematic review and analysis. J. Am. Acad. Dermatol. 2020, 86, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Garcovich, S.; De Simone, C.; Genovese, G.; Berti, E.; Cugno, M.; Marzano, A.V. Paradoxical Skin Reactions to Biologics in Patients with Rheumatologic Disorders. Front. Pharmacol. 2019, 10, 282. [Google Scholar] [CrossRef] [PubMed]
- Pérez-De-Lis, M.; Retamozo, S.; Flores-Chávez, A.; Kostov, B.; Perez-Alvarez, R.; Brito-Zerón, P.; Ramos-Casals, M. Autoimmune diseases induced by biological agents. A review of 12,731 cases (BIOGEAS Registry). Expert Opin. Drug Saf. 2017, 16, 1255–1271. [Google Scholar] [CrossRef] [PubMed]
- Eyerich, K.; Eyerich, S. Immune response patterns in non-communicable inflammatory skin diseases. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; McInnes, I.B.; Neurath, M.F. Reframing Immune-Mediated Inflammatory Diseases through Signature Cytokine Hubs. N. Engl. J. Med. 2021, 385, 628–639. [Google Scholar] [CrossRef]
- Braegelmann, C.; Niebel, D.; Wenzel, J. Targeted Therapies in Autoimmune Skin Diseases. J. Investig. Dermatol. 2022, 142, 969–975.e7. [Google Scholar] [CrossRef]
- Annunziato, F.; Romagnani, C.; Romagnani, S. The 3 major types of innate and adaptive cell-mediated effector immunity. J. Allergy Clin. Immunol. 2015, 135, 626–635. [Google Scholar] [CrossRef]
- Wenzel, J.; Tuting, T. An IFN-associated cytotoxic cellular immune response against viral, self-, or tumor antigens is a common pathogenetic feature in “interface dermatitis”. J. Investig. Dermatol. 2008, 128, 2392–2402. [Google Scholar] [CrossRef] [Green Version]
- Eyerich, S.; Onken, A.T.; Weidinger, S.; Franke, A.; Nasorri, F.; Pennino, D.; Grosber, M.; Pfab, F.; Schmidt-Weber, C.B.; Mempel, M.; et al. Mutual Antagonism of T Cells Causing Psoriasis and Atopic Eczema. N. Engl. J. Med. 2011, 365, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Hawkes, J.E.; Chan, T.C.; Krueger, J.G. Psoriasis pathogenesis and the development of novel targeted immune therapies. J. Allergy Clin. Immunol. 2017, 140, 645–653. [Google Scholar] [CrossRef] [Green Version]
- Lowes, M.A.; Kikuchi, T.; Fuentes-Duculan, J.; Cardinale, I.; Zaba, L.C.; Haider, A.S.; Bowman, E.P.; Krueger, J.G. Psoriasis Vulgaris Lesions Contain Discrete Populations of Th1 and Th17 T Cells. J. Investig. Dermatol. 2008, 128, 1207–1211. [Google Scholar] [CrossRef]
- Croxford, A.L.; Karbach, S.; Kurschus, F.C.; Wörtge, S.; Nikolaev, A.; Yogev, N.; Klebow, S.; Schüler, R.; Reissig, S.; Piotrowski, C.; et al. IL-6 Regulates Neutrophil Microabscess Formation in IL-17A-Driven Psoriasiform Lesions. J. Investig. Dermatol. 2014, 134, 728–735. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Trepicchio, W.L.; Oestreicher, J.L.; Pittman, D.; Wang, F.; Chamian, F.; Dhodapkar, M.; Krueger, J.G. Increased Expression of Interleukin 23 p19 and p40 in Lesional Skin of Patients with Psoriasis Vulgaris. J. Exp. Med. 2004, 199, 125–130. [Google Scholar] [CrossRef]
- Piskin, G.; Sylva-Steenland, R.M.R.; Bos, J.D.; Teunissen, M.B.M. In Vitro and In Situ Expression of IL-23 by Keratinocytes in Healthy Skin and Psoriasis Lesions: Enhanced Expression in Psoriatic Skin. J. Immunol. 2006, 176, 1908–1915. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deshpande, M.; Grisotto, M.; Smaldini, P.; Garcia, R.; He, Z.; Gulko, P.S.; Lira, S.A.; Furtado, G.C. Skin expression of IL-23 drives the development of psoriasis and psoriatic arthritis in mice. Sci. Rep. 2020, 10, 8259. [Google Scholar] [CrossRef]
- Lowes, M.A.; Chamian, F.; Abello, M.V.; Fuentes-Duculan, J.; Lin, S.-L.; Nussbaum, R.; Novitskaya, I.; Carbonaro, H.; Cardinale, I.; Kikuchi, T.; et al. Increase in TNF-α and inducible nitric oxide synthase-expressing dendritic cells in psoriasis and reduction with efalizumab (anti-CD11a). Proc. Natl. Acad. Sci. USA 2005, 102, 19057–19062. [Google Scholar] [CrossRef] [Green Version]
- Boyman, O.; Hefti, H.P.; Conrad, C.; Nickoloff, B.J.; Suter, M.; Nestle, F.O. Spontaneous Development of Psoriasis in a New Animal Model Shows an Essential Role for Resident T Cells and Tumor Necrosis Factor-α. J. Exp. Med. 2004, 199, 731–736. [Google Scholar] [CrossRef] [Green Version]
- Nestle, F.O.; Conrad, C.; Tun-Kyi, A.; Homey, B.; Gombert, M.; Boyman, O.; Burg, G.; Liu, Y.-J.; Gilliet, M. Plasmacytoid predendritic cells initiate psoriasis through interferon-α production. J. Exp. Med. 2005, 202, 135–143. [Google Scholar] [CrossRef]
- Chiricozzi, A.; Guttman-Yassky, E.; Suárez-Fariñas, M.; Nograles, K.E.; Tian, S.; Cardinale, I.; Chimenti, S.; Krueger, J.G. Integrative Responses to IL-17 and TNF-α in Human Keratinocytes Account for Key Inflammatory Pathogenic Circuits in Psoriasis. J. Investig. Dermatol. 2011, 131, 677–687. [Google Scholar] [CrossRef]
- Hamid, Q.; Boguniewicz, M.; Leung, D.Y. Differential in situ cytokine gene expression in acute versus chronic atopic dermatitis. J. Clin. Investig. 1994, 94, 870–876. [Google Scholar] [CrossRef]
- Lee, G.R.; Flavell, R.A. Transgenic mice which overproduce Th2 cytokines develop spontaneous atopic dermatitis and asthma. Int. Immunol. 2004, 16, 1155–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, L.S.; Robinson, N.; Xu, L. Expression of Interleukin-4 in the Epidermis of Transgenic Mice Results in a Pruritic Inflammatory Skin Disease: An Experimental Animal Model to Study Atopic Dermatitis. J. Investig. Dermatol. 2001, 117, 977–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, T.; Oh, M.H.; Oh, S.Y.; Schroeder, J.T.; Glick, A.B.; Zhu, Z. Transgenic Expression of Interleukin-13 in the Skin Induces a Pruritic Dermatitis and Skin Remodeling. J. Investig. Dermatol. 2009, 129, 742–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gittler, J.K.; Shemer, A.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Gulewicz, K.J.; Wang, C.Q.; Mitsui, H.; Cardinale, I.; Strong, C.D.G.; Krueger, J.G.; et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J. Allergy Clin. Immunol. 2012, 130, 1344–1354. [Google Scholar] [CrossRef] [Green Version]
- Banchereau, J.; Pascual, V. Type I Interferon in Systemic Lupus Erythematosus and Other Autoimmune Diseases. Immunity 2006, 25, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Hooks, J.J.; Moutsopoulos, H.M.; Geis, S.A.; Stahl, N.I.; Decker, J.L.; Notkins, A.L. Immune Interferon in the Circulation of Patients with Autoimmune Disease. N. Engl. J. Med. 1979, 301, 5–8. [Google Scholar] [CrossRef]
- Kirou, K.A.; Lee, C.; George, S.; Louca, K.; Peterson, M.G.E.; Crow, M.K. Activation of the interferon-α pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. 2005, 52, 1491–1503. [Google Scholar] [CrossRef]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef] [Green Version]
- Bennett, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and Granulopoiesis Signatures in Systemic Lupus Erythematosus Blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Rönnblom, L.E.; Alm, G.V.; Öberg, K.E. Possible induction of systemic lupus erythematosus by interferon-α treatment in a patient with a malignant carcinoid tumour. J. Intern. Med. 1990, 227, 207–210. [Google Scholar] [CrossRef]
- Niewold, T.B.; Swedler, W.I. Systemic lupus erythematosus arising during interferon-alpha therapy for cryoglobulinemic vasculitis associated with hepatitis C. Clin. Rheumatol. 2005, 24, 178–181. [Google Scholar] [CrossRef]
- Baccala, R.; Gonzalez-Quintial, R.; Blasius, A.L.; Rimann, I.; Ozato, K.; Kono, D.H.; Beutler, B.; Theofilopoulos, A.N. Essential requirement for IRF8 and SLC15A4 implicates plasmacytoid dendritic cells in the pathogenesis of lupus. Proc. Natl. Acad. Sci. USA 2013, 110, 2940–2945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lövgren, T.; Eloranta, M.-L.; Båve, U.; Alm, G.V.; Rönnblom, L. Induction of interferon-α production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 2004, 50, 1861–1872. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Raber, M.-L.; Baccala, R.; Haraldsson, K.M.; Choubey, D.; Stewart, T.A.; Kono, D.H.; Theofilopoulos, A.N. Type-I Interferon Receptor Deficiency Reduces Lupus-like Disease in NZB Mice. J. Exp. Med. 2003, 197, 777–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyagawa, F.; Tagaya, Y.; Ozato, K.; Asada, H. Essential Requirement for IFN Regulatory Factor 7 in Autoantibody Production but Not Development of Nephritis in Murine Lupus. J. Immunol. 2016, 197, 2167–2176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyagawa, F.; Tagaya, Y.; Ozato, K.; Horie, K.; Asada, H. Inflammatory monocyte-derived dendritic cells mediate autoimmunity in murine model of systemic lupus erythematosus. J. Transl. Autoimmun. 2020, 3, 100060. [Google Scholar] [CrossRef]
- Pasadyn, S.R.; Knabel, D.; Fernandez, A.P.; Warren, C.B. Cutaneous adverse effects of biologic medications. Cleve. Clin. J. Med. 2020, 87, 288–299. [Google Scholar] [CrossRef]
- Moustou, A.-E.; Matekovits, A.; Dessinioti, C.; Antoniou, C.; Sfikakis, P.P.; Stratigos, A.J. Cutaneous side effects of anti–tumor necrosis factor biologic therapy: A clinical review. J. Am. Acad. Dermatol. 2009, 61, 486–504. [Google Scholar] [CrossRef]
- Hawkes, J.E.; Yan, B.Y.; Chan, T.C.; Krueger, J.G. Discovery of the IL-23/IL-17 Signaling Pathway and the Treatment of Psoriasis. J. Immunol. 2018, 201, 1605–1613. [Google Scholar] [CrossRef]
- Zaba, L.C.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Nograles, K.E.; Guttman-Yassky, E.; Cardinale, I.; Lowes, M.A.; Krueger, J.G. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J. Allergy Clin. Immunol. 2009, 124, 1022–1030.e395. [Google Scholar] [CrossRef] [Green Version]
- Baeten, D.; Kruithof, E.; Van den Bosch, F.; Van den Bossche, N.; Herssens, A.; Mielants, H.; De Keyser, F.; Veys, E.M. Systematic safety follow up in a cohort of 107 patients with spondyloarthropathy treated with infliximab: A new perspective on the role of host defence in the pathogenesis of the disease? Ann. Rheum. Dis. 2003, 62, 829–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verea, M.M.; Del Pozo, J.; Yebra-Pimentel, M.T.; Porta, A.; Fonseca, E. Psoriasiform Eruption Induced by Infliximab. Ann. Pharmacother. 2004, 38, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Thurber, M.; Feasel, A.; Stroehlein, J.; Hymes, S.R. Pustular psoriasis induced by infliximab. J. Drugs Dermatol. 2004, 3, 439–440. [Google Scholar] [PubMed]
- Beuthien, W.; Mellinghoff, H.-U.; Von Kempis, J. Skin reaction to adalimumab. Arthritis Rheum. 2004, 50, 1690–1692. [Google Scholar] [CrossRef] [PubMed]
- Dereure, O.; Guillot, B.; Jorgensen, C.; Cohen, J.D.; Combes, B.; Guilhou, J.J. Psoriatic lesions induced by antitumour necrosis factor-alpha treatment: Two cases. Br. J. Dermatol. 2004, 151, 506–507. [Google Scholar] [CrossRef]
- Collamer, A.N.; Battafarano, D. Psoriatic Skin Lesions Induced by Tumor Necrosis Factor Antagonist Therapy: Clinical Features and Possible Immunopathogenesis. Semin. Arthritis Rheum. 2010, 40, 233–240. [Google Scholar] [CrossRef]
- Conrad, C.; Di Domizio, J.; Mylonas, A.; Belkhodja, C.; DeMaria, O.; Navarini, A.A.; Lapointe, A.-K.; French, L.E.; Vernez, M.; Gilliet, M. TNF blockade induces a dysregulated type I interferon response without autoimmunity in paradoxical psoriasis. Nat. Commun. 2018, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Harrison, M.J.; Dixon, W.G.; Watson, K.D.; King, Y.; Groves, R.; Hyrich, K.L.; Symmons, D.P.M. Rates of new-onset psoriasis in patients with rheumatoid arthritis receiving anti-tumour necrosis factor α therapy: Results from the British Society for Rheumatology Biologics Register. Ann. Rheum. Dis. 2009, 68, 209–215. [Google Scholar] [CrossRef] [Green Version]
- George, L.A.; Gadani, A.; Cross, R.K.; Jambaulikar, G.; Ghazi, L.J. Psoriasiform Skin Lesions Are Caused by Anti-TNF Agents Used for the Treatment of Inflammatory Bowel Disease. Dig. Dis. Sci. 2015, 60, 3424–3430. [Google Scholar] [CrossRef]
- Weizman, A.V.; Sharma, R.; Afzal, N.M.; Xu, W.; Walsh, S.; Stempak, J.M.; Nguyen, G.C.; Croitoru, K.; Steinhart, A.H.; Silverberg, M.S. Stricturing and Fistulizing Crohn’s Disease Is Associated with Anti-tumor Necrosis Factor-Induced Psoriasis in Patients with Inflammatory Bowel Disease. Dig. Dis. Sci. 2018, 63, 2430–2438. [Google Scholar] [CrossRef]
- Palucka, A.K.; Blanck, J.-P.; Bennett, L.; Pascual, V.; Banchereau, J. Cross-regulation of TNF and IFN-α in autoimmune diseases. Proc. Natl. Acad. Sci. USA 2005, 102, 3372–3377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Gannes, G.C.; Ghoreishi, M.; Pope, J.; Russell, A.; Bell, D.; Adams, S.; Shojania, K.; Martinka, M.; Dutz, J.P. Psoriasis and pustular dermatitis triggered by TNF-{alpha} inhibitors in patients with rheumatologic conditions. Arch Dermatol. 2007, 143, 223–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bissonnette, R.; Papp, K.; Maari, C.; Yao, Y.; Robbie, G.; White, W.I.; Le, C.; White, B. A randomized, double-blind, placebo-controlled, phase I study of MEDI-545, an anti-interferon-alfa monoclonal antibody, in subjects with chronic psoriasis. J. Am. Acad. Dermatol. 2010, 62, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.C. Atopic dermatitis-like eruption precipitated by infliximab. J. Am. Acad. Dermatol. 2003, 49, 160–161. [Google Scholar] [CrossRef]
- Flendrie, M.; Vissers, W.H.; Creemers, M.C.; de Jong, E.M.; van de Kerkhof, P.C.; van Riel, P.L. Dermatological conditions during TNF-alpha-blocking therapy in patients with rheumatoid arthritis: A prospective study. Arthritis Res. Ther. 2005, 7, R666–R676. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-H.; Song, I.-H.; Friedrich, M.; Gauliard, A.; Detert, J.; Röwert, J.; Audring, H.; Kary, S.; Burmester, G.-R.; Sterry, W.; et al. Cutaneous side-effects in patients with rheumatic diseases during application of tumour necrosis factor-α antagonists. Br. J. Dermatol. 2007, 156, 486–491. [Google Scholar] [CrossRef]
- Nakamura, M.; Lee, K.; Singh, R.; Zhu, T.H.; Farahnik, B.; Abrouk, M.; Koo, J.; Bhutani, T. Eczema as an adverse effect of anti-TNFα therapy in psoriasis and other Th1-mediated diseases: A review. J. Dermatol. Treat. 2017, 28, 237–241. [Google Scholar] [CrossRef]
- Zaba, L.C.; Cardinale, I.; Gilleaudeau, P.; Sullivan-Whalen, M.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Novitskaya, I.; Khatcherian, A.; Bluth, M.J.; Lowes, M.A.; et al. Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J. Exp. Med. 2007, 204, 3183–3194. [Google Scholar] [CrossRef]
- Quaglino, P.; Bergallo, M.; Ponti, R.; Barberio, E.; Cicchelli, S.; Buffa, E.; Comessatti, A.; Costa, C.; Terlizzi, M.; Astegiano, S.; et al. Th1, Th2, Th17 and Regulatory T Cell Pattern in Psoriatic Patients: Modulation of Cytokines and Gene Targets Induced by Etanercept Treatment and Correlation with Clinical Response. Dermatology 2011, 223, 57–67. [Google Scholar] [CrossRef]
- Malisiewicz, B.; Murer, C.; Schmid, J.P.; French, L.E.; Schmid-Grendelmeier, P.; Navarini, A.A. Eosinophilia during psoriasis treatment with TNF antagonists. Dermatology 2011, 223, 311–315. [Google Scholar] [CrossRef] [Green Version]
- Ghoreschi, K.; Thomas, P.; Breit, S.; Dugas, M.; Mailhammer, R.; Van Eden, W.; Van Der Zee, R.; Biedermann, T.; Prinz, J.; Mack, M.; et al. Interleukin-4 therapy of psoriasis induces Th2 responses and improves human autoimmune disease. Nat. Med. 2003, 9, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, E.; Maier, H.; Riedl, E.; Brüggen, M.C.; Reininger, B.; Schaschinger, M.; Bangert, C.; Guenova, E.; Stingl, G.; Brunner, P.M. Analysis of anti-tumour necrosis factor-induced skin lesions reveals strong T helper 1 activation with some distinct immunological characteristics. Br. J. Dermatol. 2018, 178, 1151–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariyasu, T.; Tanaka, T.; Fujioka, N.; Yanai, Y.; Yamamoto, S.; Yamauchi, H.; Ikegami, H.; Ikeda, M.; Kurimoto, M. Effects of interferon-alpha subtypes on the TH1/TH2 balance in peripheral blood mononuclear cells from patients with hepatitis virus infection-associated liver disorders. In Vitro Cell. Dev. Biol. Anim. 2005, 41, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Esmailzadeh, A.; Yousefi, P.; Farhi, D.; Bachmeyer, C.; Cosnes, J.; Berenbaum, F.; Duriez, P.; Aractingi, S.; Khosrotehrani, K. Predictive Factors of Eczema-Like Eruptions among Patients without Cutaneous Psoriasis Receiving Infliximab: A Cohort Study of 92 Patients. Dermatology 2009, 219, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Takase, K.; Horton, S.C.; Ganesha, A.; Das, S.; McHugh, A.; Emery, P.; Savic, S.; Buch, M.H. What is the utility of routine ANA testing in predicting development of biological DMARD-induced lupus and vasculitis in patients with rheumatoid arthritis? Data from a single-centre cohort. Ann. Rheum. Dis. 2014, 73, 1695–1699. [Google Scholar] [CrossRef] [PubMed]
- Benucci, M.; Saviola, G.; Baiardi, P.; Cammelli, E.; Manfredi, M. Anti-nucleosome antibodies as prediction factor of development of autoantibodies during therapy with three different TNFα blocking agents in rheumatoid arthritis. Clin. Rheumatol. 2008, 27, 91–95. [Google Scholar] [CrossRef]
- Moulis, G.; Sommet, A.; Lapeyre-Mestre, M.; Montastruc, J.-L. Is the risk of tumour necrosis factor inhibitor-induced lupus or lupus-like syndrome the same with monoclonal antibodies and soluble receptor? A case/non-case study in a nationwide pharmacovigilance database. Rheumatology 2014, 53, 1864–1871. [Google Scholar] [CrossRef] [Green Version]
- Jani, M.; Dixon, W.G.; Kersley-Fleet, L.; Bruce, I.N.; Chinoy, H.; Barton, A.; Lunt, M.; Watson, K.; Symmons, D.P.; Hyrich, K.L.; et al. Drug-specific risk and characteristics of lupus and vasculitis-like events in patients with rheumatoid arthritis treated with TNFi: Results from BSRBR-RA. RMD Open 2017, 3, e000314. [Google Scholar] [CrossRef] [Green Version]
- Vedove, C.D.; Del Giglio, M.; Schena, D.; Girolomoni, G. Drug-induced lupus erythematosus. Arch. Dermatol. Res. 2009, 301, 99–105. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Brito-Zerón, P.; Muñoz, S.; Soria, N.; Galiana, D.; Bertolaccini, L.; Maria-Jose, C.; Munther, A.K. Autoimmune diseases induced by TNF-targeted therapies: Analysis of 233 cases. Medicine 2007, 86, 242–251. [Google Scholar] [CrossRef]
- Jacob, C.O.; McDevitt, H.O. Tumour necrosis factor-α in murine autoimmune ‘lupus’ nephritis. Nature 1988, 331, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.O.; Fronek, Z.; Lewis, G.D.; Koo, M.; Hansen, J.A.; McDevitt, H.O. Heritable major histocompatibility complex class II-associated differences in production of tumor necrosis factor alpha: Relevance to genetic predisposition to systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 1990, 87, 1233–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Via, C.S.; Shustov, A.; Rus, V.; Lang, T.; Nguyen, P.; Finkelman, F.D. In vivo neutralization of TNF-alpha promotes humoral autoimmunity by preventing the induction of CTL. J. Immunol. 2001, 167, 6821–6826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamanakos, A.; Vergou, T.; Panopoulos, S.; Tektonidou, M.G.; Stratigos, A.J.; Sfikakis, P.P. Psoriasis as an adverse reaction to biologic agents beyond anti-TNF-α therapy. Eur. J. Dermatol. 2021, 31, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Pernet, C.; Guillot, B.; Bessis, D. Eczematous Drug Eruption after Ustekinumab Treatment. Arch. Dermatol. 2012, 148, 959–960. [Google Scholar] [CrossRef]
- Al-Janabi, A.; Foulkes, A.; Mason, K.; Smith, C.; Griffiths, C.; Warren, R. Phenotypic switch to eczema in patients receiving biologics for plaque psoriasis: A systematic review. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 1440–1448. [Google Scholar] [CrossRef]
- Ezzedine, K.; Visseaux, L.; Cadiot, G.; Brixi, H.; Bernard, P.; Reguiai, Z. Ustekinumab for skin reactions associated with anti-tumor necrosis factor-α agents in patients with inflammatory bowel diseases: A single-center retrospective study. J. Dermatol. 2019, 46, 322–327. [Google Scholar] [CrossRef]
- Wu, J.; Smogorzewski, J. Ustekinumab for the treatment of paradoxical skin reactions and cutaneous manifestations of inflammatory bowel diseases. Dermatol. Ther. 2021, 34, e14883. [Google Scholar] [CrossRef]
- Teraki, Y.; Takahashi, A.; Inoue, Y.; Takamura, S. Eyelid Dermatitis as a Side Effect of Interleukin-17A Inhibitors in Psoriasis. Acta Derm. Venereol. 2018, 98, 456–457. [Google Scholar] [CrossRef] [Green Version]
- Burlando, M.; Cozzani, E.; Russo, R.; Parodi, A. Atopic-like dermatitis after secukinumab injection: A case report. Dermatol. Ther. 2019, 32, e12751. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, M.; Megna, M.; Fabbrocini, G.; Nisticò, S.; Balato, N.; Dastoli, S.; Patruno, C. Eczematous eruption during anti-interleukin 17 treatment of psoriasis: An emerging condition. Br. J. Dermatol. 2019, 181, 604–606. [Google Scholar] [CrossRef] [PubMed]
- Caldarola, G.; Pirro, F.; Di Stefani, A.; Talamonti, M.; Galluzzo, M.; D’Adamio, S.; Magnano, M.; Bernardini, N.; Malagoli, P.; Bardazzi, F.; et al. Clinical and histopathological characterization of eczematous eruptions occurring in course of anti IL-17 treatment: A case series and review of the literature. Expert Opin. Biol. Ther. 2020, 20, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Danset, M.; Hacard, F.; Jaulent, C.; Nosbaum, A.; Berard, F.; Nicolas, J.-F.; Goujon, C. Brodalumab-associated generalized eczematous eruption in a difficult-to-treat psoriasis patient: Management without brodalumab withdrawal. Eur. J. Dermatol. 2020, 30, 741–743. [Google Scholar] [CrossRef] [PubMed]
- Brembilla, N.C.; Senra, L.; Boehncke, W.-H. The IL-17 Family of Cytokines in Psoriasis: IL-17A and Beyond. Front. Immunol. 2018, 9, 1682. [Google Scholar] [CrossRef] [Green Version]
- Crowley, J.; Warren, R.; Cather, J. Safety of selective IL -23p19 inhibitors for the treatment of psoriasis. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 1676–1684. [Google Scholar] [CrossRef] [Green Version]
- Reyn, B.; Gils, A.; Hillary, T. Eczematous eruption after guselkumab treatment for psoriasis. JAAD Case Rep. 2019, 5, 973–975. [Google Scholar] [CrossRef]
- Truong, A.; Le, S.; Kiuru, M.; Maverakis, E. Nummular dermatitis on guselkumab for palmoplantar psoriasis. Dermatol. Ther. 2019, 32, e12954. [Google Scholar] [CrossRef]
- Miyagawa, F.; Fukuda, K.; Mori, A.; Ogawa, K.; Asada, H. Recurrence of secukinumab-induced eczematous eruptions after guselkumab treatment for pustular psoriasis. J. Dermatol. 2021, 48, E498–E499. [Google Scholar] [CrossRef]
- Kromer, C.; Schön, M.P.; Mössner, R. Eczematous eruption in patients with psoriasis during risankizumab treatment. Eur. J. Dermatol. 2020, 30, 599–601. [Google Scholar] [CrossRef]
- Narla, S.; Silverberg, J.I.; Simpson, E.L. Management of inadequate response and adverse effects to dupilumab in atopic dermatitis. J. Am. Acad. Dermatol. 2022, 86, 628–636. [Google Scholar] [CrossRef]
- Beck, L.A.; Thaçi, D.; Hamilton, J.D.; Graham, N.M.; Bieber, T.; Rocklin, R.; Ming, J.E.; Ren, H.; Kao, R.; Simpson, E.; et al. Dupilumab Treatment in Adults with Moderate-to-Severe Atopic Dermatitis. N. Engl. J. Med. 2014, 371, 130–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, E.L.; Bieber, T.; Guttman-Yassky, E.; Beck, L.A.; Blauvelt, A.; Cork, M.J.; Silverberg, J.I.; Deleuran, M.; Kataoka, Y.; Lacour, J.-P.; et al. Two Phase 3 Trials of Dupilumab versus Placebo in Atopic Dermatitis. N. Engl. J. Med. 2016, 375, 2335–2348. [Google Scholar] [CrossRef] [PubMed]
- Tracey, E.H.; Elston, C.; Feasel, P.; Piliang, M.; Michael, M.; Vij, A. Erythrodermic presentation of psoriasis in a patient treated with dupilumab. JAAD Case Rep. 2018, 4, 708–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safa, G.; Paumier, V. Psoriasis induced by dupilumab therapy. Clin. Exp. Dermatol. 2019, 44, e49–e50. [Google Scholar] [CrossRef]
- Gori, N.; Caldarola, G.; Pirro, F.; De Simone, C.; Peris, K. A case of guttate psoriasis during treatment with dupilumab. Dermatol. Ther. 2019, 32, e12998. [Google Scholar] [CrossRef]
- Stout, M.; Guitart, J.; Tan, T.; Silverberg, J.I. Psoriasis-like Dermatitis Developing in a Patient with Atopic Dermatitis Treated with Dupilumab. Dermatitis 2019, 30, 376–378. [Google Scholar] [CrossRef]
- Napolitano, M.; Scalvenzi, M.; Fabbrocini, G.; Cinelli, E.; Patruno, C. Occurrence of psoriasiform eruption during dupilumab therapy for adult atopic dermatitis: A case series. Dermatol. Ther. 2019, 32, e13142. [Google Scholar] [CrossRef]
- Napolitano, M.; Ferrillo, M.; Patruno, C.; Scalvenzi, M.; D’Andrea, M.; Fabbrocini, G. Efficacy and Safety of Dupilumab in Clinical Practice: One Year of Experience on 165 Adult Patients from a Tertiary Referral Centre. Dermatol. Ther. 2021, 11, 355–361. [Google Scholar] [CrossRef]
- Jaulent, L.; Staumont-Sallé, D.; Tauber, M.; Paul, C.; Aubert, H.; Marchetti, A.; Sassolas, B.; Valois, A.; Nicolas, J.; Nosbaum, A.; et al. De novo psoriasis in atopic dermatitis patients treated with dupilumab: A retrospective cohort. J. Eur. Acad. Dermatol. Venereol. 2021, 35, e296–e297. [Google Scholar] [CrossRef]
- Brumfiel, C.M.; Patel, M.H.; Zirwas, M.J. Development of psoriasis during treatment with dupilumab: A systematic review. J. Am. Acad. Dermatol. 2022, 86, 708–709. [Google Scholar] [CrossRef]
- Guenova, E.; Skabytska, Y.; Hoetzenecker, W.; Weindl, G.; Sauer, K.; Tham, M.; Kim, K.W.; Park, J.H.; Seo, J.H.; Ignatova, D.; et al. IL-4 abrogates T(H)17 cell-mediated inflammation by selective silencing of IL-23 in antigen-presenting cells. Proc. Natl. Acad. Sci. USA 2015, 112, 2163–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, M.; Caiazzo, G.; Fabbrocini, G.; Balato, A.; Di Caprio, R.; Scala, E.; Scalvenzi, M.; Patruno, C. Increased expression of interleukin-23A in lesional skin of patients with atopic dermatitis with psoriasiform reaction during dupilumab treatment. Br. J. Dermatol. 2021, 184, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Mirza, F.; Wang, A.; Ramachandran, S.; Damsky, W.; Cohen, J. Dupilumab-induced phenotype switch from atopic dermatitis to psoriasis is characterized by de novo interleukin-17A expression: A case report. Br. J. Dermatol. 2021, 185, 432–434. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Chen, J.K.; Chiou, A.; Ko, J.; Honari, G. Assessment of the Development of New Regional Dermatoses in Patients Treated for Atopic Dermatitis with Dupilumab. JAMA Dermatol. 2019, 155, 850–852. [Google Scholar] [CrossRef]
- Soria, A.; Du-Thanh, A.; Seneschal, J.; Jachiet, M.; Staumont-Sallé, D.; Barbarot, S.; GREAT Research Group. Development or Exacerbation of Head and Neck Dermatitis in Patients Treated for Atopic Dermatitis with Dupilumab. JAMA Dermatol. 2019, 155, 1312–1315. [Google Scholar] [CrossRef]
- Buhl, T.; Sulk, M.; Nowak, P.; Buddenkotte, J.; McDonald, I.; Aubert, J.; Carlavan, I.; Deret, S.; Reiniche, P.; Rivier, M.; et al. Molecular and Morphological Characterization of Inflammatory Infiltrate in Rosacea Reveals Activation of Th1/Th17 Pathways. J. Investig. Dermatol. 2015, 135, 2198–2208. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Indications | Class | Medication | Description |
|---|---|---|---|
| Psoriasis | TNF-α inhibitors | Infliximab | chimeric mAb to TNF-α |
| Adalimumab | fully human mAb to TNF-α | ||
| Certolizumab pegol | humanized mAb to TNF-α | ||
| Etanercept | recombinant human TNF-R/IgGFc | ||
| IL-17 inhibitors | Secukinumab | fully human mAb to IL-17A | |
| Brodalumab | fully human mAb to IL-17RA | ||
| Ixekizumab | humanized mAb to IL-17A | ||
| IL-12/23p40 inhibitor | Ustekinumab | fully human mAb to IL-12/23p40 | |
| IL-23p19 inhibitors | Guselkumab | fully human mAb to IL-23p19 | |
| Risankizumab | humanized mAb to IL-23p19 | ||
| Tildrakizumab | humanized mAb to IL-23p19 | ||
| AD | IL-4Rα inhibitor | Dupilumab | fully human mAb to IL-4Rα |
| Pt No. | Author | Age | Sex | Clinical Description (Duration) | Biologic | Time of Onset | Previous Atopy | Histology | Clinical Course |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Reyn et al. (2019) | 47 | M | Psoriasis vulgaris (NR) | Guselkumab | 10 w | AD | Acanthosis, spongiosis, lymphocytic inflammatory infiltrates mixed with eosinophils | Guselkumab discontinued; Eczema resolved with tar preparation |
| 2 | Truong et al. (2019) | 40 | M | Pustular psoriasis (since childhood) | Guselkumab | 3 m | NR | Psoriasiform epidermal hyperplasia, parakeratosis, spongiosis, perivascular lymphohistiocytic infiltrates | NR |
| 3 | Miyagawa et al. (2021) | 75 | M | Pustular psoriasis (13 years) | Guselkumab | 3 m | None | Parakeratosis, spongiosis, perivascular inflammatory infiltrates consisting of lymphocytes and eosinophils | Switched from secukinumab due to eczematous eruptions; Guselkumab continued; Treated with topical corticosteroids; Eczema persisted |
| 4 | Kromer et al. (2020) | 52 | M | Psoriasis vulgaris (4 years) | Risankizumab | 3 w | Allergic rhinitis | Acanthosis, compact orthokeratosis with foci of parakeratosis, spongiosis, perivascular lymphocytic infiltrates | Guselkumab continued; Improved after treatment with topical corticosteroids |
| 5 | as above | 59 | M | Psoriasis vulgaris (26 years) | Risankizumab | 4 w | Allergic rhinitis, Asthma | NR | Switched to ustekinumab; Eczema improved |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyagawa, F. Pathogenesis of Paradoxical Reactions Associated with Targeted Biologic Agents for Inflammatory Skin Diseases. Biomedicines 2022, 10, 1485. https://doi.org/10.3390/biomedicines10071485
Miyagawa F. Pathogenesis of Paradoxical Reactions Associated with Targeted Biologic Agents for Inflammatory Skin Diseases. Biomedicines. 2022; 10(7):1485. https://doi.org/10.3390/biomedicines10071485
Chicago/Turabian StyleMiyagawa, Fumi. 2022. "Pathogenesis of Paradoxical Reactions Associated with Targeted Biologic Agents for Inflammatory Skin Diseases" Biomedicines 10, no. 7: 1485. https://doi.org/10.3390/biomedicines10071485
APA StyleMiyagawa, F. (2022). Pathogenesis of Paradoxical Reactions Associated with Targeted Biologic Agents for Inflammatory Skin Diseases. Biomedicines, 10(7), 1485. https://doi.org/10.3390/biomedicines10071485